
Scan-Centric, Frequency-Based Method for Characterizing Peaks from Direct Injection Fourier Transform Mass Spectrometry Experiments


Abstract
:1. Introduction
2. Results
2.1. Simplistically Averaged Data Have Bad Relative Intensities
2.2. m/z to Frequency
2.3. Sliding Window Density to Remove Noise
2.4. Peak Characterization Using Quadratic Fit
2.5. Breaking Up Initial Regions
2.6. Normalization of Scans
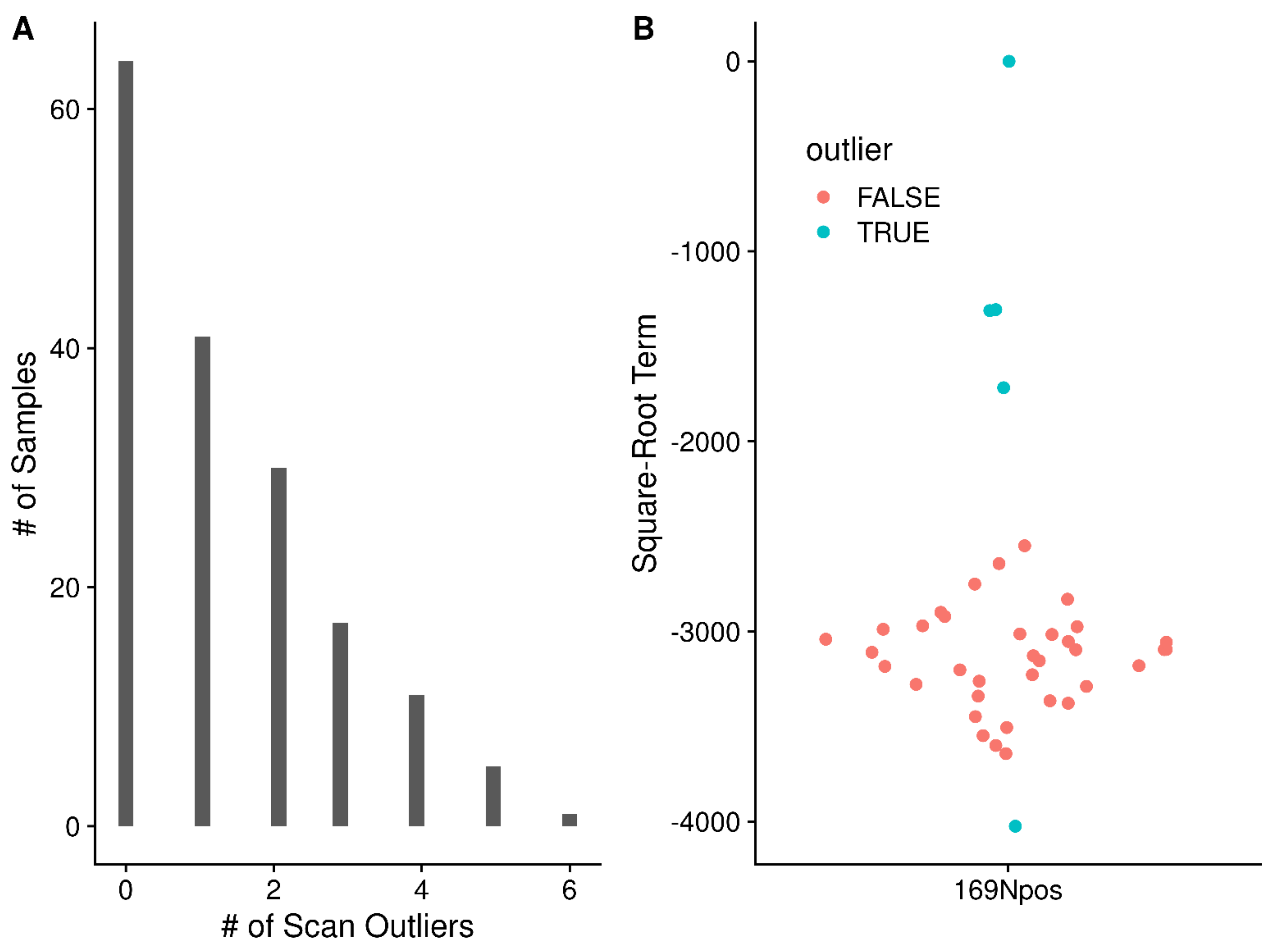
2.7. Mitigation of High Peak Density Artifacts
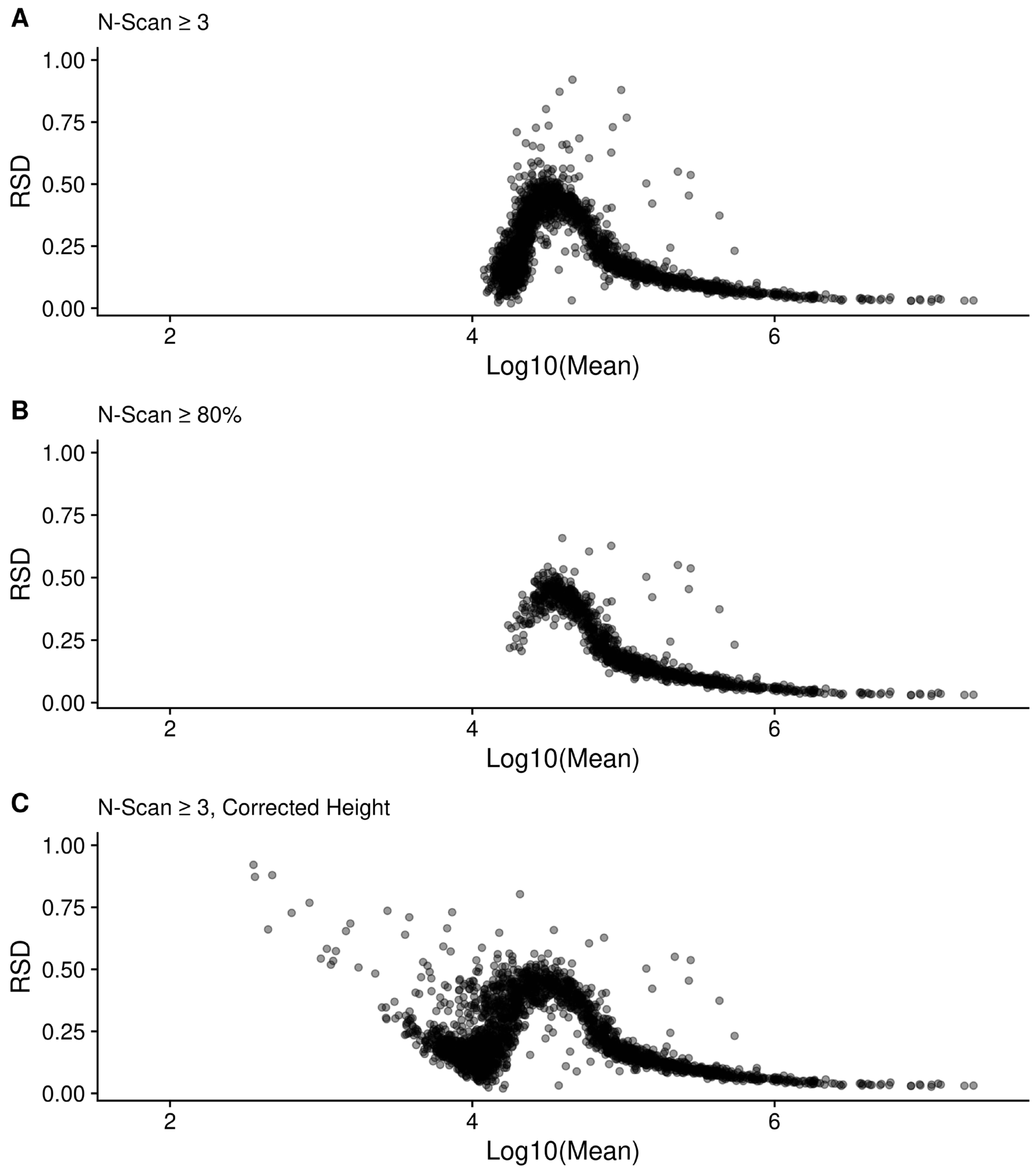
2.8. Changes in Relative Standard Deviation (RSD)
2.9. Difference to Relative Natural Abundance
2.10. Method Specific Peaks
2.11. Changes in p-Values on a Large Dataset
2.12. Quality Control and Quality Analysis
3. Discussion
4. Materials and Methods
4.1. Samples and Overall Processing
- No noise removal, no normalization (noperc_nonorm);
- Noise removal, no normalization (perc99_nonorm);
- Noise removal, single-pass normalization with all peaks (singlenorm);
- Noise removal, single-pass normalization with high ratio peaks (singlenorm_int);
- Noise removal, two-pass normalization (doublenorm);
- Noise removal, two-pass normalization (filtersd);
- Scans merged and then centroids generated by MSnbase (using combineSpectra and pickPeaks);
- Scans merged and peak-list exported by Xcalibur.
4.2. Matching Peaks
4.3. Conversion of m/z to Frequency
4.4. Frequency Intervals
4.5. Interval Range Based Data
4.6. Peak Containing Intervals
4.7. Peak Detection and Centroided Values
4.8. Scan to Scan Normalization
4.9. Full Scan-Centric Characterization
4.10. Correction of Height and Standard Deviation
4.11. Marking High Frequency Standard Deviation Peaks
4.12. Calculation of Relative Standard Deviation
4.13. Scan-Centric Peak Assignment
4.14. Consistently Assigned Lipid Spectral Feature (Corresponded Peak) Generation and Peak Intensity Normalization
4.15. Peak—Peak NAP Height Ratios
4.16. Differential Analysis of Large Dataset
4.17. Software Used
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Higashi, R.M.; Fan, T.W.-M.; Lorkiewicz, P.K.; Moseley, H.N.B.; Lane, A.N. Stable Isotope-Labeled Tracers for Metabolic Pathway Elucidation by GC-MS and FT-MS. In Mass Spectrometry in Metabolomics: Methods and Protocols; Raftery, D., Ed.; Humana Press: New York, NY, USA, 2014; Volume 1198, pp. 147–167. [Google Scholar] [CrossRef] [Green Version]
- Moseley, H.N. Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry. BMC Bioinform. 2010, 11, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreer, W.J.; Flight, R.M.; Moseley, H.N.B. A Computational Framework for High-Throughput Isotopic Natural Abundance Correction of Omics-Level Ultra-High Resolution FT-MS Datasets. Metabolites 2013, 3, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Parsons, L.R.; Su, X. AccuCor2: Isotope natural abundance correction for dual-isotope tracer experiments. Lab. Investig. 2021, 101, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.M.; Flight, R.M.; Moseley, H.N.B. Small Molecule Isotope Resolved Formula Enumeration: A Methodology for Assigning Isotopologues and Metabolite Formulas in Fourier Transform Mass Spectra. Anal. Chem. 2019, 91, 8933–8940. [Google Scholar] [CrossRef]
- Eyles, S.J.; Kaltashov, I.A. Methods to study protein dynamics and folding by mass spectrometry. Methods 2004, 34, 88–99. [Google Scholar] [CrossRef]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, T.W.-M.; Lane, A.N.; Higashi, R.M. Chloroformate derivatization for tracing the fate of Amino acids in cells and tissues by multiple stable isotope resolved metabolomics (mSIRM). Anal. Chim. Acta 2017, 976, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Chokkathukalam, A.; Jankevics, A.; Burgess, K.E.V.; Breitling, R.; Barrett, M.P. Stable Isotope-Assisted Metabolomics for Network-Wide Metabolic Pathway Elucidation. Anal. Chem. 2012, 84, 8442–8447. [Google Scholar] [CrossRef]
- Hiller, K.; Metallo, C.M.; Kelleher, J.K.; Stephanopoulos, G. Nontargeted Elucidation of Metabolic Pathways Using Stable-Isotope Tracers and Mass Spectrometry. Anal. Chem. 2010, 82, 6621–6628. [Google Scholar] [CrossRef]
- Fan, T.W.-M.; Lorkiewicz, P.K.; Sellers, K.; Moseley, H.; Higashi, R.M.; Lane, A.N. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol. Ther. 2012, 133, 366–391. [Google Scholar] [CrossRef] [Green Version]
- Moseley, H.N.; Lane, A.N.; Belshoff, A.C.; Higashi, R.M.; Fan, T.W. A novel deconvolution method for modeling UDP-N-acetyl-D-glucosamine biosynthetic pathways based on 13C mass isotopologue profiles under non-steady-state conditions. BMC Biol. 2011, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Sellers, K.; Fox, M.P.; Bousamra, M.; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Verdegem, D.; Moseley, H.N.B.; Vermaelen, W.; Sanchez, A.A.; Ghesquière, B. MAIMS: A software tool for sensitive metabolic tracer analysis through the deconvolution of 13C mass isotopologue profiles of large composite metabolites. Metabolomics 2017, 13, 123. [Google Scholar] [CrossRef]
- Jin, H.; Moseley, H.N.B. Moiety modeling framework for deriving moiety abundances from mass spectrometry measured isotopologues. BMC Bioinform. 2019, 20, 254. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Moseley, H.N. Robust Moiety Model Selection Using Mass Spectrometry Measured Isotopologues. Metabolites 2020, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.M.; Flight, R.M.; Wang, Q.J.; Higashi, R.M.; Fan, T.W.-M.; Lane, A.N.; Moseley, H.N.B. New methods to identify high peak density artifacts in Fourier transform mass spectra and to mitigate their effects on high-throughput metabolomic data analysis. Metabolomics 2018, 14, 125. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.M.; Flight, R.M.; Moseley, H.N. Deriving Lipid Classification Based on Molecular Formulas. Metabolites 2020, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.M.; Flight, R.M.; Moseley, H.N.B. Untargeted Lipidomics of Non-Small Cell Lung Carcinoma Demonstrates Differentially Abundant Lipid Classes in Cancer vs. Non-Cancer Tissue. Metabolites 2021, 11, 740. [Google Scholar] [CrossRef]
- Moseley, H.N. Error Analysis and Propagation In Metabolomics Data Analysis. Comput. Struct. Biotechnol. J. 2013, 4, e201301006. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, W.S.; Grosse, E.; Shyu, W.M. Local regression models. In Statistical Models in S; Chambers, J.M., Hastie, T.J., Eds.; Wadsworth & Brooks/Cole: Pacific Grove, CA, USA, 1992. [Google Scholar]
- Sampford, M.R. The Truncated Negative Binomial Distribution. Biometrika 1955, 42, 58. [Google Scholar] [CrossRef]
- Ledford, E.B.; Rempel, D.L.; Gross, M.L. Space charge effects in Fourier transform mass spectrometry. II. Mass calibration. Anal. Chem. 1984, 56, 2744–2748. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Borchers, H.W. Pracma: Practical Numerical Math Functions. 2021. Available online: https://CRAN.R-project.org/package=pracma (accessed on 7 December 2021).
- Flight, R.M.; Bhatt, P.S.; Moseley, H.N. Information-Content-Informed Kendall-Tau Correlation: Utilizing Missing Values. bioRvix 2022. preprint. [Google Scholar] [CrossRef]
- Truncated Normal Distribution. Wikipedia. 2022. Available online: https://en.wikipedia.org/w/index.php?title=Truncated_normal_distribution&oldid=1074943875 (accessed on 16 March 2022).
- Burkardt, J. The Truncated Normal Distribution. Available online: https://people.sc.fsu.edu/~jburkardt/presentations/truncated_normal.pdf (accessed on 22 March 2022).
- Gentleman, R.; Carey, V.J.; Huber, W.; Hahne, F. Genefilter: Methods for Filtering Genes from High-Throughput Experiments. Available online: https://bioconductor.org/packages/3.14/bioc/html/genefilter.html (accessed on 26 October 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 20 May 2021).
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Landau, W.M. The targets R package: A dynamic Make-like function-oriented pipeline toolkit for reproducibility and high-performance computing. J. Open Source Softw. 2021, 6, 2959. [Google Scholar] [CrossRef]
- Ushey, K. Renv: Project Environments. 2022. Available online: https://CRAN.R-project.org/package=renv (accessed on 28 February 2022).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 25 June 2021).
- Pedersen, T.L. Patchwork: The Composer of Plots. 2020. Available online: https://CRAN.R-project.org/package=patchwork (accessed on 17 December 2020).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, C.O. Ggridges: Ridgeline Plots in “ggplot2”. 2021. Available online: https://CRAN.R-project.org/package=ggridges (accessed on 8 January 2021).
- Pedersen, T.L. Ggforce: Accelerating “ggplot2”. 2021. Available online: https://CRAN.R-project.org/package=ggforce (accessed on 5 March 2021).
- Gatto, L.; Gibb, S.; Rainer, J. MSnbase, efficient and elegant R-based processing and visualisation of raw mass spectrometry data. J. Proteome Res. 2021, 20, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Gatto, L.; Lilley, K. MSnbase—An R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28, 288–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flight, R.M.; Moseley, H.N. Visualization Quality Control: Development of Visualization Methods for Quality Control. 2021. Available online: https://moseleybioinformaticslab.github.io/visualizationQualityControl (accessed on 3 December 2021).
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation. 2022. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 8 February 2022).
- Wickham, H.; Girlich, M. Tidyr: Tidy Messy Data. 2022. Available online: https://CRAN.R-project.org/package=tidyr (accessed on 1 February 2022).
- Vaughan, D.; Dancho, M. Furrr: Apply Mapping Functions in Parallel Using Futures. 2021. Available online: https://CRAN.R-project.org/package=furrr (accessed on 30 June 2021).
- Allaire, J.; Xie, Y.; McPherson, J.; Luraschi, J.; Ushey, K.; Atkins, A.; Wickham, H.; Cheng, J.; Chang, W.; Iannone, R. Rmarkdown: Dynamic Documents for R. 2021. Available online: https://github.com/rstudio/rmarkdown (accessed on 1 April 2022).
- Xie, Y.; Allaire, J.J.; Grolemund, G. R Markdown: The Definitive Guide; Chapman and Hall/CRC: Boca Raton, FL, USA, 2018; Available online: https://bookdown.org/yihui/rmarkdown (accessed on 1 April 2022).
- Xie, Y.; Dervieux, C.; Riederer, E. R Markdown Cookbook; Chapman and Hall/CRC: Boca Raton, FL, USA, 2020; Available online: https://bookdown.org/yihui/rmarkdown-cookbook (accessed on 1 April 2022).
- Flight, R.M.; Mitchell, J.M.; Moseley, H.N.B. Moseley Bioinformatics Lab/Manuscript.Peak Characterization. 2022. Available online: https://zenodo.org/record/6453346#.YpiialRBxPY (accessed on 1 April 2022).
- Flight, R.M.; Moseley, H.N.B. Moseley Bioinformatics Lab/FTMS.Peak Characterization: V0.1.102. Zenodo. 2022. Available online: https://zenodo.org/record/6453304#.YpiiwlRBxPY (accessed on 1 April 2022).



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Processed | Mean | Sd | Median | Mode 1 | Mode 2 | Max |
|---|---|---|---|---|---|---|---|
| 1ecf | filtersd | 0.26 | 0.09 | 0.25 | 0.26 | 1.37 | |
| 1ecf | doublenorm | 0.26 | 0.09 | 0.26 | 0.26 | 1.37 | |
| 1ecf | singlenorm_int | 0.26 | 0.10 | 0.26 | 0.26 | 1.41 | |
| 1ecf | singlenorm | 0.26 | 0.10 | 0.25 | 0.28 | 1.43 | |
| 1ecf | perc99_nonorm | 0.31 | 0.12 | 0.31 | 0.32 | 1.19 | |
| 1ecf | noperc_nonorm | 0.31 | 0.12 | 0.30 | 0.32 | 1.19 | |
| 1ecf | msnbase_only | 0.37 | 0.14 | 0.36 | 0.35 | 1.19 | |
| 2ecf | filtersd | 0.26 | 0.09 | 0.26 | 0.27 | 1.01 | |
| 2ecf | doublenorm | 0.27 | 0.10 | 0.26 | 0.27 | 1.05 | |
| 2ecf | singlenorm_int | 0.27 | 0.10 | 0.26 | 0.27 | 1.05 | |
| 2ecf | singlenorm | 0.26 | 0.10 | 0.26 | 0.27 | 1.03 | |
| 2ecf | perc99_nonorm | 0.26 | 0.11 | 0.26 | 0.28 | 1.08 | |
| 2ecf | noperc_nonorm | 0.26 | 0.11 | 0.26 | 0.29 | 1.08 | |
| 2ecf | msnbase_only | 0.29 | 0.11 | 0.29 | 0.30 | 0.99 | |
| 49lipid | filtersd | 0.25 | 0.13 | 0.23 | 0.20 | 0.37 | 1.13 |
| 49lipid | doublenorm | 0.25 | 0.14 | 0.23 | 0.19 | 0.37 | 1.13 |
| 49lipid | singlenorm_int | 0.25 | 0.14 | 0.23 | 0.20 | 0.37 | 1.13 |
| 49lipid | singlenorm | 0.25 | 0.14 | 0.23 | 0.19 | 0.37 | 1.13 |
| 49lipid | perc99_nonorm | 0.27 | 0.15 | 0.25 | 0.16 | 0.23 | 1.13 |
| 49lipid | noperc_nonorm | 0.27 | 0.15 | 0.25 | 0.16 | 0.23 | 1.13 |
| 49lipid | msnbase_only | 0.26 | 0.15 | 0.22 | 0.14 | 0.42 | 1.14 |
| 97lipid | filtersd | 0.24 | 0.14 | 0.20 | 0.16 | 0.41 | 2.05 |
| 97lipid | doublenorm | 0.24 | 0.15 | 0.20 | 0.15 | 0.41 | 2.05 |
| 97lipid | singlenorm_int | 0.24 | 0.15 | 0.20 | 0.16 | 0.41 | 2.05 |
| 97lipid | singlenorm | 0.24 | 0.15 | 0.20 | 0.15 | 0.41 | 2.04 |
| 97lipid | perc99_nonorm | 0.23 | 0.14 | 0.19 | 0.15 | 0.41 | 2.03 |
| 97lipid | noperc_nonorm | 0.23 | 0.14 | 0.19 | 0.15 | 0.41 | 2.03 |
| 97lipid | msnbase_only | 0.34 | 0.20 | 0.30 | 0.19 | 0.43 | 1.94 |
| Method | Set_Sizes | ||||
|---|---|---|---|---|---|
| Scan-centric | x | x | 2937 | ||
| Xcalibur | x | x | x | 68,244 | |
| MSnbase | x | x | 10,330 | ||
| comb_sizes | 778 | 2159 | 65,307 | 9552 |
| Method | Set_Sizes | ||||||
|---|---|---|---|---|---|---|---|
| Scan-centric | x | x | x | x | 2405 | ||
| Xcalibur | x | x | x | 1747 | |||
| MSnbase | x | x | x | 3263 | |||
| comb_sizes | 448 | 502 | 472 | 983 | 797 | 2343 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flight, R.M.; Mitchell, J.M.; Moseley, H.N.B. Scan-Centric, Frequency-Based Method for Characterizing Peaks from Direct Injection Fourier Transform Mass Spectrometry Experiments. Metabolites 2022, 12, 515. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12060515
Flight RM, Mitchell JM, Moseley HNB. Scan-Centric, Frequency-Based Method for Characterizing Peaks from Direct Injection Fourier Transform Mass Spectrometry Experiments. Metabolites. 2022; 12(6):515. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12060515
Chicago/Turabian StyleFlight, Robert M., Joshua M. Mitchell, and Hunter N. B. Moseley. 2022. "Scan-Centric, Frequency-Based Method for Characterizing Peaks from Direct Injection Fourier Transform Mass Spectrometry Experiments" Metabolites 12, no. 6: 515. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12060515