
Multi-Omics, an Integrated Approach to Identify Novel Blood Biomarkers of Alzheimer’s Disease
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Blood Collection, Biochemical Measurements, and Statistics
2.3. Apolipoprotein E Genotyping
2.4. Chemicals
2.5. Untargeted Metabolomics
2.6. Untargeted Proteomics
2.7. Protein Data Analysis
2.8. Chemometric Analysis, Plasma Metabolome and Proteome Integration
2.9. Statistics
3. Results
3.1. Cohort
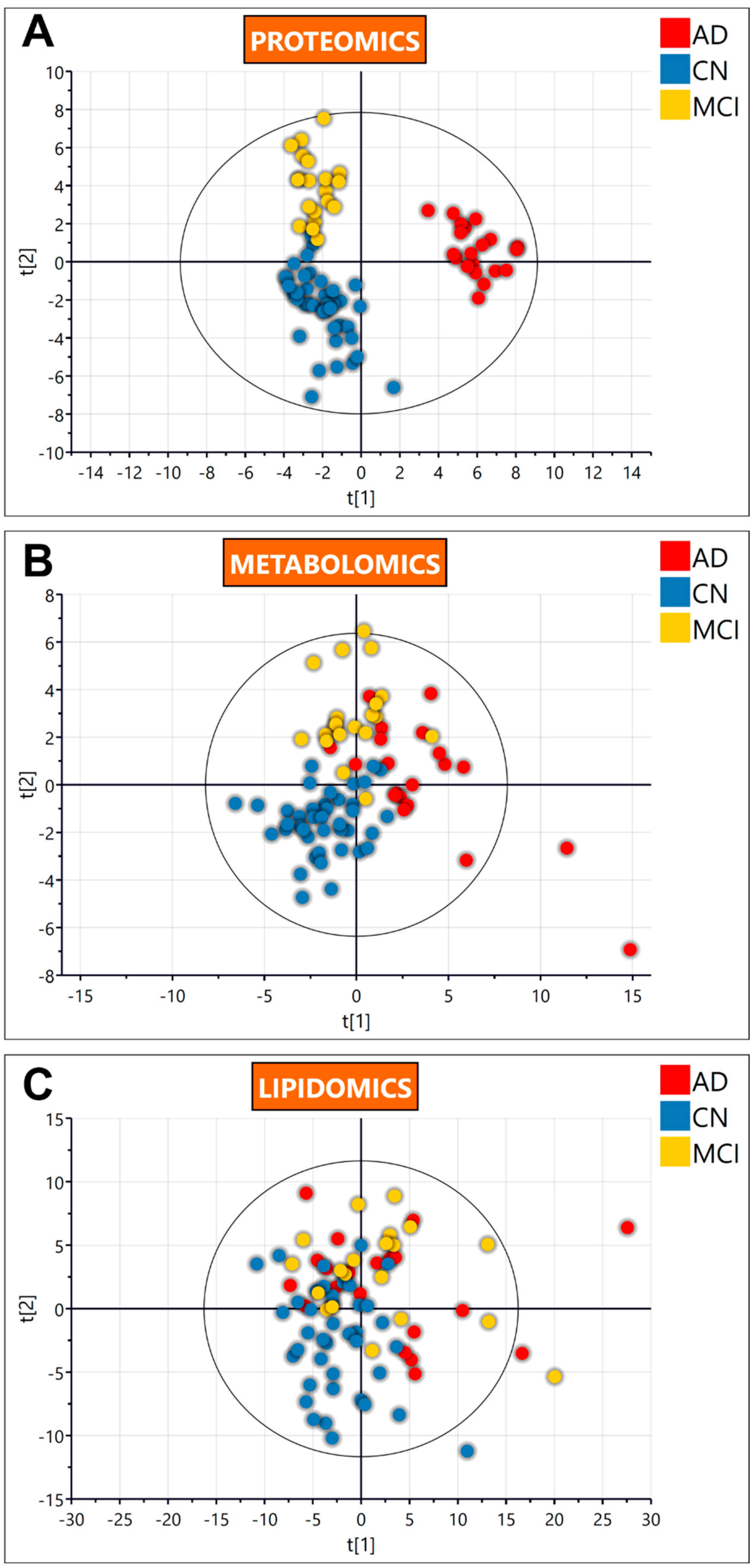
3.2. Untargeted Omics
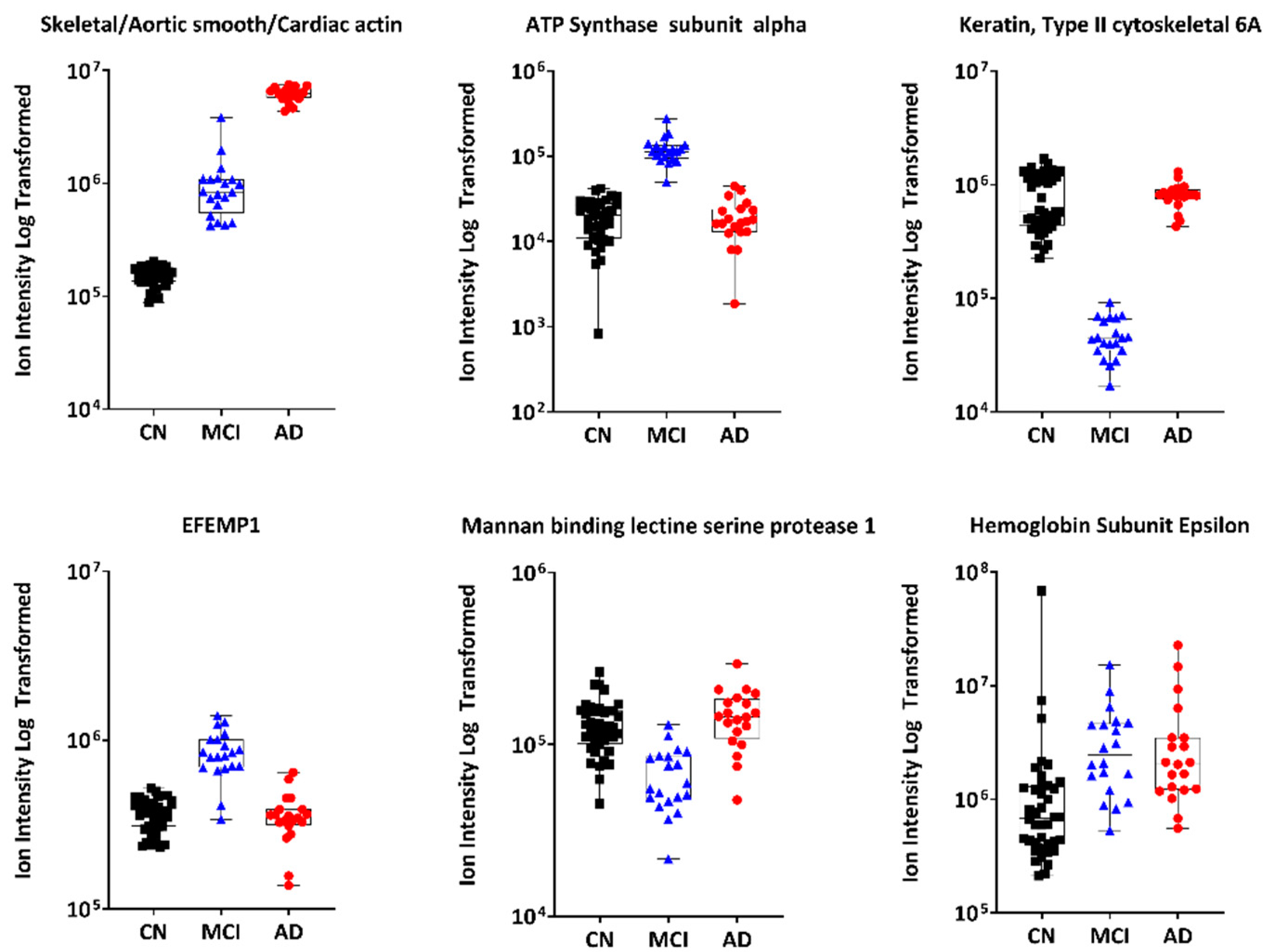
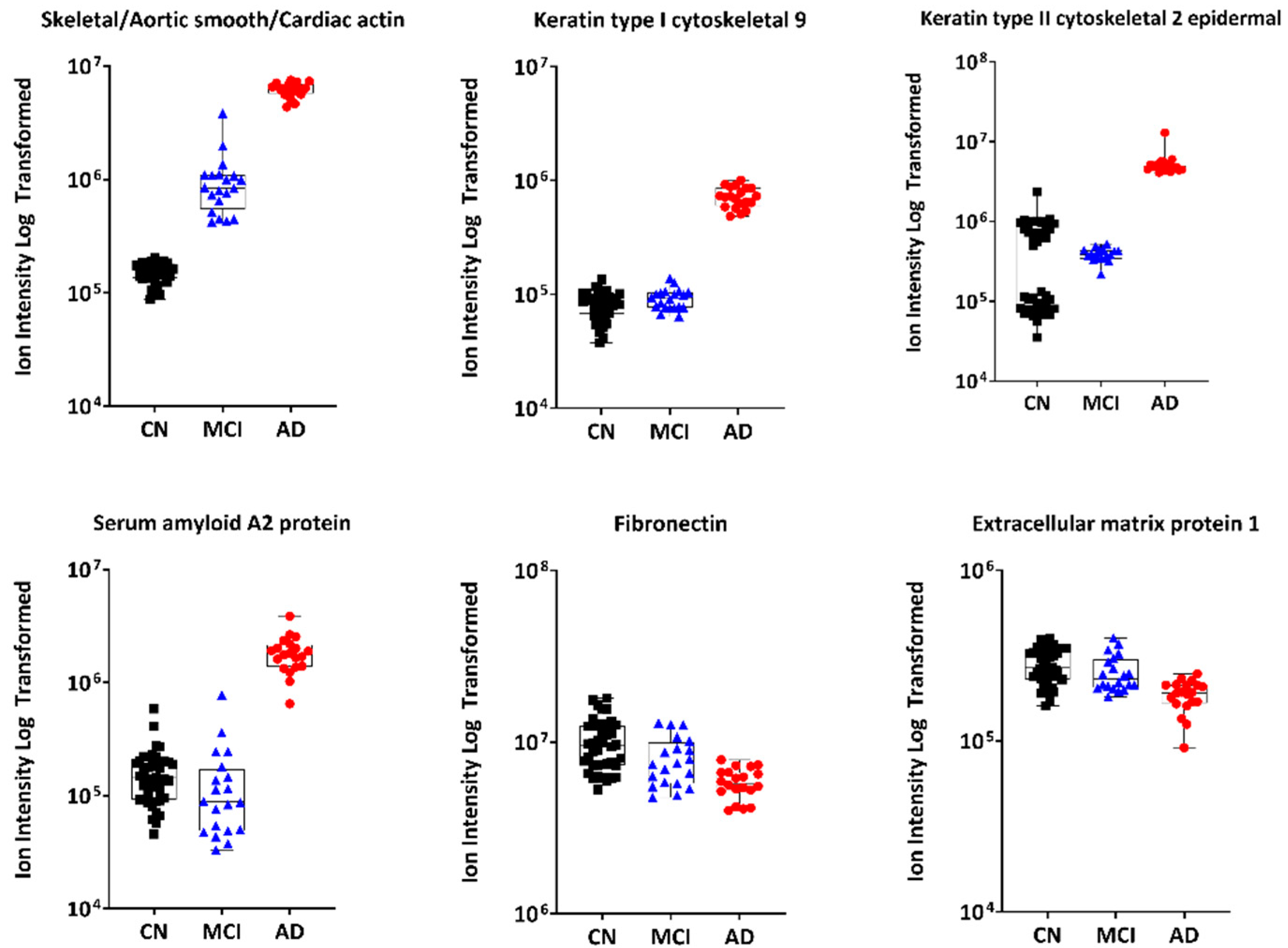
3.2.1. Untargeted Proteomics
3.2.2. Untargeted Metabolomics
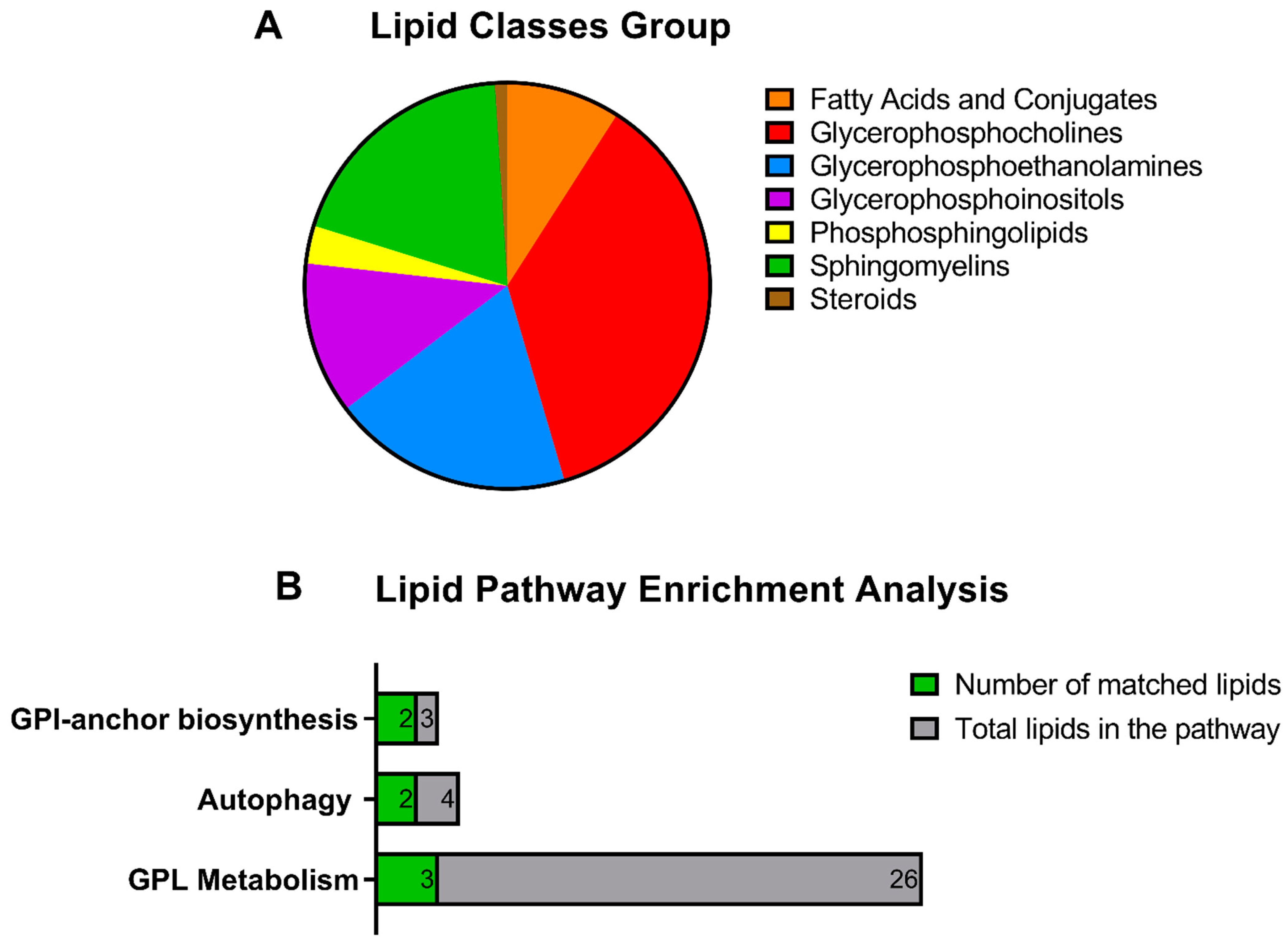
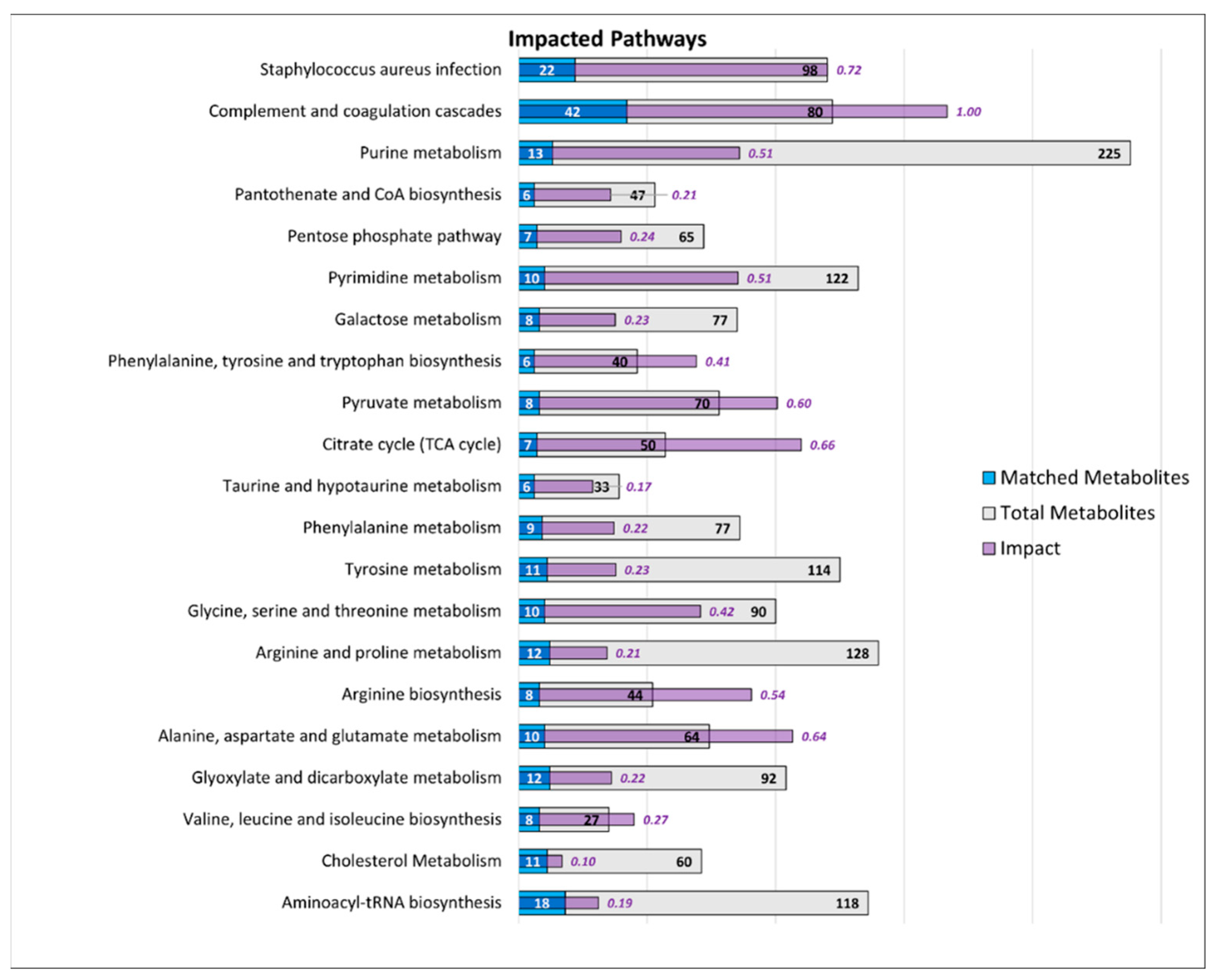
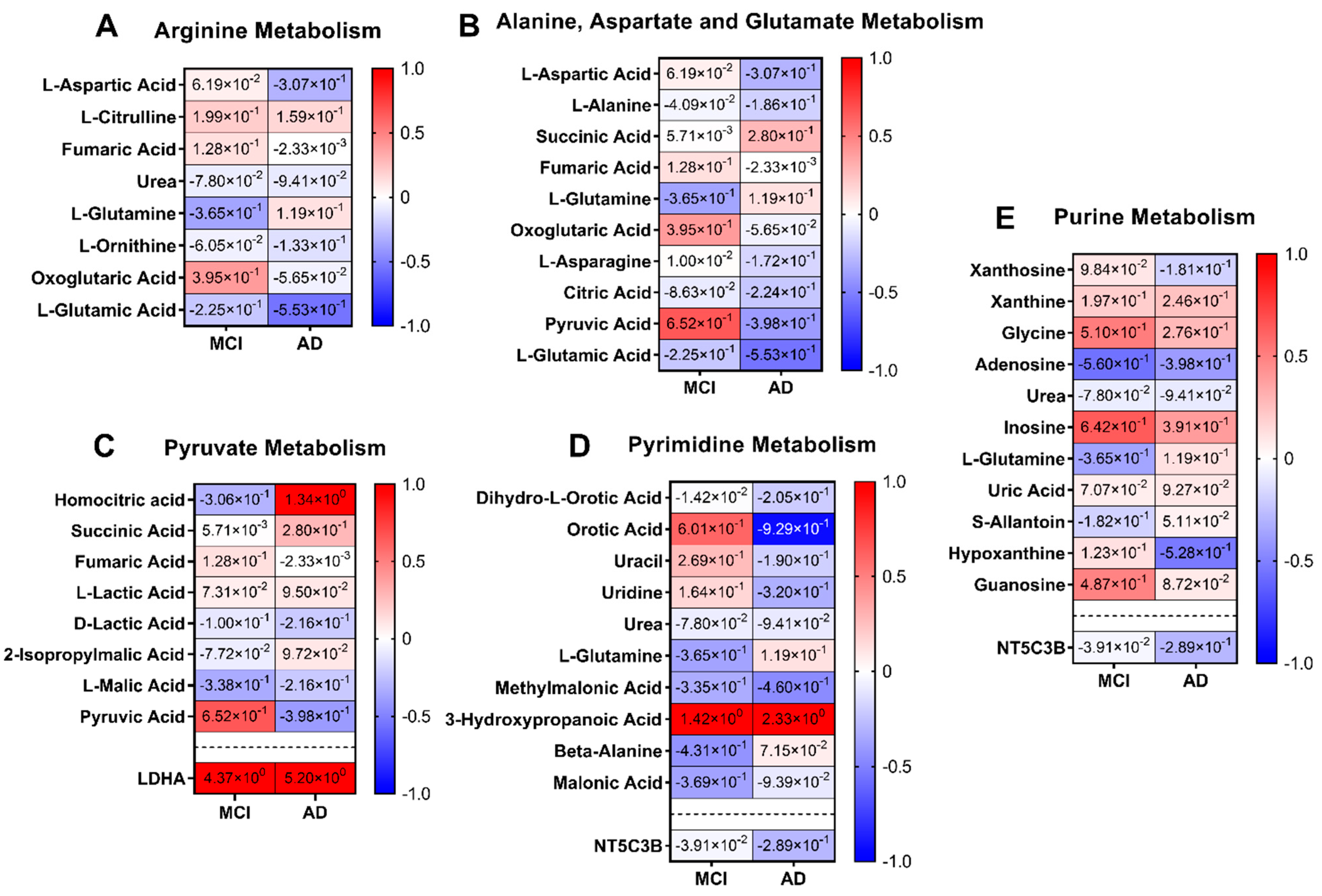
3.2.3. Integrated Pathway Analysis
Lipidomics
Pathways
4. Discussion
4.1. Lipidomics
4.2. Proteomics
4.2.1. Plasma Actin
4.2.2. Serum Amyloid A2
4.2.3. Keratin 9
4.2.4. Selenoprotein P
4.2.5. Fibronectin and Extracellular Matrix Protein 1
4.2.6. Mannan-Binding Lectin Serine Protease 1
4.3. Metabolomics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burns, A.; Byrne, E.J.; Maurer, K. Alzheimer’s disease. Lancet 2002, 360, 163–165. [Google Scholar] [CrossRef]
- Alzheimer’s Association; Thies, W.; Bleiler, L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011, 7, 208–244. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Maestre, G.E.; Arizaga, R.; Friedland, R.P.; Galasko, D.; Hall, K.; Luchsinger, J.A.; Ogunniyi, A.; Perry, E.K.; Potocnik, F.; et al. World Federation of Neurology Dementia Research Group Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008, 7, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Sloane, P.D.; Zimmerman, S.; Suchindran, C.; Reed, P.; Wang, L.; Boustani, M.; Sudha, S. The public health impact of Alzheimer’s disease, 2000–2050: Potential implication of treatment advances. Annu. Rev. Public Health 2002, 23, 213–231. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.D. The worldwide challenge of the dementias: A role for B vitamins and homocysteine? Food Nutr. Bull. 2008, 29, S143–S172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Alzheimer’s Disease International Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Anonymous. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- Grundman, M.; Petersen, R.C.; Ferris, S.H.; Thomas, R.G.; Aisen, P.S.; Bennett, D.A.; Foster, N.L.; Jack, C.R., Jr.; Galasko, D.R.; Doody, R.; et al. Alzheimer’s Disease Cooperative Study Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials. Arch. Neurol. 2004, 61, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Petersen, R.C.; Roberts, R.O.; Knopman, D.S.; Boeve, B.F.; Geda, Y.E.; Ivnik, R.J.; Smith, G.E.; Jack, C.R., Jr. Mild cognitive impairment: Ten years later. Arch. Neurol. 2009, 66, 1447–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francois, M.; Bull, C.F.; Fenech, M.F.; Leifert, W.R. Current State of Saliva Biomarkers for Aging and Alzheimer’s Disease. Curr Alzheimer Res 2019, 16, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; Lovestone, S. Blood-based biomarkers of Alzheimer’s disease: Challenging but feasible. Biomark Med. 2010, 4, 65–79. [Google Scholar] [CrossRef]
- Hampel, H.; Prvulovic, D. Are biomarkers harmful to recruitment and retention in Alzheimer’s disease clinical trials? An international perspective. J. Nutr. Health Aging 2012, 16, 346–348. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Cerebrospinal fluid biomarkers for Alzheimer’s disease. J. Alzheimers Dis. 2009, 18, 413–417. [Google Scholar] [CrossRef]
- Doecke, J.D.; Laws, S.M.; Faux, N.G.; Wilson, W.; Burnham, S.C.; Lam, C.P.; Mondal, A.; Bedo, J.; Bush, A.I.; Brown, B.; et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch. Neurol. 2012, 69, 1318–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; Macarthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancesario, G.M.; Bernardini, S. Alzheimer’s disease in the omics era. Clin. Biochem. 2018, 59, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Marksteiner, J.; Oberacher, H.; Humpel, C. Acyl-Alkyl-Phosphatidlycholines are Decreased in Saliva of Patients with Alzheimer’s Disease as Identified by Targeted Metabolomics. J. Alzheimers Dis. 2019, 68, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.N.; Huang, C.C.; Huang, K.L.; Lin, K.J.; Yen, T.C.; Kuo, H.C. A metabolomic approach to identifying biomarkers in blood of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Francois, M.; Karpe, A.; Liu, J.W.; Beale, D.; Hor, M.; Hecker, J.; Faunt, J.; Maddison, J.; Johns, S.; Doecke, J.; et al. Salivaomics as a Potential Tool for Predicting Alzheimer’s Disease During the Early Stages of Neurodegeneration. J. Alzheimers Dis. 2021, 82, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, M.S.; Francois, M.; Hecker, J.; Faunt, J.; Fenech, M.F.; Leifert, W.R. GammaH2AX is increased in peripheral blood lymphocytes of Alzheimer’s disease patients in the South Australian Neurodegeneration, Nutrition and DNA Damage (SAND) study of aging. Mutat. Res. 2018, 829–830, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Xie, Y.Z.; Cao, T.T.; Wang, Z.; Wang, T.; Li, X.; Shen, R.C.; Xu, H.; Bu, G.; Chen, X.F. A rapid and cost-effective method for genotyping apolipoprotein E gene polymorphism. Mol. Neurodegener 2016, 11, 2–016-0069-4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Jadhav, S.; Phan, L.; Tremellen, K.; Tran, C.; Beale, D. Plasma metabolic and lipidomic fingerprinting of individuals with increased intestinal permeability. Metabolites 2022, 12, 302. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Tang, K.; Karpe, A.; Beale, D.; Totsika, M.; Kenny, L.; Morrison, M.; Punyadeera, C. Chemoradiation therapy changes oral microbiome and metabolomic profiles in patients with oral cavity cancer and oropharyngeal cancer. Head Neck 2021, 43, 1521–1534. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, P.; Karpe, A.V.; Hillyer, K.E.; Nguyen, T.V.; Hewitt, J.; Beale, D.J. A multi-platform metabolomics approach to identify possible biomarkers for human faecal contamination in Greenshell™ mussels (Perna canaliculus). Sci. Total Environ. 2021, 771, 145363. [Google Scholar] [CrossRef] [PubMed]
- Beale, D.; Shah, R.; Karpe, A.; Hillyer, K.; McAuley, A.; Au, G.; Marsh, G.; Vasan, V. Metabolic profiling from an asymptomatic ferret model of SARS-CoV-2 infection. Metabolites 2021, 11, 327. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-de-Diego, R.; Tarazona, S.; Martinez-Mira, C.; Balzano-Nogueira, L.; Furio-Tari, P.; Pappas, G.J., Jr.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef]
- Acevedo, A.; Durán, C.; Ciucci, S.; Gerl, M.; Cannistraci, C. LIPEA: Lipid Pathway Enrichment Analysis. Available online: https://lipea.biotec.tu-dresden.de/home (accessed on 15 April 2021).
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, M.; Ma, K.; Gao, F.; Kim, H.W.; Rapoport, S.I.; Rao, J.S. Disturbed choline plasmalogen and phospholipid fatty acid concentrations in Alzheimer’s disease prefrontal cortex. J. Alzheimers Dis. 2011, 24, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr. Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: Molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 2001, 77, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Mankidy, R.; Ritchie, S.; Heath, D.; Wood, J.A.; Flax, J.; Goodenowe, D.B. Circulating plasmalogen levels and Alzheimer Disease Assessment Scale-Cognitive scores in Alzheimer patients. J. Psychiatry Neurosci. 2010, 35, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Kling, M.A.; Goodenowe, D.B.; Senanayake, V.; MahmoudianDehkordi, S.; Arnold, M.; Massaro, T.J.; Baillie, R.; Han, X.; Leung, Y.Y.; Saykin, A.J.; et al. Alzheimer’s Disease Metabolomics Consortium (ADMC) and the Alzheimer’s Disease Neuroimaging Initiative (ADNI) Circulating ethanolamine plasmalogen indices in Alzheimer’s disease: Relation to diagnosis, cognition, and CSF tau. Alzheimers Dement. 2020, 16, 1234–1247. [Google Scholar] [CrossRef]
- Goodenowe, D.B.; Cook, L.L.; Liu, J.; Lu, Y.; Jayasinghe, D.A.; Ahiahonu, P.W.; Heath, D.; Yamazaki, Y.; Flax, J.; Krenitsky, K.F.; et al. Peripheral ethanolamine plasmalogen deficiency: A logical causative factor in Alzheimer’s disease and dementia. J. Lipid Res. 2007, 48, 2485–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, A.M.; Barnett, J.L.; Pieroni, C.; Nixon, R.A. Increased Neuronal Endocytosis and Protease Delivery to Early Endosomes in Sporadic Alzheimer’s Disease: Neuropathologic Evidence for a Mechanism of Increased beta -Amyloidogenesis. J. Neurosci. 1997, 17, 6142–6151. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Pineda, J.A.; Vera-Lopez, K.J.; Shrivastava, P.; Chavez-Fumagalli, M.A.; Nieto-Montesinos, R.; Alvarez-Fernandez, K.L.; Goyzueta Mamani, L.D.; Davila Del-Carpio, G.; Gomez-Valdez, B.; Miller, C.L.; et al. Vascular smooth muscle cell dysfunction contribute to neuroinflammation and Tau hyperphosphorylation in Alzheimer disease. iScience 2021, 24, 102993. [Google Scholar] [CrossRef]
- Hazeldine, J.; Dinsdale, R.J.; Naumann, D.N.; Acharjee, A.; Bishop, J.R.B.; Lord, J.M.; Harrison, P. Traumatic injury is associated with reduced deoxyribonuclease activity and dysregulation of the actin scavenging system. Burns Trauma. 2021, 9, tkab001. [Google Scholar] [CrossRef] [PubMed]
- Kułakowska, A.; Tarasiuk, J.; Kapica-Topczewska, K.; Chorąży, M.; Pogorzelski, R.; Kulczyńska-Przybik, A.; Mroczko, B.; Bucki, R. Pathophysiological implications of actin-free Gc-globulin concentration changes in blood plasma and cerebrospinal fluid collected from patients with Alzheimer’s disease and other neurological disorders. Adv. Clin. Exp. Med. 2018, 27, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Sloane, J.A.; Wells, J.M.; Abraham, C.R.; Fine, R.E.; Sipe, J.D. Evidence for local production of acute phase response apolipoprotein serum amyloid A in Alzheimer’s disease brain. Neurosci. Lett. 1997, 225, 73–76. [Google Scholar] [CrossRef]
- Chung, T.; Liang, J.; Schreiber, B.M.; Sipe, J.D.; McKee, A.; Fine, R.E.; Johnson, R.J. Serum amyloid A in Alzheimer’s disease brain is predominantly localized to myelin sheaths and axonal membrane. Amyloid 2000, 7, 105–110. [Google Scholar] [CrossRef]
- Page, M.J.; Thomson, G.J.A.; Nunes, J.M.; Engelbrecht, A.; Nell, T.A.; de Villiers, W.J.S.; de Beer, M.C.; Engelbrecht, L.; Kell, D.B.; Pretorius, E. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci. Rep. 2019, 9, 3102. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, D.; Li, S.; Yu, Y.; Ye, R.D. Suppression of LPS-induced tau hyperphosphorylation by serum amyloid A. J. Neuroinflamm. 2016, 13, 28. [Google Scholar] [CrossRef] [Green Version]
- Kindy, M.S.; Yu, J.; Guo, J.T.; Zhu, H. Apolipoprotein serum amyloid A in Alzheimer’s disease. J. Alzheimer’s Dis. 1999, 1, 155–167. [Google Scholar] [CrossRef]
- Vafadar-Isfahani, B.; Ball, G.; Coveney, C.; Lemetre, C.; Boocock, D.; Minthon, L.; Hansson, O.; Miles, A.K.; Janciauskiene, S.M.; Warden, D.; et al. Identification of SPARC-like 1 protein as part of a biomarker panel for Alzheimer’s disease in cerebrospinal fluid. J. Alzheimers Dis. 2012, 28, 625–636. [Google Scholar] [CrossRef]
- Richens, J.L.; Spencer, H.L.; Butler, M.; Cantlay, F.; Vere, K.; Bajaj, N.; Morgan, K.; O’Shea, P. Rationalising the role of Keratin 9 as a biomarker for Alzheimer’s disease. Sci. Rep. 2016, 6, 22962. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, A.S.; Berry, M.J.; Bellinger, F.P. Role of selenoprotein P in Alzheimer’s disease. Ethn. Dis. 2010, 20, S1–S92. [Google Scholar]
- Zhang, Z.; Song, G. Roles of Selenoproteins in Brain Function and the Potential Mechanism of Selenium in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 646518. [Google Scholar] [CrossRef]
- Yankner, B.A.; Lu, T.; Pan, Y.; Kao, S.; Li, C.; Kohane, I.; Chan, J. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar]
- Miller, J.A.; Oldham, M.C.; Geschwind, D.H. A Systems Level Analysis of Transcriptional Changes in Alzheimer’s Disease and Normal Aging. J. Neurosci. 2008, 28, 1410–1420. [Google Scholar] [CrossRef] [Green Version]
- Rueli, R.H.L.H.; Parubrub, A.C.; Dewing, A.S.T.; Hashimoto, A.C.; Bellinger, M.T.; Weeber, E.J.; Uyehara-Lock, J.; White, L.R.; Berry, M.J.; Bellinger, F.P. Increased selenoprotein P in choroid plexus and cerebrospinal fluid in Alzheimer’s disease brain. J. Alzheimer’s Dis. 2015, 44, 379–383. [Google Scholar] [CrossRef] [Green Version]
- Lepelletier, F.-X.; Mann, D.M.A.; Robinson, A.C.; Pinteaux, E.; Boutin, H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropath. Appl. Neuro. 2017, 43, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenchhoff, J.; Poljak, A.; Song, F.; Raftery, M.; Brodaty, H.; Duncan, M.; McEvoy, M.; Attia, J.; Schofield, P.W.; Sachdev, P.S. Plasma protein profiling of mild cognitive impairment and Alzheimer’s disease across two independent cohorts. J. Alzheimer’s Dis. 2014, 43, 1355–1373. [Google Scholar] [CrossRef] [Green Version]
- Lemańska-Perek, A.; Leszek, J.; Krzyżanowska-Gołą;b, D.; Radzik, J.; Ką;tnik-Prastowska, I. Molecular Status of Plasma Fibronectin as an Additional Biomarker for Assessment of Alzheimer’s Dementia Risk. Dement. Geriatr. Cogn. 2009, 28, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Lanzrein, A.; Jobst, K.A.; Thiel, S.; Jensenius, J.C.; Sim, R.B.; Perry, V.H.; Sim, E. Mannan-binding lectin in human serum, cerebrospinal fluid and brain tissue and its role in Alzheimer’s disease. Neuroreport 1998, 9, 1491–1495. [Google Scholar] [CrossRef]
- Larvie, M.; Shoup, T.; Chang, W.; Chigweshe, L.; Hartshorn, K.; White, M.R.; Stahl, G.L.; Elmaleh, D.R.; Takahashi, K. Mannose-Binding Lectin Binds to Amyloid Protein and Modulates Inflammation. J. Biomed. Biotechnol. 2012, 2012, 929803. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Gao, M.; Wang, F.; Yun, Y.; Sun, Q.; Guo, R.; Yan, C.; Sun, X.; Li, Y. Serum metabolomic profiling in patients with Alzheimer disease and amnestic mild cognitive impairment by GC/MS. Biomed. Chromatogr. 2020, 34, e4875. [Google Scholar] [CrossRef] [PubMed]
- Vardarajan, B.; Kalia, V.; Manly, J.; Brickman, A.; Reyes-Dumeyer, D.; Lantigua, R.; Ionita-Laza, I.; Jones, D.P.; Miller, G.W.; Mayeux, R. Differences in plasma metabolites related to Alzheimer’s disease, APOE epsilon4 status, and ethnicity. Alzheimers Dement. 2020, 6, e12025. [Google Scholar]
- Zhang, X.; Hu, W.; Wang, Y.; Wang, W.; Liao, H.; Zhang, X.; Kiburg, K.V.; Shang, X.; Bulloch, G.; Huang, Y.; et al. Plasma metabolomic profiles of dementia: A prospective study of 110,655 participants in the UK Biobank. BMC Med. 2022, 20, 252. [Google Scholar] [CrossRef]
- Pena-Bautista, C.; Roca, M.; Hervas, D.; Cuevas, A.; Lopez-Cuevas, R.; Vento, M.; Baquero, M.; Garcia-Blanco, A.; Chafer-Pericas, C. Plasma metabolomics in early Alzheimer’s disease patients diagnosed with amyloid biomarker. J. Proteom. 2019, 200, 144–152. [Google Scholar] [CrossRef]
- Niedzwiecki, M.M.; Walker, D.I.; Howell, J.C.; Watts, K.D.; Jones, D.P.; Miller, G.W.; Hu, W.T. High-resolution metabolomic profiling of Alzheimer’s disease in plasma. Ann. Clin. Transl. Neurol. 2020, 7, 36–45. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | CN | MCI | AD | 1p-Value CN vs. MCI | 1p-Value CN vs. AD |
|---|---|---|---|---|---|
| Sample number | 40 | 20 | 20 | ||
| Sex (F/M) | 19/21 | 11/9 | 8/12 | ns | ns |
| Age | 75.3 | 77.8 | 78 | ns | ns |
| MMSE | 28.6 | 26.6 | 21.1 | 0.018 | <0.0001 |
| APOE ε4 allele positive (%) Homozygous ε4/ε4% | 7/40 (17.5%) 0% | 11/20 (55%) 0% | 10/20 (50%) 20% | ||
| Vitamin B12 (pmol/L) | 303.8 ± 14.5 | 402.5 ± 42.6 | 395.6 ± 27.6 | 0.02 | 0.03 |
| Folate (nmol/L) | 30.1 ± 1.5 | 34.5 ± 2.2 | 33.9 ± 2.9 | ns | ns |
| Homocysteine (µmol/L) | 14.1 ± 0.6 | 14.1 ± 0.9 | 15.6 ± 1.2 | ns | ns |
| Vitamin D3 (nmol/L) | 69.1 ± 3.6 | 89.2 ± 5.8 | 70.7 ± 4.3 | 0.006 | ns |
| CRP (mg/L) | 2.33 ± 0.39 | 1.76 ± 0.39 | 1.41 ± 0.32 | ns | ns |
| MCI Protein Biomarkers | |||
|---|---|---|---|
| Protein | AUC (95% CI) | Sensitivity (%) | Specificity (%) |
| Skeletal/aortic smooth/cardiac actin | 1 (1–1) | 100 | 100 |
| ATP synthase subunit beta | 1 (1–1) | 100 | 100 |
| Keratin type II cytoskeletal 6A | 1 (1–1) | 100 | 100 |
| EGF containing fibulin like extracellular matrix protein 1 | 0.98 (0.91–1) | 95 | 95 |
| Mannan-binding lectin serine protease 1 | 0.91 (0.84–0.99 | 95 | 82.5 |
| Hemoglobin subunit gamma 1 | 0.90 (0.82–0.99) | 92.5 | 84.2 |
| Inter alpha trypsin inhibitor heavy chain H1 | 0.89 (0.81–0.97) | 95 | 72.5 |
| Complement C4A | 0.88 (0.78–0.98) | 85 | 82.5 |
| Hemoglobin subunit epsilon | 0.82 (0.8–0.97) | 80 | 85 |
| AD Protein Biomarkers | |||
| Protein | AUC (95% CI) | Sensitivity (%) | Specificity (%) |
| Skeletal/aortic smooth/cardiac actin | 1 (1–1) | 100 | 100 |
| Keratin type I cytoskeletal 9 | 1 (1–1) | 100 | 100 |
| Keratin type II cytoskeletal 2 epidermal | 1 (1–1) | 100 | 100 |
| Serum amyloid A2 protein | 1 (1–1) | 100 | 100 |
| Fibronectin | 0.92 (0.86) | 80 | 90 |
| Extracellular matrix protein 1 | 0.92 (0.85–0.99) | 90 | 85 |
| Keratin type I cytoskeletal 16 | 0.91 (0.85–0.99) | 100 | 70 |
| Selenoprotein P | 0.89 (0.82–0.97) | 100 | 67.5 |
| Apolipoprotein A | 0.86 (0.78–0.96) | 95 | 70 |
| MCI Metabolite Biomarkers | |||
|---|---|---|---|
| Metabolite | AUC (95% CI) | Sensitivity (%) | Specificity (%) |
| N-Acetyl-alpha-D-glucosamine-1-phosphate | 0.86 (0.77–0.95) | 90 | 70 |
| D-Mannose | 0.85 (0.76–0.94) | 100 | 62.5 |
| Maleic acid | 0.84 (0.75–0.95) | 95 | 70 |
| L-Norleucine | 0.84 (0.74–0.96) | 85 | 77.5 |
| Myo-inositol | 0.84 (0.74–0.94) | 90 | 72.5 |
| L-Glutamine | 0.84 (0.74–096) | 80 | 80 |
| Creatinine-1 | 0.84 (0.74–0.95) | 95 | 72.5 |
| Isopentyl acetate | 0.84 (0.75–0.95) | 70 | 90 |
| Itaconic acid | 0.84 (0.75–0.95) | 70 | 90 |
| AD Metabolite Biomarkers | |||
| Metabolite | AUC (95% CI) | Sensitivity (%) | Specificity (%) |
| Hypoxanthine | 0.86 (0.75–0.98) | 90 | 80 |
| L-Glutamic acid | 0.83 (0.61–0.89) | 80 | 75 |
| Epinephrine | 0.83 (0.73–0.93) | 70 | 85 |
| 3-4-Dihydroxyphenylglycol | 0.83 (0.73–0.94) | 70 | 85 |
| D-sedoheptulose-7-phosphate | 0.83 (0.73–0.93) | 100 | 57.5 |
| N-acetyl-alpha-D-glucosamine-1-phosphate | 0.82 (0.72–0.93) | 85 | 72.5 |
| Uridine | 0.81 (0.71–0.93) | 75 | 82.5 |
| Methylmalonic acid | 0.81 (0.71–0.93) | 75 | 77.5 |
| Erythrose-4-phosphate | 0.81 (0.71–0.93) | 87 | 72.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
François, M.; Karpe, A.V.; Liu, J.-W.; Beale, D.J.; Hor, M.; Hecker, J.; Faunt, J.; Maddison, J.; Johns, S.; Doecke, J.D.; et al. Multi-Omics, an Integrated Approach to Identify Novel Blood Biomarkers of Alzheimer’s Disease. Metabolites 2022, 12, 949. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12100949
François M, Karpe AV, Liu J-W, Beale DJ, Hor M, Hecker J, Faunt J, Maddison J, Johns S, Doecke JD, et al. Multi-Omics, an Integrated Approach to Identify Novel Blood Biomarkers of Alzheimer’s Disease. Metabolites. 2022; 12(10):949. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12100949
Chicago/Turabian StyleFrançois, Maxime, Avinash V. Karpe, Jian-Wei Liu, David J. Beale, Maryam Hor, Jane Hecker, Jeff Faunt, John Maddison, Sally Johns, James D. Doecke, and et al. 2022. "Multi-Omics, an Integrated Approach to Identify Novel Blood Biomarkers of Alzheimer’s Disease" Metabolites 12, no. 10: 949. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12100949