
Lipoprotein Proteomics and Aortic Valve Transcriptomics Identify Biological Pathways Linking Lipoprotein(a) Levels to Aortic Stenosis

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results
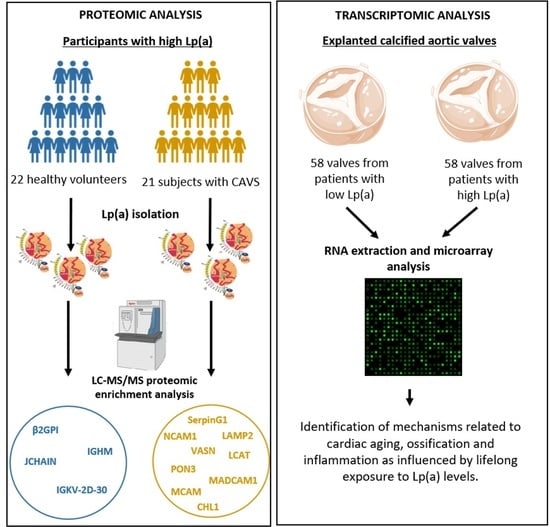
2.1. Proteomic Analysis of Lp(a) Proteome from Patients with Versus without CAVS
2.2. Transcriptomic Analysis of Explanted Calcified Valves from Patients with CAVS
3. Discussion
4. Materials and Methods
4.1. Study Participants
4.2. Lipoprotein Isolation
4.3. Assessment of Lp(a) Proteome by Nanolc-MS/MS
4.4. Transcriptomic Analysis of Explanted Calcified Valves from Patients with CAVS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nkomo, V.T.; Gardin, J.M.; Skelton, T.N.; Gottdiener, J.S.; Scott, C.G.; Enriquez-Sarano, M. Burden of valvular heart diseases: A population-based study. Lancet 2006, 368, 1005–1011. [Google Scholar] [CrossRef]
- Vahanian, A.; Alfieri, O.; Andreotti, F.; Antunes, M.J.; Baron-Esquivias, G.; Baumgartner, H.; Borger, M.A.; Carrel, T.P.; De Bonis, M.; Evangelista, A.; et al. Guidelines on the management of valvular heart disease (version 2012): The Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur. J. Cardiothorac. Surg. 2012, 42, S1–S44. [Google Scholar]
- Eveborn, G.W.; Schirmer, H.; Heggelund, G.; Lunde, P.; Rasmussen, K. The evolving epidemiology of valvular aortic stenosis. the Tromso study. Heart 2013, 99, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Helske, S.; Kupari, M.; Lindstedt, K.A.; Kovanen, P.T. Aortic valve stenosis: An active atheroinflammatory process. Curr. Opin. Lipidol. 2007, 18, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll. Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Katz, R.; Wong, N.D.; Kronmal, R.; Takasu, J.; Shavelle, D.M.; Probstfield, J.L.; Bertoni, A.G.; Budoff, M.J.; O’Brien, K.D. Features of the metabolic syndrome and diabetes mellitus as predictors of aortic valve calcification in the Multi-Ethnic Study of Atherosclerosis. Circulation 2006, 113, 2113–2119. [Google Scholar] [CrossRef] [Green Version]
- Aronow, W.S.; Schwartz, K.S.; Koenigsberg, M. Correlation of serum lipids, calcium, and phosphorus, diabetes mellitus and history of systemic hypertension with presence or absence of calcified or thickened aortic cusps or root in elderly patients. Am. J. Cardiol. 1987, 59, 998–999. [Google Scholar] [CrossRef]
- Mohler, E.R.; Sheridan, M.J.; Nichols, R.; Harvey, W.P.; Waller, B.F. Development and progression of aortic valve stenosis: Atherosclerosis risk factors—A causal relationship? A clinical morphologic study. Clin. Cardiol. 1991, 14, 995–999. [Google Scholar] [CrossRef]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, B.J.; Boekholdt, S.M.; Dube, M.P.; Rheaume, E.; Wareham, N.J.; Khaw, K.T.; Sandhu, M.S.; Tardif, J.C. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: A prospective Mendelian randomization study and replication in a case-control cohort. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olkowicz, M.; Debski, J.; Jablonska, P.; Dadlez, M.; Smolenski, R.T. Application of a new procedure for liquid chromatography/mass spectrometry profiling of plasma amino acid-related metabolites and untargeted shotgun proteomics to identify mechanisms and biomarkers of calcific aortic stenosis. J. Chromatogr. A 2017, 1517, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Gil-Dones, F.; Darde, V.M.; Alonso-Orgaz, S.; Lopez-Almodovar, L.F.; Mourino-Alvarez, L.; Padial, L.R.; Vivanco, F.; Barderas, M.G. Inside human aortic stenosis: A proteomic analysis of plasma. J. Proteom. 2012, 75, 1639–1653. [Google Scholar] [CrossRef]
- Ljungberg, J.; Janiec, M.; Bergdahl, I.A.; Holmgren, A.; Hultdin, J.; Johansson, B.; Naslund, U.; Siegbahn, A.; Fall, T.; Soderberg, S. Proteomic Biomarkers for Incident Aortic Stenosis Requiring Valvular Replacement. Circulation 2018, 138, 590–599. [Google Scholar] [CrossRef]
- Schlotter, F.; Halu, A.; Goto, S.; Blaser, M.C.; Body, S.C.; Lee, L.H.; Higashi, H.; DeLaughter, D.M.; Hutcheson, J.D.; Vyas, P.; et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation 2018, 138, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Martin-Rojas, T.; Gil-Dones, F.; Lopez-Almodovar, L.F.; Padial, L.R.; Vivanco, F.; Barderas, M.G. Proteomic profile of human aortic stenosis: Insights into the degenerative process. J. Proteome Res. 2012, 11, 1537–1550. [Google Scholar] [CrossRef]
- Martin-Rojas, T.; Mourino-Alvarez, L.; Alonso-Orgaz, S.; Rosello-Lleti, E.; Calvo, E.; Lopez-Almodovar, L.F.; Rivera, M.; Padial, L.R.; Lopez, J.A.; de la Cuesta, F.; et al. iTRAQ proteomic analysis of extracellular matrix remodeling in aortic valve disease. Sci. Rep. 2015, 5, 17290. [Google Scholar] [CrossRef] [Green Version]
- Heuschkel, M.A.; Skenteris, N.T.; Hutcheson, J.D.; van der Valk, D.D.; Bremer, J.; Goody, P.; Hjortnaes, J.; Jansen, F.; Bouten, C.V.C.; van den Bogaerdt, A.; et al. Integrative Multi-Omics Analysis in Calcific Aortic Valve Disease Reveals a Link to the Formation of Amyloid-Like Deposits. Cells 2020, 9, 2164. [Google Scholar] [CrossRef] [PubMed]
- Bosse, Y.; Miqdad, A.; Fournier, D.; Pepin, A.; Pibarot, P.; Mathieu, P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ. Cardiovasc. Genet. 2009, 2, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Helgadottir, A.; Thorleifsson, G.; Gretarsdottir, S.; Stefansson, O.A.; Tragante, V.; Thorolfsdottir, R.B.; Jonsdottir, I.; Bjornsson, T.; Steinthorsdottir, V.; Verweij, N.; et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat. Commun. 2018, 9, 987. [Google Scholar] [CrossRef] [Green Version]
- Theriault, S.; Gaudreault, N.; Lamontagne, M.; Rosa, M.; Boulanger, M.C.; Messika-Zeitoun, D.; Clavel, M.A.; Capoulade, R.; Dagenais, F.; Pibarot, P.; et al. A transcriptome-wide association study identifies PALMD as a susceptibility gene for calcific aortic valve stenosis. Nat. Commun. 2018, 9, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein (a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [Green Version]
- van der Hoek, Y.Y.; Wittekoek, M.E.; Beisiegel, U.; Kastelein, J.J.; Koschinsky, M.L. The apolipoprotein(a) kringle IV repeats which differ from the major repeat kringle are present in variably-sized isoforms. Hum. Mol. Genet. 1993, 2, 361–366. [Google Scholar] [CrossRef]
- Erqou, S.; Thompson, A.; Di Angelantonio, E.; Saleheen, D.; Kaptoge, S.; Marcovina, S.; Danesh, J. Apolipoprotein(a) isoforms and the risk of vascular disease: Systematic review of 40 studies involving 58,000 participants. J. Am. Coll. Cardiol. 2010, 55, 2160–2167. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Witztum, J.L. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr. Opin. Lipidol. 2008, 19, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Mohty, D.; Pibarot, P.; Despres, J.P.; Cote, C.; Arsenault, B.; Cartier, A.; Cosnay, P.; Couture, C.; Mathieu, P. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Hafiane, A.; Thanassoulis, G.; Ott, L.; Filwood, N.; Cerruti, M.; Gourgas, O.; Shum-Tim, D.; Al Kindi, H.; de Varennes, B.; et al. Lipoprotein(a) Induces Human Aortic Valve Interstitial Cell Calcification. JACC Basic Transl. Sci. 2017, 2, 358–371. [Google Scholar] [CrossRef]
- Torzewski, M.; Ravandi, A.; Yeang, C.; Edel, A.; Bhindi, R.; Kath, S.; Twardowski, L.; Schmid, J.; Yang, X.; Franke, U.F.W.; et al. Lipoprotein(a) Associated Molecules are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2017, 2, 229–240. [Google Scholar] [CrossRef]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bosse, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lepine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin Derived From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef]
- Nsaibia, M.J.; Mahmut, A.; Boulanger, M.C.; Arsenault, B.J.; Bouchareb, R.; Simard, S.; Witztum, J.L.; Clavel, M.A.; Pibarot, P.; Bosse, Y.; et al. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J. Intern. Med. 2016, 280, 509–517. [Google Scholar] [CrossRef]
- Von Zychlinski, A.; Kleffmann, T.; Williams, M.J.; McCormick, S.P. Proteomics of Lipoprotein(a) identifies a protein complement associated with response to wounding. J. Proteom. 2011, 74, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, R.; Girard, A.; Perrot, N.; Guertin, J.; Mitchell, P.L.; Couture, C.; Gotti, C.; Bourassa, S.; Poggio, P.; Mass, E.; et al. A Comparative Analysis of the Lipoprotein(a) and Low-Density Lipoprotein Proteomic Profiles Combining Mass Spectrometry and Mendelian Randomization. CJC Open 2020, in press. [Google Scholar]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Pinero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutierrez-Sacristan, A.; Deu-Pons, J.; Centeno, E.; Garcia-Garcia, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Ikeda, Y.; Imai, Y.; Kumagai, H.; Nosaka, T.; Morikawa, Y.; Hisaoka, T.; Manabe, I.; Maemura, K.; Nakaoka, T.; Imamura, T.; et al. Vasorin, a transforming growth factor beta-binding protein expressed in vascular smooth muscle cells, modulates the arterial response to injury in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 10732–10737. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, A.L.; Chaussain, C.; Broutin, I.; Rochefort, G.Y.; Schrewe, H.; Gaucher, C. From Vascular Smooth Muscle Cells to Folliculogenesis: What About Vasorin? Front Med. 2018, 5, 335. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.A.; Petrosino, J.M.; Manring, H.R.; Wright, P.; Shettigar, V.; Kilic, A.; Janssen, P.M.L.; Ziolo, M.T.; Accornero, F. TGF-beta1 affects cell-cell adhesion in the heart in an NCAM1-dependent mechanism. J. Mol. Cell Cardiol. 2017, 112, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Ono, K.; Iwanaga, Y.; Tamaki, Y.; Kojima, Y.; Horie, T.; Nishi, H.; Kinoshita, M.; Kuwabara, Y.; Hasegawa, K.; et al. Neural cell adhesion molecule is a cardioprotective factor up-regulated by metabolic stress. J. Mol. Cell Cardiol. 2010, 48, 1157–1168. [Google Scholar] [CrossRef] [Green Version]
- Bardin, N.; Anfosso, F.; Masse, J.M.; Cramer, E.; Sabatier, F.; Le Bivic, A.; Sampol, J.; Dignat-George, F. Identification of CD146 as a component of the endothelial junction involved in the control of cell-cell cohesion. Blood 2001, 98, 3677–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardin, N.; Blot-Chabaud, M.; Despoix, N.; Kebir, A.; Harhouri, K.; Arsanto, J.P.; Espinosa, L.; Perrin, P.; Robert, S.; Vely, F.; et al. CD146 and its soluble form regulate monocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 746–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebir, A.; Harhouri, K.; Guillet, B.; Liu, J.W.; Foucault-Bertaud, A.; Lamy, E.; Kaspi, E.; Elganfoud, N.; Vely, F.; Sabatier, F.; et al. CD146 short isoform increases the proangiogenic potential of endothelial progenitor cells in vitro and in vivo. Circ. Res. 2010, 107, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Duan, H.; Qian, Y.; Feng, L.; Wu, Z.; Wang, F.; Feng, J.; Yang, D.; Qin, Z.; Yan, X. Macrophagic CD146 promotes foAm. cell formation and retention during atherosclerosis. Cell Res. 2017, 27, 352–372. [Google Scholar] [CrossRef] [Green Version]
- Connor, E.M.; Eppihimer, M.J.; Morise, Z.; Granger, D.N.; Grisham, M.B. Expression of mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in acute and chronic inflammation. J. Leukoc. Biol. 1999, 65, 349–355. [Google Scholar] [CrossRef]
- Hillenbrand, R.; Molthagen, M.; Montag, D.; Schachner, M. The close homologue of the neural adhesion molecule L1 (CHL1): Patterns of expression and promotion of neurite outgrowth by heterophilic interactions. Eur. J. Neurosci. 1999, 11, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Holm, J.; Hillenbrand, R.; Steuber, V.; Bartsch, U.; Moos, M.; Lubbert, H.; Montag, D.; Schachner, M. Structural featuRes. of a close homologue of L1 (CHL1) in the mouse: A new member of the L1 family of neural recognition molecules. Eur. J. Neurosci. 1996, 8, 1613–1629. [Google Scholar] [CrossRef]
- Frints, S.G.; Marynen, P.; Hartmann, D.; Fryns, J.P.; Steyaert, J.; Schachner, M.; Rolf, B.; Craessaerts, K.; Snellinx, A.; Hollanders, K.; et al. CALL interrupted in a patient with non-specific mental retardation: Gene dosage-dependent alteration of murine brain development and behavior. Hum. Mol. Genet. 2003, 12, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Migita, O.; Toru, M.; Arinami, T. An association between a missense polymorphism in the close homologue of L1 (CHL1, CALL) gene and schizophrenia. Mol. Psychiatry 2002, 7, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; Von Figura, K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Jansen, I.D.C.; Tigchelaar-Gutter, W.; Hogervorst, J.M.A.; de Vries, T.J.; Saftig, P.; Everts, V. LAMP-2 Is Involved in Surface Expression of RANKL of Osteoblasts In Vitro. Int. J. Mol. Sci. 2020, 21, 6110. [Google Scholar] [CrossRef]
- Mavrogeni, S.; Markousis-Mavrogenis, G.; Markussis, V.; Kolovou, G. The Emerging Role of Cardiovascular Magnetic Resonance Imaging in the Evaluation of Metabolic Cardiomyopathies. Horm. Metab. Res. 2015, 47, 623–632. [Google Scholar] [CrossRef]
- Shagdarsuren, E.; Bidzhekov, K.; Djalali-Talab, Y.; Liehn, E.A.; Hristov, M.; Matthijsen, R.A.; Buurman, W.A.; Zernecke, A.; Weber, C. C1-esterase inhibitor protects against neointima formation after arterial injury in atherosclerosis-prone mice. Circulation 2008, 117, 70–78. [Google Scholar] [CrossRef] [Green Version]
- ter Weeme, M.; Vonk, A.B.; Kupreishvili, K.; van Ham, M.; Zeerleder, S.; Wouters, D.; Stooker, W.; Eijsman, L.; Van Hinsbergh, V.W.; Krijnen, P.A.; et al. Activated complement is more extensively present in diseased aortic valves than naturally occurring complement inhibitors: A sign of ongoing inflammation. Eur. J. Clin. Investig. 2010, 40, 4–10. [Google Scholar] [CrossRef]
- Saeedi, R.; Li, M.; Frohlich, J. A review on lecithin:cholesterol acyltransferase deficiency. Clin. Biochem. 2015, 48, 472–475. [Google Scholar] [CrossRef]
- Holleboom, A.G.; Kuivenhoven, J.A.; Vergeer, M.; Hovingh, G.K.; van Miert, J.N.; Wareham, N.J.; Kastelein, J.J.; Khaw, K.T.; Boekholdt, S.M. Plasma levels of lecithin:cholesterol acyltransferase and risk of future coronary artery disease in apparently healthy men and women: A prospective case-control analysis nested in the EPIC-Norfolk population study. J. Lipid Res. 2010, 51, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Tani, S.; Takahashi, A.; Nagao, K.; Hirayama, A. Association of lecithin-cholesterol acyltransferase activity measured as a serum cholesterol esterification rate and low-density lipoprotein heterogeneity with cardiovascular risk: A cross-sectional study. Heart Vessels 2016, 31, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Priyanka, K.; Singh, S.; Gill, K. Paraoxonase 3: Structure and Its Role in Pathophysiology of Coronary Artery Disease. Biomolecules 2019, 9, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Melnichenko, A.A.; Orekhov, A.N.; Bobryshev, Y.V. Paraoxonase and atherosclerosis-related cardiovascular diseases. Biochimie 2017, 132, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, Y.; Schaefer, E.J.; Brewer, H.B., Jr. Activation of human post heparin lipoprotein lipase by apolipoprotein H (beta 2-glycoprotein I). Biochem. Biophys. Res. Commun. 1980, 95, 1168–1172. [Google Scholar] [CrossRef]
- Kaltoft, M.; Langsted, A.; Nordestgaard, B.G. Triglycerides and remnant cholesterol associated with risk of aortic valve stenosis: Mendelian randomization in the Copenhagen General Population Study. Eur. Heart J. 2020, 41, 2288–2299. [Google Scholar] [CrossRef]
- Hasunuma, Y.; Matsuura, E.; Makita, Z.; Katahira, T.; Nishi, S.; Koike, T. Involvement of beta 2-glycoprotein I and anticardiolipin antibodies in oxidatively modified low-density lipoprotein uptake by macrophages. Clin. Exp. Immunol. 1997, 107, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Kochl, S.; Fresser, F.; Lobentanz, E.; Baier, G.; Utermann, G. Novel interaction of apolipoprotein(a) with beta-2 glycoprotein I mediated by the kringle IV domain. Blood 1997, 90, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Chonn, A.; Semple, S.C.; Cullis, P.R. Beta 2 glycoprotein I is a major protein associated with very rapidly cleared liposomes in vivo, suggesting a significant role in the immune clearance of “non-self” particles. J. Biol. Chem. 1995, 270, 25845–25849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zioncheck, T.F.; Powell, L.M.; Rice, G.C.; Eaton, D.L.; Lawn, R.M. Interaction of recombinant apolipoprotein(a) and lipoprotein(a) with macrophages. J. Clin. Investig. 1991, 87, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Sanghera, D.K.; Wagenknecht, D.R.; McIntyre, J.A.; Kamboh, M.I. Identification of structural mutations in the fifth domain of apolipoprotein H (beta 2-glycoprotein I) which affect phospholipid binding. Hum. Mol. Genet. 1997, 6, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Bosse, Y.; Feitosa, M.F.; Despres, J.P.; Lamarche, B.; Rice, T.; Rao, D.C.; Bouchard, C.; Perusse, L.; Vohl, M.C. Detection of a major gene effect for LDL peak particle diameter and association with apolipoprotein H gene haplotype. Atherosclerosis 2005, 182, 231–239. [Google Scholar] [CrossRef]
- Hoekstra, M.; Chen, H.Y.; Rong, J.; Dufresne, L.; Yao, J.; Guo, X.; Tsai, M.Y.; Tsimikas, S.; Post, W.S.; Vasan, R.S.; et al. Genome-Wide Association Study Highlights APOH as a Novel Locus for Lipoprotein(a) Levels-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 458–464. [Google Scholar]
- Ehrenstein, M.R.; Notley, C.A. The importance of natural IgM: Scavenger, protector and regulator. Nat. Rev. Immunol. 2010, 10, 778–786. [Google Scholar] [CrossRef]
- Sjoberg, B.G.; Su, J.; Dahlbom, I.; Gronlund, H.; Wikstrom, M.; Hedblad, B.; Berglund, G.; de Faire, U.; Frostegard, J. Low levels of IgM antibodies against phosphorylcholine-A potential risk marker for ischemic stroke in men. Atherosclerosis 2009, 203, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Brilakis, E.S.; Lennon, R.J.; Miller, E.R.; Witztum, J.L.; McConnell, J.P.; Kornman, K.S.; Berger, P.B. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J. Lipid Res. 2007, 48, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, P.X.; Goodyear, C.S.; Chang, M.K.; Witztum, J.L.; Silverman, G.J. The autoreactivity of anti-phosphorylcholine antibodies for atherosclerosis-associated neo-antigens and apoptotic cells. J. Immunol. 2003, 170, 6151–6157. [Google Scholar] [CrossRef] [Green Version]
- Tuominen, A.; Miller, Y.I.; Hansen, L.F.; Kesaniemi, Y.A.; Witztum, J.L.; Horkko, S. A natural antibody to oxidized cardiolipin binds to oxidized low-density lipoprotein, apoptotic cells, and atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2096–2102. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kondo, H.; Hojo, Y. Granzyme B as a novel factor involved in cardiovascular diseases. J. Cardiol. 2011, 57, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohukainen, P.; Napakangas, J.; Ohtonen, P.; Ruskoaho, H.; Taskinen, P.; Peltonen, T.; Rysa, J. Expression and Localization of Granzymes and Perforin in Human Calcific Aortic Valve Disease. J. Heart Valve Dis. 2015, 24, 612–620. [Google Scholar]
- Hendel, A.; Cooper, D.; Abraham, T.; Zhao, H.; Allard, M.F.; Granville, D.J. Proteinase inhibitor 9 is reduced in human atherosclerotic lesion development. Cardiovasc. Pathol. 2012, 21, 28–38. [Google Scholar] [CrossRef]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. High lipoprotein(a) and high risk of mortality. Eur. Heart J. 2019, 40, 2760–2770. [Google Scholar] [CrossRef]
- Zenin, A.; Tsepilov, Y.; Sharapov, S.; Getmantsev, E.; Menshikov, L.I.; Fedichev, P.O.; Aulchenko, Y. Identification of 12 genetic loci associated with human healthspan. Commun. Biol. 2019, 2, 41. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, B.J.; Pelletier, W.; Kaiser, Y.; Perrot, N.; Couture, C.; Khaw, K.T.; Wareham, N.J.; Bosse, Y.; Pibarot, P.; Stroes, E.S.G.; et al. Association of Long-term Exposure to Elevated Lipoprotein(a) Levels With Parental Life Span, Chronic Disease-Free Survival, and Mortality Risk: A Mendelian Randomization Analysis. JAMA Netw. Open 2020, 3, e200129. [Google Scholar] [CrossRef] [Green Version]
- Timmers, P.R.; Mounier, N.; Lall, K.; Fischer, K.; Ning, Z.; Feng, X.; Bretherick, A.D.; Clark, D.W.; eQTLGen Consortium; Shen, X.; et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. Elife 2019, 8, e39856. [Google Scholar] [CrossRef]
- Despres, A.A.; Perrot, N.; Poulin, A.; Tastet, L.; Shen, M.; Chen, H.Y.; Bourgeois, R.; Trottier, M.; Tessier, M.; Guimond, J.; et al. Lipoprotein(a), Oxidized Phospholipids, and Aortic Valve Microcalcification Assessed by 18F-Sodium Fluoride Positron Emission Tomography and Computed Tomography. CJC Open 2019, 1, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnuolo, R.; Marcovina, S.M.; Boffa, M.B.; Koschinsky, M.L. Inhibition of plasminogen activation by apo(a): Role of carboxyl-terminal lysines and identification of inhibitory domains in apo(a). J. Lipid Res. 2014, 55, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, R.; Devillers, R.; Perrot, N.; Despres, A.A.; Boulanger, M.C.; Mitchell, P.L.; Guertin, J.; Couture, P.; Boffa, M.B.; Scipione, C.A.; et al. Interaction of Autotaxin With Lipoprotein(a) in Patients With Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2020, 5, 888–897. [Google Scholar] [CrossRef]
- Theriault, S.; Dina, C.; Messika-Zeitoun, D.; Le Scouarnec, S.; Capoulade, R.; Gaudreault, N.; Rigade, S.; Li, Z.; Simonet, F.; Lamontagne, M.; et al. Genetic Association Analyses Highlight IL6, ALPL, and NAV1 As 3 New Susceptibility Genes Underlying Calcific Aortic Valve Stenosis. Circ. Genom. Precis Med. 2019, 12, e002617. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | Control Lp(a) (n = 22) | CAVS Lp(a) (n = 21) | p-Value |
|---|---|---|---|
| Age | 61.2 ± 15.1 | 71.1 ± 4.4 | <0.0001 |
| Men, % (n) | 68.2 (15) | 57.1 (12) | 0.454 |
| Smoking, %, (n) | 42.8 (9) | 66.7 (14) | 0.121 |
| BMI, kg/m2 | 27.6 ± 5.3 | 30.3 ± 5.8 | 0.056 |
| Diabetes, %, (n) | 40.9 (9) | 42.9(9) | 0.897 |
| Blood pressure, mmHg | |||
| Systolic | 129 ± 30.9 | 133.6 ± 18.7 | 0.601 |
| Diastolic | 77.8 ± 19.5 | 72.4 ± 9.8 | 0.148 |
| Cardiovascular disease, %, (n) | 54.5 (12) | 57.1 (12) | 0.864 |
| Statin use, %, (n) | 59.1 (13) | 85.7 (18) | 0.052 |
| Lipoprotein(a), nmol/L | 202.1 ± 84 | 198.3 ± 81.8 | 0.759 |
| Gene | Protein | CAVS/Control Ratio | Limma p-Value |
|---|---|---|---|
| LCAT | lecithin–cholesterol acyltransferase | 3.4007 | 0.0029 |
| NCAM1 | neural cell adhesion molecule 1 | 2.9069 | 0.0070 |
| VASN | Vasorin | 3.4835 | 0.0072 |
| β2GPI | beta-2-glycoprotein 1 | 0.4060 | 0.0087 |
| PON3 | paraoxonase 3 | 2.3675 | 0.0108 |
| SERPING1 | serpin family G member 1 | 2.0398 | 0.0109 |
| LAMP2 | lysosomal-associated membrane protein 2 | 2.1678 | 0.0270 |
| IGHM | immunoglobulin heavy constant mu | 0.2424 | 0.0285 |
| IGKV2D-30 | immunoglobulin kappa variable 2D-30 | 0.3026 | 0.0343 |
| MCAM | melanoma cell adhesion molecule | 2.9838 | 0.0356 |
| MADCAM1 | mucosal vascular addressin cell adhesion molecule 1 | 2.4532 | 0.0392 |
| CHL1 | cell adhesion molecule L1-like | 1.7736 | 0.0427 |
| JCHAIN;IGJ | joining chain of multimeric IgA and IgM | 0.5021 | 0.0479 |
| Clinical Characteristics | Low Lp(a) (n = 59) | High Lp(a) (n = 59) | p-Value |
|---|---|---|---|
| Age, years | 73.0 ± 6.6 | 72.8 ± 6.9 | 0.85 |
| Men, % (n) | 61.0 (36) | 61.0 (36) | 1 |
| Smoking, %, (n) | 5.1 (3) | 5.1 (3) | 1 |
| BMI, kg/m2 | 29.9 ± 5.1 | 30.4 ± 6.4 | 0.61 |
| Diabetes, %, (n) | 45.8 (27) | 37.3 (22) | 0.35 |
| Blood pressure, mmHg | |||
| Systolic | 134.2 ± 19.8 | 134.5 ± 20.4 | 0.71 |
| Diastolic | 73.1 ± 9.8 | 73.1 ± 9.6 | 0.51 |
| Cardiovascular disease, % (n) | 57.6 (34) | 74.6 (44) | 0.051 |
| NYHA severity, %, (n) | |||
| 1 | 13.6 (8) | 18.6 (11) | |
| 2 | 47.5 (28) | 50.8 (30) | |
| 3 | 37.3 (22) | 27.1 (16) | |
| 4 | 1.7 (1) | 3.4 (2) | |
| Statin use, %, (n) | 83.1 (49) | 84.7 (49) | 1 |
| Lipoprotein(a), nmol/L | 29.2 ± 28.6 | 217.6 ± 93.1 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourgeois, R.; Bourgault, J.; Despres, A.-A.; Perrot, N.; Guertin, J.; Girard, A.; Mitchell, P.L.; Gotti, C.; Bourassa, S.; Scipione, C.A.; et al. Lipoprotein Proteomics and Aortic Valve Transcriptomics Identify Biological Pathways Linking Lipoprotein(a) Levels to Aortic Stenosis. Metabolites 2021, 11, 459. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11070459
Bourgeois R, Bourgault J, Despres A-A, Perrot N, Guertin J, Girard A, Mitchell PL, Gotti C, Bourassa S, Scipione CA, et al. Lipoprotein Proteomics and Aortic Valve Transcriptomics Identify Biological Pathways Linking Lipoprotein(a) Levels to Aortic Stenosis. Metabolites. 2021; 11(7):459. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11070459
Chicago/Turabian StyleBourgeois, Raphaëlle, Jérôme Bourgault, Audrey-Anne Despres, Nicolas Perrot, Jakie Guertin, Arnaud Girard, Patricia L. Mitchell, Clarisse Gotti, Sylvie Bourassa, Corey A. Scipione, and et al. 2021. "Lipoprotein Proteomics and Aortic Valve Transcriptomics Identify Biological Pathways Linking Lipoprotein(a) Levels to Aortic Stenosis" Metabolites 11, no. 7: 459. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11070459