
Perturbation-Based Modeling Unveils the Autophagic Modulation of Chemosensitivity and Immunogenicity in Breast Cancer Cells

 ,
,  ,
, 
 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
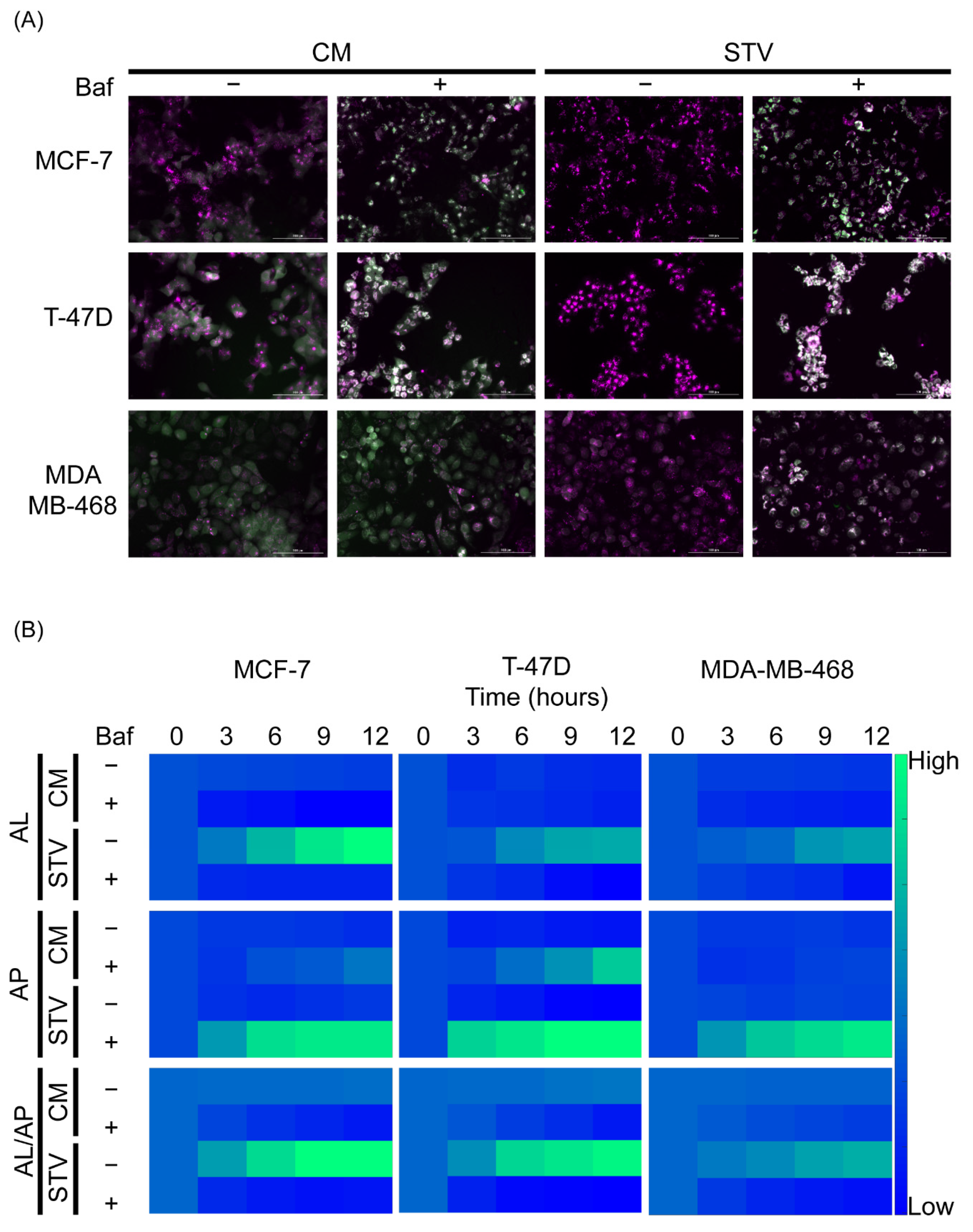
2.1. The Autophagolysosome/Autophagosome Ratio of an mCHR-GFP Tandem LC3 Sensor Reports Dynamic Changes of Autophagic Flux in Response to Starvation-Induction and Bafilomycin-Mediated Degradation Blockade in Breast Cancer Cell Lines
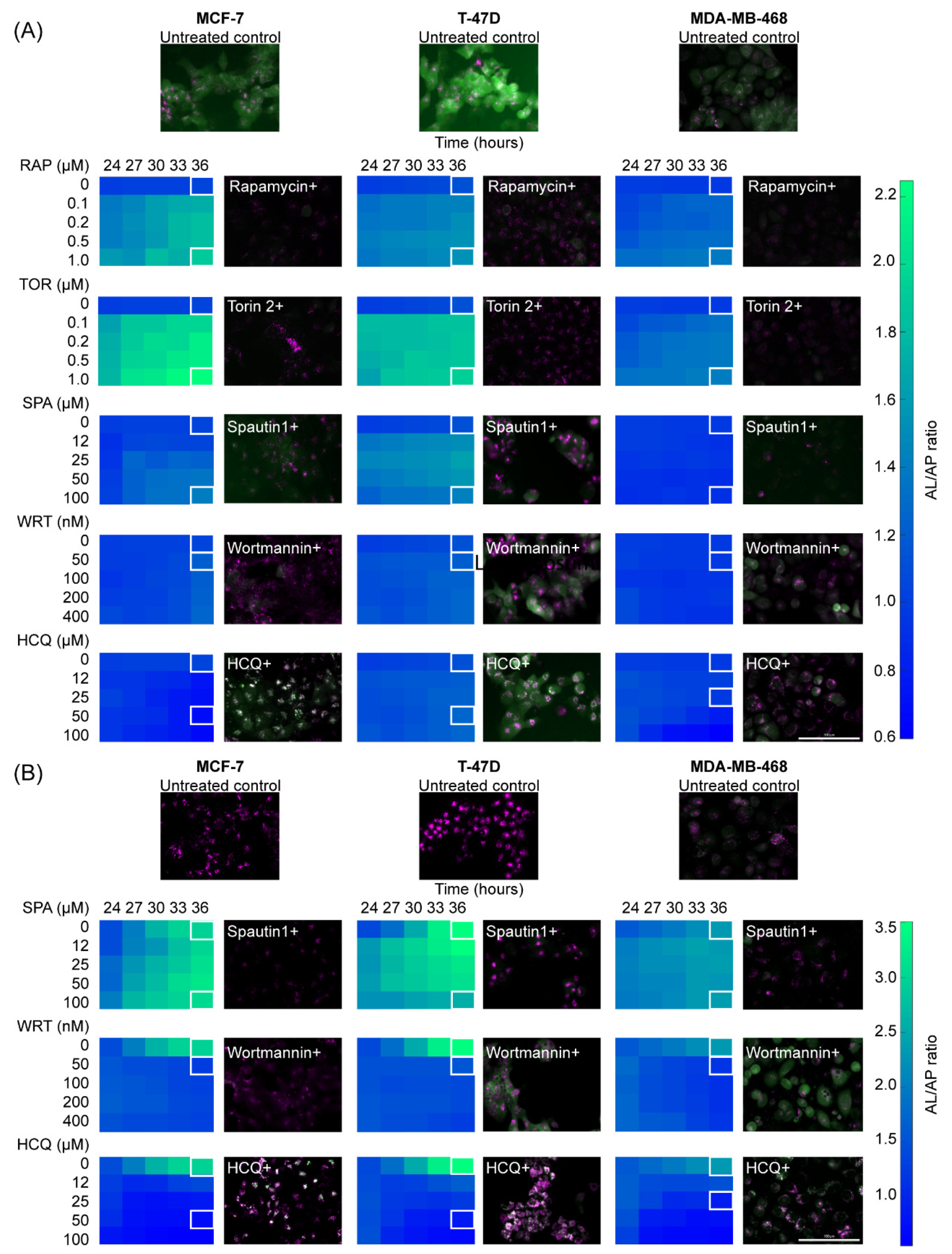
2.2. Breast Cancer Cell Lines Display a Heterogeneous Response to Drugs That Induce Known Perturbations of Autophagic Flux
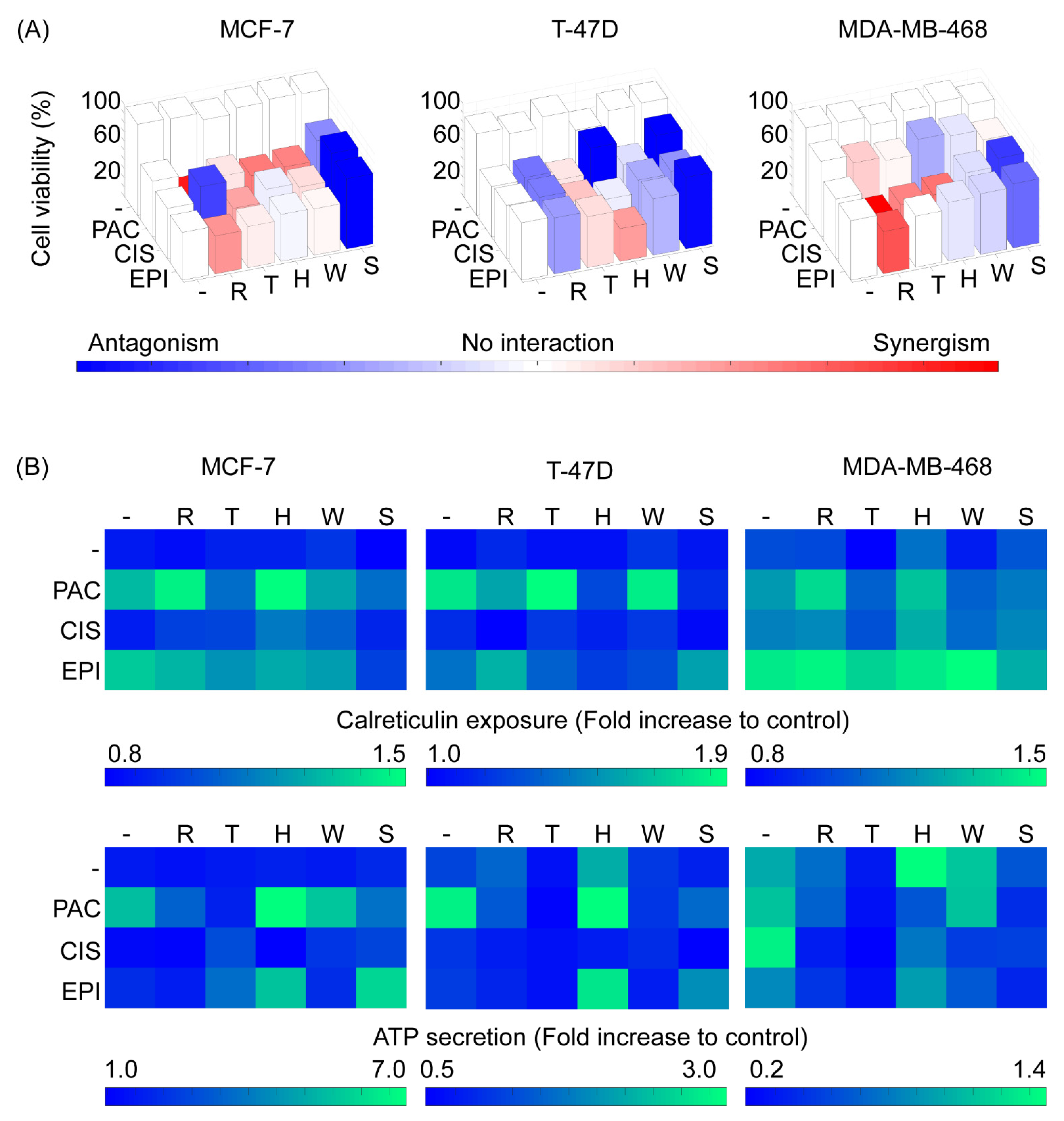
2.3. Cell Line-Specific Profiles of Cytotoxicity and Cell Death Immunogenicity Arise from the Interactions between Autophagy Regulators and Chemotherapeutic Perturbations
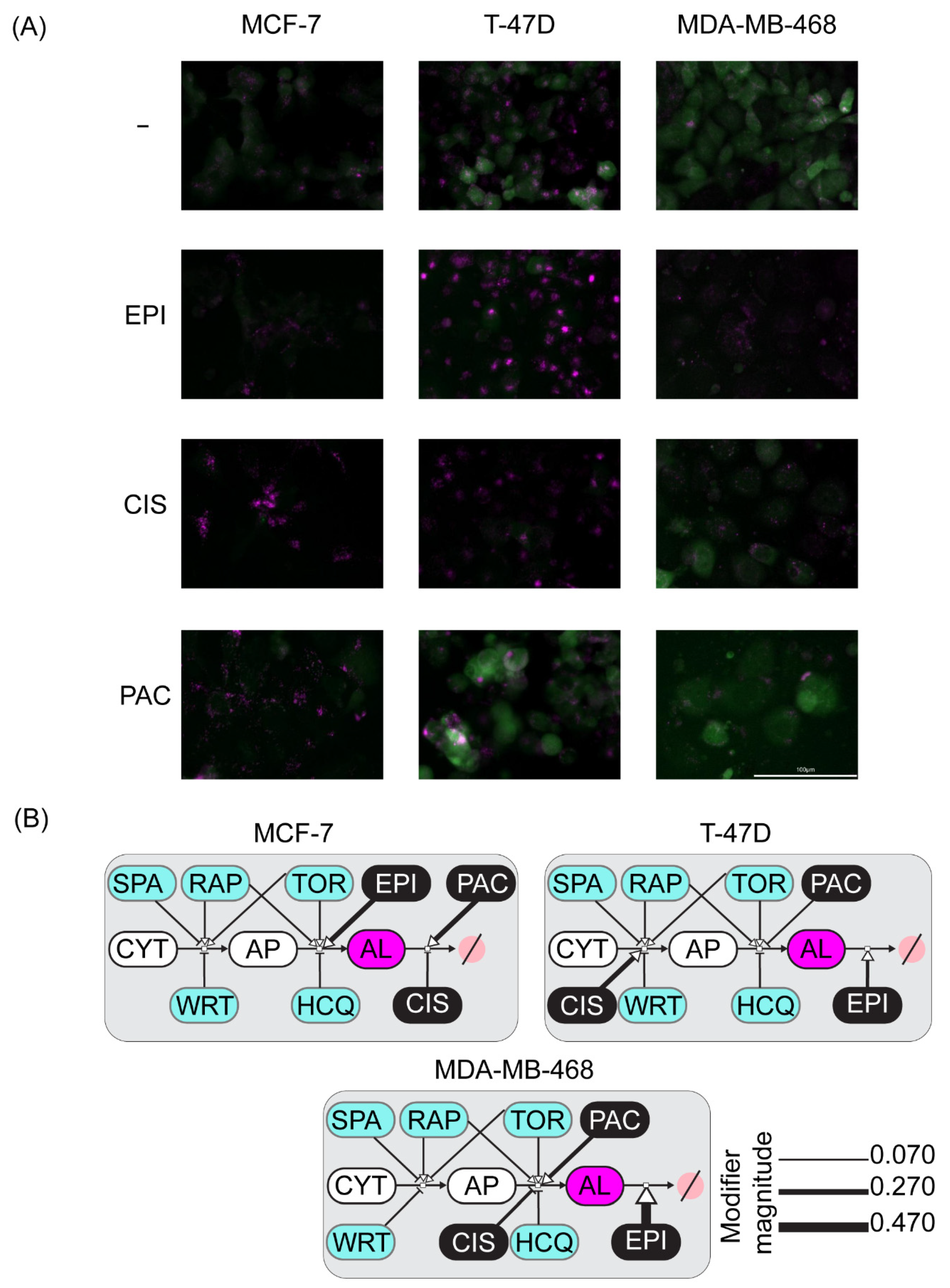
2.4. Perturbation-Based Dynamic Modeling of the Autophagic Sensor Indicates Cell-Line Specific Modes of Action of Chemotherapeutic Agents on the Autophagic Flux
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. mCherry-EGFP-LC3B Retroviral Vectors Assembly
4.3. Generation and Sorting of Stable mCherry-EGFP-LC3B Cell Lines
4.4. Autophagy Modulators and Chemotherapeutic Drugs
4.5. Autophagic Flux Kinetic Assay
4.6. Live Cell Image Analysis
4.7. Cytotoxicity Assay
4.8. Calreticulin Measurement
4.9. ATP Measurement
4.10. Perturbation-Based Mathematical Modeling
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Female Breast Cancer—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 12 July 2021).
- Guía de Práctica Clínica Para el Tratamiento del Cáncer de Mama; Caja Costarricense del Seguro Social: San José, Costa Rica, 2012; pp. 1–164.
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coley, H.M. Mechanisms and consequences of chemotherapy resistance in breast cancer. Eur. J. Cancer Suppl. 2009, 7, 3–7. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Qin, L.; Wang, Z.; Tao, L.; Wang, Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010, 6, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Senovilla, L.; Vitale, I.; Martins, I.; Tailler, M.; Pailleret, C.; Michaud, M.; Galluzzi, L.; Adjemian, S.; Kepp, O.; Niso-Santano, M.; et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012, 337, 1678–1684. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [Green Version]
- Ladoire, S.; Enot, D.; Andre, F.; Zitvogel, L.; Kroemer, G. Immunogenic cell death-related biomarkers: Impact on the survival of breast cancer patients after adjuvant chemotherapy. Oncoimmunology 2016, 5, e1082706. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Lee, J.G.; Shin, J.H.; Shim, H.S.; Lee, C.Y.; Kim, D.J.; Kim, Y.S.; Chung, K.Y. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir. Res. 2015, 16, 138. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fader, C.M.; Aguilera, M.O.; Colombo, M.I. ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy 2012, 8, 1741–1756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martins, I.; Ma, Y.; Kepp, O.; Galluzzi, L.; Kroemer, G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy 2013, 9, 1624–1625. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An autophagy-driven pathway of ATP secretion supports the aggressive phenotype of BRAFV600E inhibitor-resistant metastatic melanoma cells. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.D.; Xie, B.; Wu, X.J.; Li, J.J.; Ding, Y.; Wen, X.Z.; Zhang, X.; Zhu, S.G.; Liu, W.; Zhang, X.S.; et al. Late-stage inhibition of autophagy enhances calreticulin surface exposure. Oncotarget 2016, 7, 80842–80854. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Tang, D. HMGB1-dependent and -independent autophagy. Autophagy 2014, 10, 1873–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Kovaleva, V.; Mora, R.; Park, Y.J.; Plass, C.; Chiramel, A.I.; Bartenschlager, R.; Doḧner, H.; Stilgenbauer, S.; Pscherer, A.; Lichter, P.; et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res. 2012, 72, 1763–1772. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Zhou, C.; Zhong, W.; Zhou, J.; Sheng, F.; Fang, Z.; Wei, Y.; Chen, Y.; Deng, X.; Xia, B.; Lin, J. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012, 8, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Sarmah, D.T.; Bairagi, N.; Chatterjee, S. Tracing the footsteps of autophagy in computational biology. Brief. Bioinform. 2020, 22, bbaa286. [Google Scholar] [CrossRef]
- Jegga, A.G.; Schneider, L.; Ouyang, X.; Zhang, J. Systems biology of the autophagy-lysosomal pathway. Autophagy 2011, 7, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holczer, M.; Márton, M.; Kurucz, A.; Bánhegyi, G.; Kapuy, O. A comprehensive systems biological study of autophagy-apoptosis crosstalk during endoplasmic reticulum stress. Biomed Res. Int. 2015, 2015, 319589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Ware, K.; Ding, Y.; Kim, S.; Sheth, M.; Rao, S.; Chan, W.; Armstrong, A.; Eward, W.; Jolly, M.; et al. An Integrative Systems Biology and Experimental Approach Identifies Convergence of Epithelial Plasticity, Metabolism, and Autophagy to Promote Chemoresistance. J. Clin. Med. 2019, 8, 205. [Google Scholar] [CrossRef] [Green Version]
- Robnik-Šikonja, M.; Bohanec, M. Perturbation-Based Explanations of Prediction Models. In Human and Machine Learning; Zhou, J., Chen, F., Eds.; Springer International Publishing AG: Cham, Switzerland, 2018; pp. 159–175. [Google Scholar]
- Molina-Mora, J.A.; Kop-Montero, M.; Quirós-Fernández, I.; Quiros, S.; Crespo-Mariño, J.L.; Mora-Rodríguez, R.A. A hybrid mathematical modeling approach of the metabolic fate of a fluorescent sphingolipid analogue to predict cancer chemosensitivity. Comput. Biol. Med. 2018, 97, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Mauvezin, C.; Nagy, P.; Juhász, G.; Neufeld, T.P. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Garson, K.; Li, L.; Vanderhyden, B.C. Optimization of lentiviral vector production using polyethylenimine-mediated transfection. Oncol. Lett. 2015, 9, 55–62. [Google Scholar] [CrossRef]
- Kim, L.C.; Cook, R.S.; Chen, J. MTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, C.; Kirubakaran, S.; Zhang, X.; Hur, W.; Liu, Y.; Kwiatkowski, N.P.; Wang, J.; Westover, K.D.; Gao, P.; et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res. 2013, 73, 2574–2586. [Google Scholar] [CrossRef] [Green Version]
- Duffy, A.; Le, J.; Sausville, E.; Emadi, A. Autophagy modulation: A target for cancer treatment development. Cancer Chemother. Pharmacol. 2015, 75, 439–447. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, M.B.E.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M.A. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef] [Green Version]
- Agrotis, A.; Pengo, N.; Burden, J.J.; Ketteler, R. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 2019, 15, 976–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamacher-Brady, A.; Stein, H.A.; Turschner, S.; Toegel, I.; Mora, R.; Jennewein, N.; Efferth, T.; Eils, R.; Brady, N.R. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. J. Biol. Chem. 2011, 286, 6587–6601. [Google Scholar] [CrossRef] [Green Version]
- McIsaac, R.S.; Petti, A.A.; Bussemaker, H.J.; Botstein, D. Perturbation-based analysis and modeling of combinatorial regulation in the yeast sulfur assimilation pathway. Mol. Biol. Cell 2012, 23, 2993–3008. [Google Scholar] [CrossRef]
- Lau, T.S.; Chan, L.K.Y.; Man, G.C.W.; Wong, C.H.; Lee, J.H.S.; Yim, S.F.; Cheung, T.H.; McNeish, I.A.; Kwong, J. Paclitaxel induces immunogenic cell death in ovarian cancer via TLR4/IKK2/SNARE-dependent exocytosis. Cancer Immunol. Res. 2020, 8, 1099–1111. [Google Scholar] [CrossRef]
- Song, Y.; Li, W.; Peng, X.; Xie, J.; Li, H.; Tan, G. Inhibition of autophagy results in a reversal of taxol resistance in nasopharyngeal carcinoma by enhancing taxol-induced caspase-dependent apoptosis. Am. J. Transl. Res. 2017, 9, 1934–1942. [Google Scholar]
- Kim, H.J.; Lee, S.G.; Kim, Y.J.; Park, J.E.; Lee, K.Y.; Yoo, Y.H.; Kim, J.M. Cytoprotective role of autophagy during paclitaxel-induced apoptosis in Saos-2 osteosarcoma cells. Int. J. Oncol. 2013, 42, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Gong, F.; Chen, Y.; Jiang, Y.; Liu, J.; Yu, M.; Zhang, S.; Wang, M.; Xiao, G.; Liao, H. Autophagy promotes paclitaxel resistance of cervical cancer cells: Involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis. 2014, 5, e1367. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Liu, J.H.; Zhang, J.; Zhang, N.; Wang, Z.H. Blockade of autophagy aggravates endoplasmic reticulum stress and improves paclitaxel cytotoxicity in human cervical cancer cells. Cancer Res. Treat. 2015, 47, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, R.A.; Banman, S.L.; Hemmerling, D.R.; Odsen, R.; Simmen, T.; Simmonds, A.J.; Underhill, D.A.; Goping, I.S. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene 2013, 32, 736–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.F.; Hu, P.C.; Wang, Y.; Xu, X.L.; Rushworth, G.M.; Zhang, Z.; Wei, L.; Zhang, J.W. Paclitaxel induces autophagy in gastric cancer BGC823 cells. Ultrastruct. Pathol. 2017, 41, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.F.; Lin, Y.C.; Tsai, T.F.; Chen, H.E.; Chou, K.Y.; Hwang, T.I.S. Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des. Devel. Ther. 2017, 11, 1517–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Ji, F.; Liu, Y.; He, M.; Zhang, Z.; Yang, J.; Wang, N.; Zhong, C.; Jin, Q.; Ye, X.; et al. Cisplatin-induced autophagy protects breast cancer cells from apoptosis by regulating yes-associated protein. Oncol. Rep. 2017, 38, 3668–3676. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, L.; Zhou, H.; Wang, W.; Luo, Y.; Yang, H.; Yi, H. Inhibition of autophagy promotes cisplatin-induced apoptotic cell death through Atg5 and Beclin 1 in A549 human lung cancer cells. Mol. Med. Rep. 2018, 17, 6859–6865. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Xie, H.; Lu, W.; Li, F.; Li, J.; Guo, Z. Chloroquine sensitizes hepatocellular carcinoma cells to chemotherapy via blocking autophagy and promoting mitochondrial dysfunction. Int. J. Clin. Exp. Pathol. 2017, 10, 10056–10065. [Google Scholar]
- Sun, W.L.; Chen, J.; Wang, Y.P.; Zheng, H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011, 7, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Chittaranjan, S.; Bortnik, S.; Dragowska, W.H.; Xu, J.; Abeysundara, N.; Leung, A.; Go, N.E.; DeVorkin, L.; Weppler, S.A.; Gelmon, K.; et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 2014, 20, 3159–3173. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.I.; Whang, E.E.; Donner, D.B.; Du, J.; Lorch, J.; He, F.; Jiang, X.; Price, B.D.; Moore, F.D.; Ruan, D.T. Autophagy induction with RAD001 enhances chemosensitivity and radiosensitivity through met inhibition in papillary thyroid cancer. Mol. Cancer Res. 2010, 8, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- N’Diaye, E.N.; Kajihara, K.K.; Hsieh, I.; Morisaki, H.; Debnath, J.; Brown, E.J. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009, 10, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Naviaux, R.K.; Costanzi, E.; Haas, M.; Verma, I.M. The pCL vector system: Rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 1996, 70, 5701–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaysher, S.; Cree, I.A. Cell Sensitivity Assay: The ATP-based Tumor Chemosensitivity Assays. In Cancer Cell Culture; Cree, I.A., Ed.; Springer Science+Business Media: New York, NY, USA, 2011; pp. 247–257. [Google Scholar]
- Hoops, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiros-Fernandez, I.; Figueroa-Protti, L.; Arias-Arias, J.L.; Brenes-Cordero, N.; Siles, F.; Mora, J.; Mora-Rodríguez, R.A. Perturbation-Based Modeling Unveils the Autophagic Modulation of Chemosensitivity and Immunogenicity in Breast Cancer Cells. Metabolites 2021, 11, 637. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11090637
Quiros-Fernandez I, Figueroa-Protti L, Arias-Arias JL, Brenes-Cordero N, Siles F, Mora J, Mora-Rodríguez RA. Perturbation-Based Modeling Unveils the Autophagic Modulation of Chemosensitivity and Immunogenicity in Breast Cancer Cells. Metabolites. 2021; 11(9):637. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11090637
Chicago/Turabian StyleQuiros-Fernandez, Isaac, Lucía Figueroa-Protti, Jorge L. Arias-Arias, Norman Brenes-Cordero, Francisco Siles, Javier Mora, and Rodrigo Antonio Mora-Rodríguez. 2021. "Perturbation-Based Modeling Unveils the Autophagic Modulation of Chemosensitivity and Immunogenicity in Breast Cancer Cells" Metabolites 11, no. 9: 637. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11090637