
NMR Metabolomics Reveal Urine Markers of Microbiome Diversity and Identify Benzoate Metabolism as a Mediator between High Microbial Alpha Diversity and Metabolic Health

 , , , , , ,
, , , , , ,
Abstract
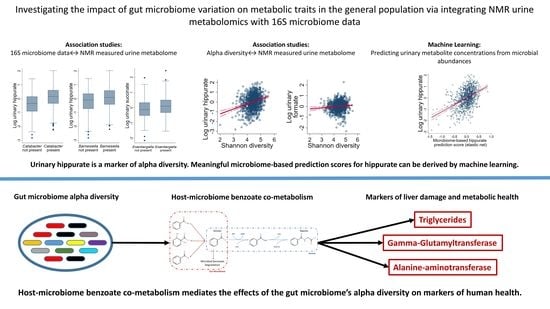
:
1. Introduction
2. Results
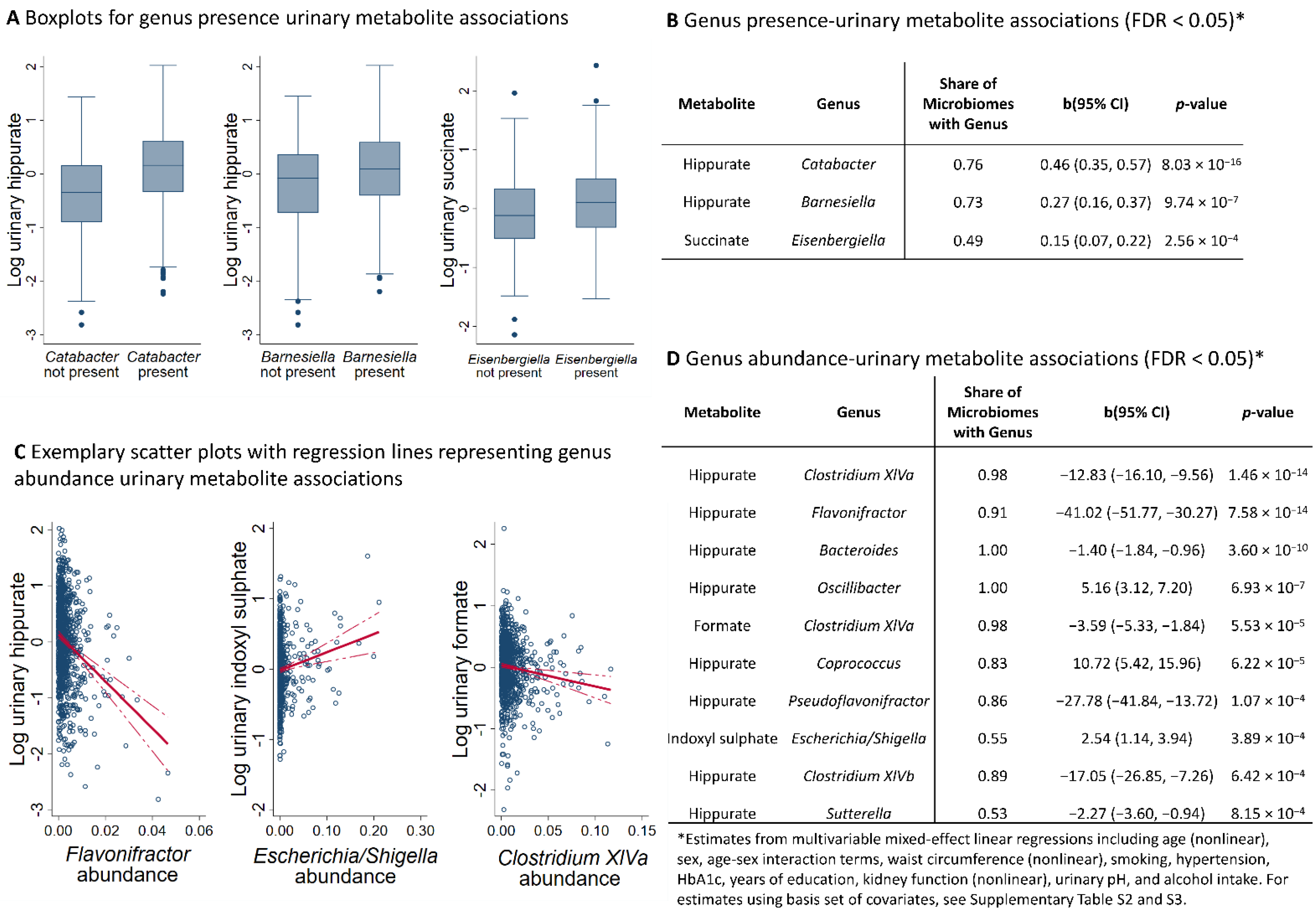
2.1. NMR Metabolomics Revealed Genus Metabolite Associations
2.2. NMR Metabolomics Revealed Markers of Microbial Alpha Diversity
2.3. Microbiome-Based Predictions Scores for Urinary Hippurate Mediated the Associations of SHANNON Diversity to Markers of Metabolic Health
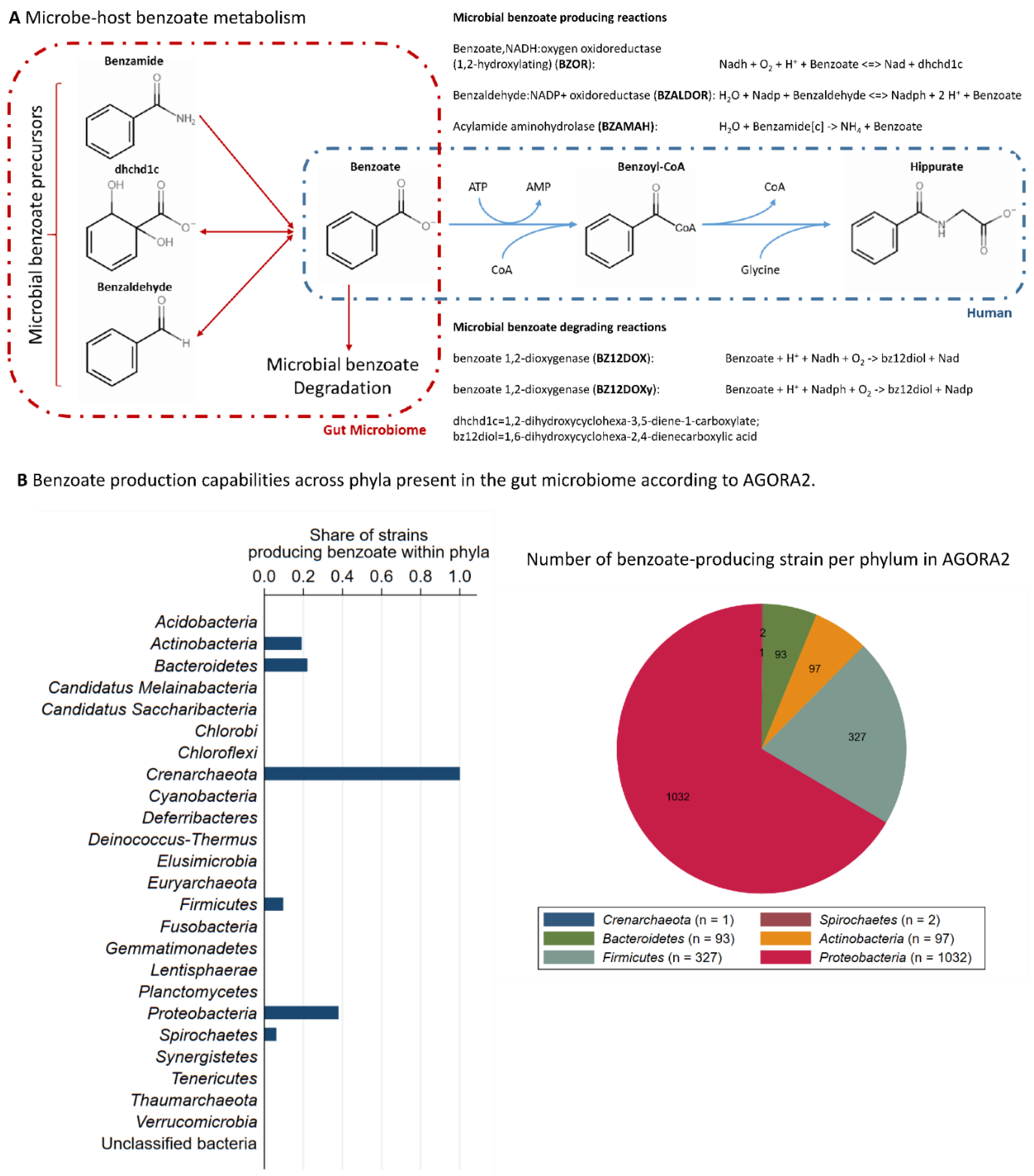
2.4. Functional Annotations of an Independent Published Dataset Indicated a Direct Relationship between Microbial Diversity and Benzoate Metabolism
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Assays and Phenotypes
4.3. 16S rRNA Gene Sequencing and Taxonomic Assignments
4.4. NMR Measurements in SHIP-TREND
4.5. Data Normalisation and Outlier Detection
4.6. Statistical Analyses in SHIP-TREND
4.7. Statistical Analyses on Yachida et al.’s Metagenome Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.; Blaser, M.J. The Human Microbiome: At the Interface of Health and Disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Hollister, E.B.; Gao, C.; Versalovic, J. Compositional and Functional Features of the Gastrointestinal Microbiome and Their Effects on Human Health. Gastroenterology 2014, 146, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the Human Gut Microbiome and Host Metabolism. Nat. Commun. 2019, 10, 4505. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, J.G.; Chain, F.; Martín, R.; Bermúdez-Humarán, L.G.; Courau, S.; Langella, P. Beneficial Effects on Host Energy Metabolism of Short-Chain Fatty Acids and Vitamins Produced by Commensal and Probiotic Bacteria. Microb. Cell Fact. 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Puertollano, E.; Kolida, S.; Yaqoob, P. Biological Significance of Short-Chain Fatty Acid Metabolism by the Intestinal Microbiome. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 139–144. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The Role of Short-Chain Fatty Acids in Health and Disease. In Advances in Immunology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 121, pp. 91–119. ISBN 978-0-12-800100-4. [Google Scholar]
- Buret, A.G.; Allain, T.; Motta, J.-P.; Wallace, J.L. Effects of Hydrogen Sulfide on the Microbiome: From Toxicity to Therapy. Antioxid. Redox Signal. 2021, 36, 211–219. [Google Scholar] [CrossRef]
- Blachier, F.; Andriamihaja, M.; Larraufie, P.; Ahn, E.; Lan, A.; Kim, E. Production of Hydrogen Sulfide by the Intestinal Microbiota and Epithelial Cells and Consequences for the Colonic and Rectal Mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G125–G135. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile Acids and the Gut Microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Thomsen, M.; Vitetta, L. Interaction of Gut Microbiota with Dysregulation of Bile Acids in the Pathogenesis of Nonalcoholic Fatty Liver Disease and Potential Therapeutic Implications of Probiotics. J. Cell Biochem. 2019, 120, 2713–2720. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.W.; Hazen, S.L. Microbiome, Trimethylamine N-Oxide, and Cardiometabolic Disease. Transl. Res. 2017, 179, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [CrossRef] [Green Version]
- Sandrini, S.; Aldriwesh, M.; Alruways, M.; Freestone, P. Microbial Endocrinology: Host–Bacteria Communication within the Gut Microbiome. J. Endocrinol. 2015, 225, R21–R34. [Google Scholar] [CrossRef] [Green Version]
- Bonder, M.J.; Kurilshikov, A.; Tigchelaar, E.F.; Mujagic, Z.; Imhann, F.; Vila, A.V.; Deelen, P.; Vatanen, T.; Schirmer, M.; Smeekens, S.P.; et al. The Effect of Host Genetics on the Gut Microbiome. Nat. Genet. 2016, 48, 1407–1412. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Rühlemann, M.C.; Hermes, B.M.; Bang, C.; Doms, S.; Moitinho-Silva, L.; Thingholm, L.B.; Frost, F.; Degenhardt, F.; Wittig, M.; Kässens, J.; et al. Genome-Wide Association Study in 8956 German Individuals Identifies Influence of ABO Histo-Blood Groups on Gut Microbiome. Nat. Genet. 2021, 53, 147–155. [Google Scholar] [CrossRef]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Raygoza Garay, J.A.; Finnicum, C.T.; Liu, X.; et al. Large-Scale Association Analyses Identify Host Factors Influencing Human Gut Microbiome Composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors Affecting the Composition of the Gut Microbiota, and Its Modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleman, F.D.D.; Valenzano, D.R. Microbiome Evolution during Host Aging. PLoS Pathog. 2019, 15, e1007727. [Google Scholar] [CrossRef] [PubMed]
- Prehn-Kristensen, A.; Zimmermann, A.; Tittmann, L.; Lieb, W.; Schreiber, S.; Baving, L.; Fischer, A. Reduced Microbiome Alpha Diversity in Young Patients with ADHD. PLoS ONE 2018, 13, e0200728. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of Metatranscription in the Inflammatory Bowel Disease Gut Microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]
- Hirano, A.; Umeno, J.; Okamoto, Y.; Shibata, H.; Ogura, Y.; Moriyama, T.; Torisu, T.; Fujioka, S.; Fuyuno, Y.; Kawarabayasi, Y.; et al. Comparison of the Microbial Community Structure between Inflamed and Non-Inflamed Sites in Patients with Ulcerative Colitis: Dysbiosis in Ulcerative Colitis. J. Gastroenterol. Hepatol. 2018, 33, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Stanislawski, M.A.; Dabelea, D.; Lange, L.A.; Wagner, B.D.; Lozupone, C.A. Gut Microbiota Phenotypes of Obesity. npj Biofilms Microbiom. 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Zhu, J.; Le Roy, C.I.; Mompeo, O.; Young, K.; Rebholz, C.M.; Selvin, E.; North, K.E.; Mohney, R.P.; Bell, J.T.; et al. Serum Metabolites Reflecting Gut Microbiome Alpha Diversity Predict Type 2 Diabetes. Gut Microb. 2020, 11, 1632–1642. [Google Scholar] [CrossRef]
- Frost, F.; Weiss, F.U.; Sendler, M.; Kacprowski, T.; Rühlemann, M.; Bang, C.; Franke, A.; Völker, U.; Völzke, H.; Lamprecht, G.; et al. The Gut Microbiome in Patients With Chronic Pancreatitis Is Characterized by Significant Dysbiosis and Overgrowth by Opportunistic Pathogens. Clin. Trans. Gastroenterol. 2020, 11, e00232. [Google Scholar] [CrossRef]
- Pietzner, M.; Budde, K.; Rühlemann, M.; Völzke, H.; Homuth, G.; Weiss, F.U.; Lerch, M.M.; Frost, F. Exocrine Pancreatic Function Modulates Plasma Metabolites Through Changes in Gut Microbiota Composition. J. Clin. Endocrinol. Metab. 2021, 106, e2290–e2298. [Google Scholar] [CrossRef]
- Frost, F.; Kacprowski, T.; Rühlemann, M.; Weiss, S.; Bang, C.; Franke, A.; Pietzner, M.; Aghdassi, A.A.; Sendler, M.; Völker, U.; et al. Carrying Asymptomatic Gallstones Is Not Associated with Changes in Intestinal Microbiota Composition and Diversity but Cholecystectomy with Significant Dysbiosis. Sci. Rep. 2021, 11, 6677. [Google Scholar] [CrossRef]
- Frost, F.; Storck, L.J.; Kacprowski, T.; Gärtner, S.; Rühlemann, M.; Bang, C.; Franke, A.; Völker, U.; Aghdassi, A.A.; Steveling, A.; et al. A Structured Weight Loss Program Increases Gut Microbiota Phylogenetic Diversity and Reduces Levels of Collinsella in Obese Type 2 Diabetics: A Pilot Study. PLoS ONE 2019, 14, e0219489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, F.; Kacprowski, T.; Rühlemann, M.; Pietzner, M.; Bang, C.; Franke, A.; Nauck, M.; Völker, U.; Völzke, H.; Dörr, M.; et al. Long-Term Instability of the Intestinal Microbiome Is Associated with Metabolic Liver Disease, Low Microbiota Diversity, Diabetes Mellitus and Impaired Exocrine Pancreatic Function. Gut 2021, 70, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Wilmanski, T.; Rappaport, N.; Earls, J.C.; Magis, A.T.; Manor, O.; Lovejoy, J.; Omenn, G.S.; Hood, L.; Gibbons, S.M.; Price, N.D. Blood Metabolome Predicts Gut Microbiome α-Diversity in Humans. Nat. Biotechnol. 2019, 37, 1217–1228. [Google Scholar] [CrossRef]
- Volzke, H.; Alte, D.; Schmidt, C.O.; Radke, D.; Lorbeer, R.; Friedrich, N.; Aumann, N.; Lau, K.; Piontek, M.; Born, G.; et al. Cohort Profile: The Study of Health in Pomerania. Int. J. Epidemiol. 2011, 40, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertel, J.; Rotter, M.; Frenzel, S.; Zacharias, H.U.; Krumsiek, J.; Rathkolb, B.; Hrabe de Angelis, M.; Rabstein, S.; Pallapies, D.; Brüning, T.; et al. Dilution Correction for Dynamically Influenced Urinary Analyte Data. Anal. Chim. Acta 2018, 1032, 18–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertel, J.; Heinken, A.; Martinelli, F.; Thiele, I. Integration of Constraint-Based Modeling with Fecal Metabolomics Reveals Large Deleterious Effects of Fusobacterium Spp. on Community Butyrate Production. Gut Microb. 2021, 13, 1915673. [Google Scholar] [CrossRef]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated Analyses of Microbiome and Longitudinal Metabolome Data Reveal Microbial-Host Interactions on Sulfur Metabolism in Parkinson’s Disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and Metabolomic Analyses Reveal Distinct Stage-Specific Phenotypes of the Gut Microbiota in Colorectal Cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Heinken, A.; Acharya, G.; Ravcheev, D.A.; Hertel, J.; Nyga, M.; Okpala, O.E.; Hogan, M.; Magnúsdóttir, S.; Martinelli, F.; Preciat, G.; et al. AGORA2: Large Scale Reconstruction of the Microbiome Highlights Wide-Spread Drug-Metabolising Capacities. Systems Biology. 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.11.09.375451v1 (accessed on 28 January 2022).
- Wishart, D.S. Quantitative Metabolomics Using NMR. TrAC Trends Anal. Chem. 2008, 27, 228–237. [Google Scholar] [CrossRef]
- Wu, S.; Wang, M.; Zhang, M.; He, J.-Q. Metabolomics and Microbiomes for Discovering Biomarkers of Antituberculosis Drugs-Induced Hepatotoxicity. Arch. Biochem. Biophys. 2022, 716, 109118. [Google Scholar] [CrossRef]
- Xu, H.; Tamrat, N.E.; Gao, J.; Xu, J.; Zhou, Y.; Zhang, S.; Chen, Z.; Shao, Y.; Ding, L.; Shen, B.; et al. Combined Signature of the Urinary Microbiome and Metabolome in Patients With Interstitial Cystitis. Front. Cell Infect. Microbiol. 2021, 11, 711746. [Google Scholar] [CrossRef] [PubMed]
- Yaskolka Meir, A.; Tuohy, K.; von Bergen, M.; Krajmalnik-Brown, R.; Heinig, U.; Zelicha, H.; Tsaban, G.; Rinott, E.; Kaplan, A.; Aharoni, A.; et al. The Metabolomic-Gut-Clinical Axis of Mankai Plant-Derived Dietary Polyphenols. Nutrients 2021, 13, 1866. [Google Scholar] [CrossRef]
- Laforest-Lapointe, I.; Becker, A.B.; Mandhane, P.J.; Turvey, S.E.; Moraes, T.J.; Sears, M.R.; Subbarao, P.; Sycuro, L.K.; Azad, M.B.; Arrieta, M.-C. Maternal Consumption of Artificially Sweetened Beverages during Pregnancy Is Associated with Infant Gut Microbiota and Metabolic Modifications and Increased Infant Body Mass Index. Gut Microb. 2021, 13, 1857513. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, H.; Li, Y.; Huang, S.; Zhang, L.; Cao, C.; Baker, P.N.; Tong, C.; Zheng, P.; Qi, H. Altered Gut Bacterial and Metabolic Signatures and Their Interaction in Gestational Diabetes Mellitus. Gut Microb. 2020, 12, 1840765. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Gu, X.; Li, L.-X.; Li, M.; Li, B.; Cui, X.; Zuo, X.-L. Microbial and Metabolomic Profiles in Correlation with Depression and Anxiety Co-Morbidities in Diarrhoea-Predominant IBS Patients. BMC Microbiol. 2020, 20, 168. [Google Scholar] [CrossRef]
- Jeffery, I.B.; Das, A.; O’Herlihy, E.; Coughlan, S.; Cisek, K.; Moore, M.; Bradley, F.; Carty, T.; Pradhan, M.; Dwibedi, C.; et al. Differences in Fecal Microbiomes and Metabolomes of People With vs Without Irritable Bowel Syndrome and Bile Acid Malabsorption. Gastroenterology 2020, 158, 1016–1028.e8. [Google Scholar] [CrossRef]
- Dewulf, E.M.; Cani, P.D.; Claus, S.P.; Fuentes, S.; Puylaert, P.G.; Neyrinck, A.M.; Bindels, L.B.; de Vos, W.M.; Gibson, G.R.; Thissen, J.-P.; et al. Insight into the Prebiotic Concept: Lessons from an Exploratory, Double Blind Intervention Study with Inulin-Type Fructans in Obese Women. Gut 2013, 62, 1112–1121. [Google Scholar] [CrossRef]
- Prochazkova, M.; Budinska, E.; Kuzma, M.; Pelantova, H.; Hradecky, J.; Heczkova, M.; Daskova, N.; Bratova, M.; Modos, I.; Videnska, P.; et al. Vegan Diet Is Associated With Favorable Effects on the Metabolic Performance of Intestinal Microbiota: A Cross-Sectional Multi-Omics Study. Front. Nutr. 2021, 8, 783302. [Google Scholar] [CrossRef]
- West, K.A.; Kanu, C.; Maric, T.; McDonald, J.A.K.; Nicholson, J.K.; Li, J.V.; Johnson, M.R.; Holmes, E.; Savvidou, M.D. Longitudinal Metabolic and Gut Bacterial Profiling of Pregnant Women with Previous Bariatric Surgery. Gut 2020, 69, 1452–1459. [Google Scholar] [CrossRef]
- Wijeyesekera, A.; Wagner, J.; De Goffau, M.; Thurston, S.; Rodrigues Sabino, A.; Zaher, S.; White, D.; Ridout, J.; Peters, M.J.; Ramnarayan, P.; et al. Multi-Compartment Profiling of Bacterial and Host Metabolites Identifies Intestinal Dysbiosis and Its Functional Consequences in the Critically Ill Child. Crit. Care Med. 2019, 47, e727–e734. [Google Scholar] [CrossRef]
- Pietzke, M.; Meiser, J.; Vazquez, A. Formate Metabolism in Health and Disease. Mol. Metab. 2020, 33, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.R.; Winter, M.G.; Duerkop, B.A.; Spiga, L.; Furtado de Carvalho, T.; Zhu, W.; Gillis, C.C.; Büttner, L.; Smoot, M.P.; Behrendt, C.L.; et al. Microbial Respiration and Formate Oxidation as Metabolic Signatures of Inflammation-Associated Dysbiosis. Cell Host Microb. 2017, 21, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClean, S.; Callaghan, M. Burkholderia Cepacia Complex: Epithelial Cell–Pathogen Confrontations and Potential for Therapeutic Intervention. J. Med. Microbiol. 2009, 58, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, A.; O’Donoghue, M.; Feeney, A.; Sleator, R.D. Acinetobacter Baumannii: An Emerging Opportunistic Pathogen. Virulence 2012, 3, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Schaal, M.; Jahn, D.; Schmidt-Hohagen, K. Metabolism the Difficile Way: The Key to the Success of the Pathogen Clostridioides Difficile. Front. Microbiol. 2019, 10, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.L.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete Genome Sequence of Pseudomonas Aeruginosa PAO1, an Opportunistic Pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- McGrath, K.H.; Pitt, J.; Bines, J.E. Small Intestinal Bacterial Overgrowth in Children with Intestinal Failure on Home Parenteral Nutrition. JGH Open 2019, 3, 394–399. [Google Scholar] [CrossRef]
- Sims, G.E.; Kim, S.-H. Whole-Genome Phylogeny of Escherichia Coli/Shigella Group by Feature Frequency Profiles (FFPs). Proc. Natl. Acad. Sci. USA 2011, 108, 8329–8334. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.-H.; Ma, M.-C.; Liao, M.-T.; Zheng, C.-M.; Lu, K.-C.; Liao, C.-H.; Hou, Y.-C.; Liu, W.-C.; Lu, C.-L. Indoxyl Sulfate, a Tubular Toxin, Contributes to the Development of Chronic Kidney Disease. Toxins 2020, 12, 684. [Google Scholar] [CrossRef]
- Brial, F.; Chilloux, J.; Nielsen, T.; Vieira-Silva, S.; Falony, G.; Andrikopoulos, P.; Olanipekun, M.; Hoyles, L.; Djouadi, F.; Neves, A.L.; et al. Human and Preclinical Studies of the Host–Gut Microbiome Co-Metabolite Hippurate as a Marker and Mediator of Metabolic Health. Gut 2021, 70, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.R.; Cox, I.J.; Walker, D.G.; Cobbold, J.F.; Taylor-Robinson, S.D.; Marshall, S.E.; Orchard, T.R. Differences in Gut Microbial Metabolism Are Responsible for Reduced Hippurate Synthesis in Crohn’s Disease. BMC Gastroenterol. 2010, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallister, T.; Jackson, M.A.; Martin, T.C.; Zierer, J.; Jennings, A.; Mohney, R.P.; MacGregor, A.; Steves, C.J.; Cassidy, A.; Spector, T.D.; et al. Hippurate as a Metabolomic Marker of Gut Microbiome Diversity: Modulation by Diet and Relationship to Metabolic Syndrome. Sci. Rep. 2017, 7, 13670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, M.; Lomash, A.; Kapoor, S.; Pandey, R.; Chauhan, N.S. Mapping of the Benzoate Metabolism by Human Gut Microbiome Indicates Food-Derived Metagenome Evolution. Sci. Rep. 2021, 11, 5561. [Google Scholar] [CrossRef]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [Green Version]
- Nishijima, S.; Suda, W.; Oshima, K.; Kim, S.-W.; Hirose, Y.; Morita, H.; Hattori, M. The Gut Microbiome of Healthy Japanese and Its Microbial and Functional Uniqueness. DNA Res. 2016, 23, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Winter, T.; Friedrich, N.; Lamp, S.; Schäfer, C.; Schattschneider, M.; Bollmann, S.; Brümmer, D.; Riemann, K.; Petersmann, A.; Nauck, M. The Integrated Research Biobank of the University Medicine Greifswald. Open J. Bioresour. 2020, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Frost, F.; Kacprowski, T.; Rühlemann, M.; Bülow, R.; Kühn, J.-P.; Franke, A.; Heinsen, F.-A.; Pietzner, M.; Nauck, M.; Völker, U.; et al. Impaired Exocrine Pancreatic Function Associates With Changes in Intestinal Microbiota Composition and Diversity. Gastroenterology 2019, 156, 1010–1015. [Google Scholar] [CrossRef]
- Hertel, J.; Friedrich, N.; Wittfeld, K.; Pietzner, M.; Budde, K.; Van der Auwera, S.; Lohmann, T.; Teumer, A.; Völzke, H.; Nauck, M.; et al. Measuring Biological Age via Metabonomics: The Metabolic Age Score. J. Proteome Res. 2016, 15, 400–410. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Hicks, R.; Tingley, D. Causal Mediation Analysis. Stata J. 2011, 11, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Heinken, A.; Magnúsdóttir, S.; Fleming, R.M.T.; Thiele, I. DEMETER: Efficient Simultaneous Curation of Genome-Scale Reconstructions Guided by Experimental Data and Refined Gene Annotations. Bioinformatics 2021, 37, 3974–3975. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SHIP-TREND with Faecal Samples (n = 3637) | SHIP-TREND with Faecal Samples and NMR Metabolite Measurements (n = 951) | |||
|---|---|---|---|---|
| Variable | Missing Values, % | Mean (SD) or Share, % | Missing Values, % | Mean (SD) or Share, % |
| Age, years | 0.00 | 51.33 (14.94) | 0.00 | 50.21 (13.63) |
| Female, % | 0.00 | 51.69% | 0.00 | 56.68% |
| Body mass index, kg/m2 | 0.16 | 28.02 (5.15) | 0.00 | 27.37 (4.57) |
| Waist circumference, cm | 0.27 | 90.79 (14.35) | 0.00 | 88.08 (12.82) |
| Current smoking, % | 0.25 | 26.82% | 0.11 | 21.79% |
| Average alcohol consumption over the last 30 d, g/d | 0.91 | 8.83 (13.79) | 0.63 | 8.56 (13.31) |
| Diabetes a | 0.16 | 11.54% | 0.00 | 2.73% |
| Hypertonia b | 0.33 | 46.43% | 0.11 | 39.58% |
| HbA1c, % | 0.19 | 5.34 (0.83) | 0.11 | 5.19 (0.56) |
| eGFR, mL/min | 0.16 | 89.73 (18.81) | 0.00 | 92.12 (17.12) |
| White blood cell count, Gpt/L | 1.95 | 6.08 (2.70) | 0.21 | 5.73 (1.48) |
| Triglycerides, mmol/L | 0.16 | 1.64 (1.24) | 0.00 | 1.42 (0.86) |
| Ratio of TC/HDL-C | 0.16 | 4.03 (1.26) | 0.00 | 3.93 (1.14) |
| Fibrinogen, g/L | 2.64 | 3.07 (0.74) | 0.95 | 3.02 (0.73) |
| CRP (high sensitive), mg/L | 4.67 | 2.52 (3.93) | 3.36 | 2.29 (3.67) |
| GGT, μkat/L | 0.19 | 0.70 (0.80) | 0.00 | 0.65 (0.63) |
| ALAT, μkat/L | 0.22 | 0.45 (0.30) | 0.11 | 0.44 (0.29) |
| ASAT, μkat/L | 0.30 | 0.33 (0.19) | 0.21 | 0.32 (0.17) |
| Urinary Hippurate (n = 951) | Shannon Entropy (n = 3637) b | Microbiome-Based Hippurate Prediction Score (n = 3637) b | ||||
|---|---|---|---|---|---|---|
| Marker | b (95% CI) * | p-Value * | b (95% CI) * | p-Value * | b (95% CI) * | p-Value * |
| Log hs-CRP | −0.09 (−0.17, −0.01) | 2.40 × 10−2 | −0.01 (−0.03, 0.00) | 7.55 × 10−2 | −0.01 (−0.02, −0.00) | 1.65 × 10−2 |
| Fibrinogen | −0.02 (−0.09, 0.04) | 4.59 × 10−1 | −0.00 (−0.02, 0.01) | 6.27 × 10−1 | 0.01 (−0.00, 0.02) | 2.05 × 10−1 |
| White blood cell count | −0.15 (−0.28, −0.03) | 1.68 × 10−2 | −0.00 (−0.01, 0.00) | 1.76 × 10−1 | −0.00 (−0.01, 0.00) | 3.76 × 10−1 |
| Triglycerides | −0.02 (−0.09, 0.05) | 5.24 × 10−1 | −0.03 (−0.04, −0.02) | 3.83 × 10−6 | −0.03 (−0.04, −0.02) | 3.35 × 1014 |
| Ratio of TC/HDL-C | 0.04 (−0.05, 0.12) | 3.95 × 10−1 | −0.01 (−0.02, 0.00) | 1.58 × 10−1 | −0.00 (−0.01, 0.01) | 8.08 × 10−1 |
| Baseline glucose | −0.03 (−0.09, 0.02) | 1.87 × 10−1 | 0.00 (−0.01, 0.01) | 9.32 × 10−1 | −0.00 (−0.01, 0.00) | 1.67 × 10−1 |
| HbA1c | 0.004 (−0.04, 0.05) | 8.45 × 10−1 | 0.01 (−0.01, 0.03) | 2.13 × 10−1 | 0.01 (−0.00, 0.02) | 1.69 × 10−1 |
| Log GGT | −0.05 (−0.09, −0.01) | 2.05 × 10−2 | −0.07 (−0.10, 0.04) | 1.64 × 10−7 | −0.10 (−0.12, 0.08) | 4.13 × 1022 |
| Log ALAT | −0.03 (−0.06, 0.01) | 1.07 × 10−1 | −0.04 (−0.07, 0.01) | 9.85 × 10−3 | −0.07 (−0.09, 0.04) | 4.77 × 10−9 |
| Log ASAT | −0.03 (−0.06, 0.00) | 6.29 × 10−2 | −0.02 (−0.06, 0.01) | 1.62 × 10−1 | −0.06 (−0.08, 0.03) | 4.33 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hertel, J.; Fässler, D.; Heinken, A.; Weiß, F.U.; Rühlemann, M.; Bang, C.; Franke, A.; Budde, K.; Henning, A.-K.; Petersmann, A.; et al. NMR Metabolomics Reveal Urine Markers of Microbiome Diversity and Identify Benzoate Metabolism as a Mediator between High Microbial Alpha Diversity and Metabolic Health. Metabolites 2022, 12, 308. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12040308
Hertel J, Fässler D, Heinken A, Weiß FU, Rühlemann M, Bang C, Franke A, Budde K, Henning A-K, Petersmann A, et al. NMR Metabolomics Reveal Urine Markers of Microbiome Diversity and Identify Benzoate Metabolism as a Mediator between High Microbial Alpha Diversity and Metabolic Health. Metabolites. 2022; 12(4):308. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12040308
Chicago/Turabian StyleHertel, Johannes, Daniel Fässler, Almut Heinken, Frank U. Weiß, Malte Rühlemann, Corinna Bang, Andre Franke, Kathrin Budde, Ann-Kristin Henning, Astrid Petersmann, and et al. 2022. "NMR Metabolomics Reveal Urine Markers of Microbiome Diversity and Identify Benzoate Metabolism as a Mediator between High Microbial Alpha Diversity and Metabolic Health" Metabolites 12, no. 4: 308. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12040308