Ubiquitomics: An Overview and Future


1
Target Discovery Institute, Nuffield Department of Medicine, University of Oxford, Oxford OX3 7FZ, UK
2
St Anne’s College, University of Oxford, Oxford OX2 6HS, UK
3
Centre for Medicines Discovery, Nuffield Department of Medicine, University of Oxford, Oxford OX3 7FZ, UK
4
Chinese Academy of Medical Sciences Oxford Institute (CAMS), Nuffield Department of Medicine, University of Oxford, Oxford OX3 7FZ, UK
*
Author to whom correspondence should be addressed.
Biomolecules 2020, 10(10), 1453; https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101453
Submission received: 23 September 2020
/
Revised: 13 October 2020
/
Accepted: 14 October 2020
/
Published: 17 October 2020
(This article belongs to the Special Issue Looking Back and Ahead: Emerging Concepts in Ubiquitin and UBLs)
Abstract
:Covalent attachment of ubiquitin, a small globular polypeptide, to protein substrates is a key post-translational modification that determines the fate, function, and turnover of most cellular proteins. Ubiquitin modification exists as mono- or polyubiquitin chains involving multiple ways how ubiquitin C-termini are connected to lysine, perhaps other amino acid side chains, and N-termini of proteins, often including branching of the ubiquitin chains. Understanding this enormous complexity in protein ubiquitination, the so-called ‘ubiquitin code’, in combination with the 1000 enzymes involved in controlling ubiquitin recognition, conjugation, and deconjugation, calls for novel developments in analytical techniques. Here, we review different headways in the field mainly driven by mass spectrometry and chemical biology, referred to as “ubiquitomics”, aiming to understand this system’s biological diversity.
1. Ubiquitous and Complex
Protein ubiquitination is a post-translational modification (PTM) that involves the reversible attachment of ubiquitin to amino acid side chains, most commonly a lysine, on the target protein [1]. Ubiquitin conjugation onto substrates is carried out by a series of three enzymatic steps, with a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3) acting in a sequential manner. The modification can be removed by deubiquitinases (DUBs). Ubiquitin was first named for its ubiquitous expression across eukaryotes [2]. This name is more apt than ever, with ubiquitin and ubiquitin-like proteins contributing to regulation of almost all aspects of cell activity [3].
Ubiquitin-mediated biological processes are complex, owing to the highly variable nature of the ubiquitin modification. Attached ubiquitin can form adducts as mono- or polyubiquitin chains, the latter on different ubiquitin lysine residues, often in combination to form branched ubiquitin chains [4]. The variety in ubiquitination signals is achieved through a large number of ubiquitin ligases and DUBs that present numerous modes of action. There are 2 E1 enzymes, 40 E2 enzymes, 600 E3 ligases [5], and just under 100 DUBs [6] in the human genome giving a large scope for potential modification by the different enzymes. Signalling networks orchestrated by ubiquitin shows that E3s and DUBs exhibit redundancy and multiplicity. In addition, E3 ligases can interact with more than one E2 [7] so there is vast potential for different E2/E3 pairs to target different substrates and assemble diverse polyubiquitin chain structures.
The combinatorial possibilities of ubiquitin modification raise questions about their biological significance, but also challenges the limits of present analytical techniques. This review charts out recent progress that has been made in the field of ubiquitomics, and how these advances could help unravelling modes of ubiquitin modifications and subsequent biological effects.
2. Mapping Ubiquitination Sites on Protein Substrates
In addition to biochemical and antibody-based methods [8], mass spectrometry (MS)-based proteomics has enabled the rise of “ubiquitomics”, a term first coined in 2007 [9]. A ubiquitome refers to the set of proteins that are modified by ubiquitin and the associated ubiquitin chain topologies found under these conditions, although it is not possible to determine both of these aspects in the same experiment. MS-based proteomic tools for the high throughput detection of proteins which are modified with ubiquitin, referred to here as ubiquitin site profiling, have greatly increased our understanding of the ubiquitome. A difficulty that besets ubiquitin site profiling (or, indeed, the study of most PTMs) is the low stoichiometry of modification, thus an enrichment step is required prior to MS analysis. The groundwork for MS-based site specific detection of ubiquitination sites was laid in 2003, with 110 ubiquitination sites reported on 72 proteins (Figure 1A) [10]. This study relied on expression and enrichment of tagged ubiquitin in yeast followed by MS detection of the diGlycine (GG) remnant of ubiquitin, which remains conjugated to ubiquitinated proteins by an isopeptide bond to the N of a lysine side chain (K-GG) after trypsinisation.
In the decade following the study published in 2003, most ubiquitin site profiling studies relied on expression and pulldown of various forms of tagged ubiquitin, such as HA- or His-Ub [11]. While this pulldown strategy enriches ubiquitinated proteins, it suffers from skewed protein ubiquitination due to the presence of the tag, high background, and lack of definitive ubiquitination site identification. For example, Danielson et al. identified 5756 proteins following pulldown of tagged ubiquitin, but ubiquitination could only be confirmed on 471 of these proteins [12]. Other methods for investigating ubiquitin site profiling included use of biotin tagged ubiquitin [13,14], and, more recently, replacement of endogenous ubiquitin with a copy that has His-tag that remains after trypsinisation [15]. An alternative methodology, Combined Fractional Diagonal Chromatography (COFRADIC), involves monitoring chemical acetylation of lysine residues in the presence and absence of a non-specific DUB, from which >7500 ubiquitination sites were identified [16].
The biggest revolution in ubiquitin site profiling came in 2010 with the development of an antibody that that could recognise the K-GG remnant and specifically enrich for ubiquitinated material after trypsinisation [17]. The first large sale application of this antibody in 2011 enabled the identification of 19,000 sites [18]. The K-GG purification strategy has been built on in the last decade with astounding success with routine detection of 4000 ubiquitination sites or more in an experiment. Further optimisation has pushed this to >10,000 sites in a single run [19]. Most ubiquitin site profiling experiments rely on a commercial anti-K-GG antibody from Cell Signalling Technology.
A decade after the development of the K-GG antibody, the field is beginning to advance beyond this approach (Table 1). The K-GG antibody enrichment has its limitations. Firstly, each antibody raised against the K-GG epitope exhibits bias due to the amino acid context of the K-GG site [20]. Secondly, the K-GG antibody fails to enrich non-lysine ubiquitination modification (discussed below). Finally, the K-GG epitope is also generated following trypsinisation of ISG15- and NEDD8-modified proteins, and, while K-GG sites attributable to these modifications are low [18], these still have a confounding effect on strictly defining ubiquitination sites. One solution to these problems has been the generation of an antibody which recognises the 13-mer LysC digestion fragment of ubiquitin, referred to as the UbiSite approach [21]. LysC is typically not used alone in proteomic workflows due to its limited cleavage affinity. Additionally, detection of the 13-mer LysC fragment of ubiquitin attached to the tryptic peptide fragment derived from the substrate would be challenging due to the complex MS2 fragmentation pattern this would generate. The method therefore also uses trypsin following the pulldown to fully digest the material and generate GG-modified peptides, which are more amenable to MS detection. Across each biological triplicate, around 30,000 sites are detected and the paper reports 64,000 sites including conditions with proteasome inhibition (Figure 1A).
Methods such as stable isotope labelling of amino acids in cell culture (SILAC) [25] and tandem mass tagging (TMT) [26] have enabled multiplexing in proteomics, allowing for comparison of more conditions, decreasing sample requirements, and MS run time relative to label free quantitation approaches. Many ubiquitin site profiling experiments have used SILAC to compare 2–3 conditions such as with and without a stimulus [27] or between active and inactive ubiquitin machinery components [18]. TMT tagging permits comparison of up to 11 conditions, allowing experiments such as a detailed time course of the ubiquitination events that occur during mitophagy [28]. Performing TMT labelling on the anti-K-GG coated beads after pulldown of the GG-modified peptides enables selective labelling of the peptide N-terminus and allows TMT contaminants to be washed away, increasing the number of identified ubiquitination sites relative to in-solution TMT labelling and reducing sample requirement to sub-milligram levels. This methodology is referred to as UbiFast [29].
There are now proteomic workflows that enable the analysis of multiple “PTMomes” in single experiments through sequential pulldowns of different modifications after trypsinisation. Measurement of acetylomes and phosphoproteomes from the same sample as the ubiquitome show the interplay between PTMs and how they act together to co-ordinate cellular events [29,30,31,32]. Ubiquitome datasets increasingly include a matching proteome [21,29,32,33]. An increase in the MS quantitation value of a GG-modified peptide is more commonly explained by an increase in the abundance of its corresponding protein, so a matching proteome is necessary to tease apart differentially ubiquitinated (or otherwise modified) proteins and distinguish regulatory versus degradative ubiquitin modifications.
Two recent preprints show the application of Data-Independent Acquisition (DIA) mass spectrometry for ubiquitomics [22,23]. Although not yet peer reviewed, these studies have pushed the limits on ubiquitin site profiling, reporting 90,000 and 110,000 sites, respectively (Figure 1A). Both approaches used K-GG ubiquitin remnant enrichment combined with DIA MS to achieve deep ubiquitome coverage. Due to recent advances in software development such as DIA-NN [34], DIA MS allows improved quantitation of low abundance peptides and overcomes the stochastic nature of top-N fragmentation in Data-Dependent Acquisition (DDA) [35], which suffers from dynamic range limitations. DIA has been successfully applied to other PTMs, such as phosphorylation [36], and it seems likely that DIA MS will become common in proteomic workflows. However, DIA has limitations and the fact that there is little overlap (50%) between DIA and DDA ubiquitomes is concerning and shows that more work must be done to fully embrace it. There are other alternatives to DDA such as BoxCar [37] which increases the dynamic range of MS1 quantitation, but this method has seen limited use in the MS field.
3. Lessons from Ubiquitin Site Profiling
Ubiquitomics studies have generated a large amount of data about ubiquitination sites. While many of these papers serve to present methodological landmarks, there is insight that can be gained about the global functioning of the ubiquitin system from these datasets. There are several databases (Table 2) which have pooled information about ubiquitination sites, amongst other PTMs. This information can serve as a starting point for investigating ubiquitination of a particular protein. The PhosphositePlus database provides general information about ubiquitination, with Figure 1B highlighting the most frequently ubiquitinated domains and Figure 1D showing that substrates are generally only ubiquitinated a few times on average. However, there are outliers, with some proteins being extensively ubiquitinated, although the biological significance of this is not yet clear [21].
The interplay between ubiquitination and other PTMs has been investigated with this global data. A 2012 study exploring coevolution of PTM sites within proteins showed that ubiquitination sites frequently coevolve with phosphorylation and acetylation sites [39]. For example, phosphorylation of protein such as cyclins during the cell cycle recruits cullin-RING E3 ligases, leading to cyclin degradation by the proteasome [40]. However, this analysis only employed 600 ubiquitination sites, so new investigations with deeper ubiquitome data may show new trends. In the Protein Lysine Modification Database from 2017 there are 24,487 co-occurrences of acetylation and ubiquitination on the same residue [41]. Studies that profile both acetylation and ubiquitin sites have found a negative correlation between the two modifications on proteins [29]. This suggests that acetylation of a lysine residue could physically block ubiquitination at that site, this being particularly relevant in the case of epigenetic regulations [42]. Acetylation of K6 on ubiquitin itself has been shown to inhibit the assembly of K48-linked ubiquitin [43].
Ubiquitination shows no clear consensus sequence, unlike other PTMs including phosphorylation [46] and N-linked glycosylation [47]. The target specificity for E3 ligases is determined by more complex factors, such as the phosphorylation or glycosylation state of the protein [48]. The target specificity of DUBs is even less clear. However, it is still possible that the abundance of amino acids around a particular lysine will affect the likelihood that ubiquitination occurs at that residue. Accordingly, it has been possible to train machine learning models for the prediction of ubiquitination sites based on amino acid composition around the modified lysine [49]. Whilst a few early learning algorithms used on the order of 200 sites [50], this has been increasing in the last decade [49] with the UbiProber model [51] trained on 11,500 sites for humans, mice, and S. cerevisiae proteins. The model supports cross-species prediction, in which the training data from one organism is used to predict ubiquitination sites in another. Recently, a deep learning model was trained with over 50,000 ubiquitination sites producing one of the best performances to date, with a prediction accuracy of almost 90% [52]. While ubiquitin site profiling supersedes use of these models for investigating ubiquitination in model organisms, these models may provide a prediction of the ubiquitome for other less studied species.
Ubiquitin site profiling under different conditions has given an unprecedented way to study the function and enzymes of the ubiquitin system. Firstly, it has been possible to dissect the substrates of E3 ubiquitin ligases and DUBs, and secondly, many new ubiquitination events and modified proteins have been identified following changes in environmental conditions and stress. The chemical and genetic ablation of ubiquitin system enzymes followed by ubiquitin site profiling is a key method for inferring specific ubiquitination substrates and has been applied extensively for E3 ligases and DUBs (Table 3). Comparison between conditions with either genetic removal, knockdown, inhibition, overexpression, or reconstitution with a non-catalytically active E3 ligase or DUB can reveal how the activity of the protein affects the global ubiquitome. The differences in the resulting ubiquitomes can then be used to predict substrates and processes affected by that protein. However, it is not possible to directly infer substrates from these types of experiments as the activity of the E3 ligase or DUB may affect other components of the ubiquitin system [53] and ubiquitin site profiling experiments provide no information on ubiquitin topology. It remains difficult to validate putative DUB or E3 targets as bona fide substrates of the particular enzyme due to the low stoichiometry of ubiquitination and the pleiotropic effect of ubiquitination. There are a range of experiments for defining E3- and DUB-substrate pairs and networks [54], but the ubiquitomics approach is still very powerful because it shows the direct effect of alteration of the DUB or E3 ligase activity on the ubiquitome.
The assignment of the substrates of DUBs is becoming increasingly important as these enzymes are considered “druggable”. Specific DUB inhibitors are becoming more common and allow for the chemical alteration of DUB activity in cells. This is interesting in drug/target discovery setting, as the alteration of the ubiquitome is the end goal for DUB inhibitor use in cells. This has been exemplified in vivo by the success with USP7 inhibitors for cancer treatment in mice [23,55]. USP7 inhibition causes degradation of MDM2 due to stabilisation of proteasome-targeting ubiquitin chains, which in turn stabilises the tumour suppressor p53. The mapping of DUB substrates will be important for providing new angles to target proteins such as c-MYC which do not have surface features that are suitable for inhibitor development, which may represent as much as 80% of the proteome [56]. c-MYC exemplifies a classically “undruggable” protein [57], however its protein level is regulated by USP28 activity [58] raising the possibility of USP28 inhibition to target c-MYC levels [59,60]. This highlights DUBs as promising alternative drug targets in cancer and other diseases [61,62].
4. Limitations of Ubiquitin Site Profiling
Improvements in MS and technology have been driving forward proteomics, with mass spectrometers having greatly increased sensitivity and acquisition speed. However, measurement of low abundance peptides and modifications is still challenging due to the limitations in dynamic range [77]. Even following a K-GG pulldown, GG-modified peptides are still low abundance and typical data-dependent acquisition (DDA) MS workflows are not optimised for the detection of such peptides. In a DDA mode, a mass spectrometer first scans all ions (MS1), then selects the top-N peaks based on abundance for fragmentation and a rescan (MS2). There have been attempts to alter the DDA workflow for detecting ubiquitination such as selecting the top-N peaks for fragmentation, followed by lowest-N ions in the next cycle, which was found to increase measured GG-sites with use of less starting material [78]. One of the ways to compensate for the low abundance of GG peptides and to increase ubiquitome depth has been to use a large amount of starting material. The seminal UbiSite paper used up to 50 mg of protein starting material per condition in triplicate [21], and the CST PTMScan Ubiquitin Remnant Motif Kit recommends using 10–20 mg of protein input per pulldown. There have been titrations reported with varying amount of starting material [29,78] and new sample preparation methods that require less material [29]. For instance, the latest technology from CST, the PTMScan HS Ubiquitin/SUMO Remnant Motif kit (59322), requires only 1 mg of starting material by implementing SDS lysis and S-Trap columns in the workflow.
To reduce the need for large amounts of sample material and increase the sensitivity of detection, going beyond the DDA approach is a clear avenue for ubiquitomics. Targeted proteomics has been applied for quantitation of known ubiquitination sites following mitophagy where sample material was extremely limited [79]. However, the two above-mentioned recent studies on development of DIA ubiquitomics methods show great promise for the ubiquitomics field. For instance, in Steger et al. 30,000 sites were detected in a single run with only 500 g of protein input [23]. In the future, DIA mass spectrometry may become sensitive enough that ubiquitin remnant enrichment may not be necessary, and stochiometric information on PTM sites can be obtained. Finally, most ubiquitin site profiling studies have used Collision-Induced Fragmentation for MS2 analysis. However, other fragmentation strategies, such as Electron Transfer Dissociation, can provide alternative coverage of K-GG sites [80,81], which may give deeper or higher confidence ubiquitome data.
Spatial proteomics has begun to globally map protein localisation in cells, but the mapping of PTMs in each compartment has been lagging. The Human Cell Atlas [82] and information from other techniques [83] suggest that up to 50% of proteins are found in multiple cellular compartments. In each compartment exists a unique set of DUBs [6] and E3 ligases, suggesting that a protein located in multiple compartments could exhibit different ubiquitination landscapes in each compartment. This has been shown in principle for a protein located in both the nucleus and cytosol, where ubiquitination in each compartment occurs on different residues and leads to different outcomes [84]. While there have been enrichments of organelles in some ubiquitomic experiments [31,32,70], there have been no comparisons between organelles, which may be an interesting direction of research for the ubiquitomics field.
Almost all ubiquitin site profiling has focused on lysine modification. However, there are more and more examples of non-lysine ubiquitination in a variety of contexts [85] and there are few ubiquitomics methods for mapping these modifications. A few E3 ligases in humans have been discovered to possess esterification activity, ubiquitinating serine and threonine residues [86,87]. Several studies have now mapped N-terminally ubiquitinated proteins [16,21] through a variety of approaches. The most notable of these is the UbiSite method, which can enrich for ubiquitinated material regardless of the modified residue. Treatment with proteasome inhibitor did not cause accumulation of N-terminal ubiquitin modification, suggesting this chain type has a role outside of proteasome degradation [21]. Systematic mapping of serine/threonine ubiquitination sites is yet to be reported.
Despite the large number of identified ubiquitination sites, with over 120,000 sites in the PhosphositePlus database as of May 2020 [24], there are few identified E3- and DUB-substrates pairs or networks. A recent database contained 1806 E3-substrate pairs based on high-throughput databases of protein interactions, and this information was used to train a E3-substrate predictor [88]. However, there are many studies which profile E3- and DUB-substrate pairs and there is a need to aggregate this information in databases to form a more complete picture of the ubiquitin system. There is still extremely limited functional annotation of ubiquitination sites. Ubiquitomics suffers from the same difficulties as phosphoproteomics, where most reported phosphorylation sites either do not have an associated kinase or have unknown function [89]. There are over 235,000 human phosphorylation sites in the PhosphositePlus database as of May 2020, but only 8097 tsites have a reported function. This problem is even more pronounced for ubiquitomics. Among the 100,000 human ubiquitination sites, only 105 have an annotated function in the database. Whilst this as much shows the limitation of the database, it shows the massive task that the field will face in deciding which of these sites have real biological function and deciphering their roles.
Over half of known ubiquitination sites are only reported in 1 out of the 14 studies that have been compiled by the PhosphoSitePlus database (Figure 1C). Many of these sites have been detected in an unphysiological context under conditions such as proteasome and DUB inhibition, resulting in sites which may not be ubiquitinated in a physiological setting. Proteasome inhibition causes ubiquitination levels of proteins to increase, but this does not cause their accumulation, as the inhibition of protein degradation prevents protein synthesis [21]. False positive assignment of ubiquitination sites due to errors in mass spectrometry have long been known, including issues with false discovery rates and spectral assignment making it difficult to localise ubiquitination sites to a particular residue [90]. Artefactual modifications arising from the sample preparation can be mistaken for characteristic +114.08 Da modification of the GG remnant [91]. A combination of these errors and aggregation of ubiquitome data from multiple sources leads to an accumulation of false positives in the databases, which will be difficult to screen out.
5. Proteomics with Activity-Based Probe Profiling
Activity-based probes (ABPs) enable the profiling of active enzymes in a cell, and have been developed for many classes of proteins, especially proteases [92]. A classic ABP contains a specificity domain that targets them to their target enzyme, a chemical warhead that reacts irreversibly with the active site of the enzyme, and an identification moiety that enables the visualization or purification of the probe-enzyme species. Specific ABPs exist for E3 ligases, DUBs, and for the investigation of ubiquitin-like proteins and associated enzymes. The combination of ubiquitin site profiling and ABPs targeting the ubiquitin activitome increases the power of both approaches. ABPs can inform on the relative cellular activity of an E3 ligase or DUB, allowing for specific cellular conditions or inhibitor conditions to be selected and the corresponding effect on the ubiquitome assessed. The first DUB probe was developed in 2001 [93], and the last 20 years has seen an explosion in different DUB probe chemistries [94]. Additionally, there are now probes for the HECT [95] and RING [96] domain E3 ligases. DUB ABPs contain three elements: an epitope tag for pulldown and visualization of the probe, a recognition element (typically ubiquitin), and a chemical warhead that reacts irreversibly with the catalytic residue of active DUBs (Figure 2A). These probes have allowed the development of several MS-based methods to further investigate the biology of DUBs (Figure 2B).
The first use of ABPs has simply been to catalogue active DUBs in cells. There are two broad classes of DUBs [6], both of which have specific ABPs. DUBs with catalytic cysteines are most successfully targeted with a Ub-Propargylamine ABP [97], and metalloDUBs, which contain a Zn in their active site, can be targeted by a Ub-probe with a zinc-chelating group as a reactive warhead [98]. The depth of cysteine-targeting DUB ABPs has recently enabled the cataloguing of active cysteine-based DUBs from mammalian tissue culture; proteomic analysis showed that, out of 78 expressed DUBs, 74 reacted with the Ub-Propargylamine ABP [99]. The limited depth of E3 ligase ABPs precludes their use for this type of cataloguing experiments. For example, the E1-E2-E3 ligase cascading probe identified both E1 enzymes, around 20 E2 enzymes and only 2 E3 ligases, out of 600 total E3 ligases [100].
ABPs have been used to identify new DUBs in mammals and other species. This has been exemplified by the recent discovery of ZUFSP1 with cysteine-targeting ABPs, a member of a distinct class of DUBs [101,102]. ZUFSP1 was originally thought to be an inactive DUB as it seemed to lack a complete catalytic triad. However, the ABP reacted with the catalytic cysteine which could be identified in MS2 spectra and shown to play a role in DNA repair signalling. ABPs have been used to discover active DUBs in other species, including plasmodium [103] and Leishmania [104].
Ubiquitomes are dynamically regulated by the activity of E3 ligases and DUBs [105], and ubiquitin profiling experiments fail to capture the underlying range of enzymes, the “activitome”, that might be shaping the ubiquitome. Whilst comparison of ubiquitin under different conditions can show which proteins are ubiquitinated during a cellular response, this does not give information on what enzymes are restructuring the ubiquitome. ABPs can be used to investigate which DUBs and E3s change activity during cellular events. For example, UCH-L5 activity was found to increase during Salmonella infection [106], and OTUD3 activity was found to increase during viral infection [107]. In a recent paper with the first RING-targeting E3 ABP, the authors compared normal growth conditions with EGF stimulation, finding increased activity of E3 ligases known to be associated with EGFR signalling [96]. These studies show the potential of ABPs for discovering novel roles of E3 ligases and DUBs in coordinating ubiquitin signalling networks [108].
Variants of the ubiquitin ABPs have allowed a closer inspection of DUB specificity. Di-ubiquitin probes with a non-hydrolysable linkage have been engineered to resemble different ubiquitin chain types [109]. The highly specific probe, Met1-linked diUb probe, exhibits exclusive affinity for OTULIN, known to cleave linear ubiquitin. Using this probe, with and without a pan-DUB inhibitor, it was possible to identify SNX27 as an interactor of OTULIN, enabling the probe to be used to perform a co-IP-like experiment [110]. Finally, DUBs are rising as therapeutic targets. However, the first DUB inhibitors showed poor selectivity [111]. DUB ABPs are now being used extensively to ascertain the selectivity of DUB inhibitors. The development of the specific USP7 inhibitor involved ABP profiling, where cell lysate was incubated with inhibitors followed by ABP profiling [55]. This showed that only the reactivity of USP7 with the probe was affected by the inhibitor. Furthermore, cells can be treated with inhibitors prior to lysis followed by ABP profiling to show target engagement. Currently, this is the main method for studying target engagement of small molecules in a cellular context, allowing the development of much more selective and therefore safer treatments.
6. Ubiquitin Chain Topology and the Ubiquitin Code Hypothesis
Focusing on ubiquitination sites and activity alone misses out on the complex nature of ubiquitin chain topology. Ubiquitin can be modified extensively, most importantly on its own lysine residues, allowing the formation of polyubiquitin chains. Ubiquitin has seven internal lysine residues and can also be modified with ubiquitin at its N-terminus, allowing for eight basic ubiquitin chain types [4]. However, it has become increasingly clear that there exists a wide possible range of ubiquitin chain structures. A single ubiquitin monomer in the chain can be ubiquitinated multiple times, generating hybrid chains with multiple types of ubiquitin linkage [112]. There are important quantitative questions in the field: What modifications exist on ubiquitin itself? What is the abundance of each ubiquitin chain linkage? What proportion of ubiquitin chains in the cell are branched? What proteins are modified by each chain type? MS tools have been instrumental in the research around these questions (Figure 3).
The huge variety in possible ubiquitin chain topologies and modifications has given rise to the idea of the “ubiquitin code”. This idea was first formalised by Komander and Rape in 2012 [113], suggesting that “ubiquitylation can act as a code to store and transmit information” through the assembly of different ubiquitin chain types and modifications. There are many signals associated with different ubiquitin chain structures [114]. The picture is further complicated by the existence of hybrid ubiquitin/Ubl chains, again hugely increasing the diversity of possible structures [115]. Borrowing from the language of the histone code hypothesis [116], E3 ligases act as “writers”, DUBs as “erasers”, and ubiquitin binding proteins as “readers”.
Beyond showing the existence of the varied ubiquitin chain topology, proteomic tools have contributed to ideas of the ubiquitin code hypothesis. Proteomic MS approaches have been the starting point for discovering new readers of the ubiquitin code (Figure 3B). Studies of each possible linkage of diUb species [117] and longer ubiquitin chains [118] show that chain types have specific interactomes, supporting the existence of distinct ubiquitin binding proteins for each chain type. Structural biology has shown that each ubiquitin chain type has a distinct conformation, again lending to the idea that each chain type has unique binders [4]. A recent review postulated that the variety of ubiquitin-controlled processes and chain types suggests that more types of ubiquitin binding domain should exist [119]. While branched ubiquitin chains are common, there are no known binders of any type of ubiquitin branch point.
Ubiquitin can be modified with many more PTMs, further increasing the complexity of the ubiquitin code. MS analysis of ubiquitin through pulldown studies of various PTMs (Figure 3A) have shown that ubiquitin can undergo extensive modification [120], including phosphorylation, acetylation, SUMOylation, deamidation, and ADP-ribosylation. In human ubiquitin, there are eight phosphorylation sites, the best studied of which is pS65, involved in mitophagy signalling [121]. Acetylation of K6 has been reported to negatively regulate formation of K11, K48 and K63-linked ubiquitin chains [43]. Other modifications reflect inhibition or subversion of the ubiquitin system by infectious agents. The glutamine (G40) of ubiquitin was reported to be deamidated by the deaminidase CHBP (Cif homologue from Burkholderia pseudomallei) during Burkholderia infection [122]. This blocks the synthesis and elongation of polyubiquitin chains, which dysregulates the eukaryotic ubiquitination machinery. Finally, Legionella pneumophila ADP-ribosylates ubiquitin in order to conjugate ubiquitin to Rab small GTPases, enabling ubiquitination independent of the eukaryotic E1 and E2 enzymes [123].
The ability to purify endogenously expressed ubiquitin chains with Tandem Ubiquitin Binding Entities (TUBEs) was a major step forward in studying ubiquitomes [124]. Since their development in 2009, there has a been a large expansion in types of TUBEs and there are binders for Met1-, K27-, K48- and K63-linked chains [125]. Several proteomic workflows have been used to identify proteins modified with these chain types [126,127]. However, the use of TUBEs is still limited by the pulldown of non-ubiquitinated material and the lack of precise ubiquitination site information. A combination of TUBE pulldown followed by trypsinisation and a K-GG pulldown would allow site specific modification data for different chain types, adding another layer of depth to the ubiquitome. This has been attempted in yeast with a K63 TUBE followed by a K-GG pulldown, identifying almost 1000 sites that are likely modified by K63-linked chains [38]. However, this method required lysate containing over 200 mg of protein, so it will be difficult to scale this up for mammalian tissue culture.
Ubiquitin chain type abundance has been estimated with bottom-up targeted proteomics approaches using heavy labelled synthetic GG-modified ubiquitin peptides as a reference for ubiquitin absolute quantitation (Ub-AQUA) [128], using Parallel Reaction Monitoring (PRM) to ascertain the abundance of different modified ubiquitin tryptic peptides. This showed that all chain types are present in yeast. This method has been applied in human cells, again showing the presence of all chain types [129]. Ub-Prot is a recent method to examine both lengths and linkage types of ubiquitin chains in yeast [130]. By pulling down ubiquitin chains with a TUBE that is resistant to trypsin, followed by trypsinisation, Ub-Prot enables isolation of ubiquitin chains from cells. By separating out the purified ubiquitin chains on an SDS-PAGE gel then preparing different regions of the gel lane for Ub-AQUA PRM, it is possible to estimate the abundance of different chain types at different molecular weights. This in turn can be used to infer the length of typical ubiquitin chains of different linkage type. K48-, K6- and K29-linked chains have between two and seven ubiquitin monomers, compared with K63- and K11-linked chains containing typically no more than two ubiquitin monomers (Figure 3C).
However, Ub-AQUA cannot provide information about the abundance of hybrid ubiquitin chains with multiple linkage types and branch points. There have been several examples of hybrid ubiquitin chains in biological processes, including Met1/K63-linked chains in MyD88-driven inflammation [131], K11/K63-linked being required for MHC I internalisation [132,133], and K11/K48-linked chains enhancing proteasomal degradation [134]. Some E3 ligases generate hybrid chains in vitro [114]. Ubiquitin Chain Restriction (UbiCRest) analysis has shown that hybrid or branched chains can be found on other substrates [135]. However, there is still speculation about the prevalence of hybrid chains in the cell, and their potential involvement in other processes.
MS analysis offers a higher vantage point. While complete trypsinisation of ubiquitin chains destroys their architecture, limited trypsinolysis preserves some of the structure of the chains and enables middle down MS. This has been applied to ubiquitin chains generated in vitro [136,137]. Two separate MS-based methods have estimated that around 10–20% of ubiquitin chains contain a branch point in cells. Purification of ubiquitin chains from cells with TUBEs followed by limited trypsinolysis and middle down MS showed that 1–2% of ubiquitin monomers are branched in ubiquitin chains, increasing to 4% on inhibition of the proteasome [138]. Ub-Clipping is a powerful new technique for probing both modifications of ubiquitin and chain architecture (Figure 3D) [139]. This method involves use of an engineered viral protease from Foot and Mouth Virus, Lb, that can cleave ubiquitin at its C-terminus, between R74 and G75, leaving behind a GG remnant on a substrate protein. If a ubiquitin is found to have a GG remnant following Lb treatment by intact-MS, that ubiquitin must itself have been ubiquitinated. Two GG remnants imply the ubiquitin must be a branch point in a ubiquitin chain. By combining TUBEs with Ub-Clipping, the authors found that 4–7% of ubiquitin is modified with two separate GG remnants, suggesting that 10–20% of ubiquitin chains contain a branch point. Ub-Clipping has been used to study the ubiquitin chain architectures and modifications that occur in mitophagy, revealing that only ubiquitin monomers at the end of ubiquitin chains or monoubiquitin modifications are phosphorylated rather than monomers within chains [32].
7. Translational Ubiquitomics
As discussed above, the ubiquitin system is involved in most cellular processes. Additionally, it has been reported to be deregulated in certain diseases, most notably in cancer and neurodegeneration. There are many mutations that affect the ubiquitin processing machinery. Mutations and genetic alterations in the K63-specific deubiquitylating enzyme CYLD have been found in patients with cylindromatosis, multiple familial trichoepithelioma, and Brooke–Spiegler syndrome [140]. Inactivating mutations of the deISGylating enzyme USP18 are the cause of type I interferonopathies and auto-inflammation events that lead to the life-threatening pseudo-TORCH syndrome [141]. Mutations in E3 ligases that control cell signalling, DNA repair, and cell cycle progression are common in cancer [142]. For example, loss of activity in the E3 ligase pVHL (an inhibitor of hypoxia signalling) is found in almost all cases of renal carcinoma, due to loss of the allele, mutation, or methylation in its promoter [143]. Beyond cancer, mutations in the E3 ligase Parkin are associated with Parkinson’s disease [144].
Studying how these genetic alterations alter the ubiquitome in patient samples could prove very informative and indeed lead to more effective forms of therapy. Mouse tissue has been used successfully for K-GG profiling studies [20,145]. However, since the required amount of starting material for performing ubiquitin site profiling studies is relatively high and the cellular material coming from patients is normally low, this has been challenging so far. For example, K-GG-based ubiquitin site profiling has been used to describe the ubiquitin landscape in platelets derived from six human donors following stimulation by collagen-related peptide (CRP-XL) [73]. Higher throughput MS methods and a decrease in the amount of sample required will help drive translational ubiquitomics. UbiFast allowed the identification of differentially ubiquitinated proteins in basal and luminal human breast cancer xenografts using as little as 500 g of protein per condition [29]. The developments in DIA ubiquitomics also allow the use of much less protein input [23]. DUB ABPs have been used on human breast cancer tissue samples and showed increased activity of UCHL1, suggesting this enzyme as a therapeutic target in these cancers [108].
8. Discussion
The study of cellular ubiquitomics provides unprecedented details into cell biological processes controlled by ubiquitin and its interplay with other post-translational modifications. The topology of ubiquitin chains clearly dictates the fate of cognate substrates, as formulated by the ubiquitin code hypothesis. However, it is not yet clear to what extent the variability in chain structures reflects distinct versus redundant information.
Novel breakthroughs in ubiquitin biology are interlinked with technical advances, and constant developments in chemical and analytics tools are permitting deeper and faster profiling analysis. This includes higher-throughput proteomics permitting many tens to hundreds of samples to be analysed in single experiments. New MS platforms, developments in liquid chromatography, and new MS approaches such as DIA reflect technical efforts towards such a goal.
We have more ubiquitomics data than ever. The compilation of this data and studies is a starting point, but there is little functional annotation of these sites and there is no comprehensive database of DUB and E3 substrates. To better understand ubiquitination in the disease setting, the generation of a database compiling disease ubiquitomics data, mutations in ubiquitinated sites in disease, and their associated modifiers could be a useful resource for diagnostics and for the generation of newer therapies targeting the ubiquitin system.
Implementing the knowledge gained from ubiquitomics into the clinic represents the ultimate challenge for the field. The study of the ubiquitome in disease could be helpful to identify biomarkers, resistance mechanisms, and potential co-treatment opportunities. Since DUB activity can reflect aberrant signalling, studying the DUB “activitome” in disease could also help to better understand its aetiology and unveil new DUB targets. ABPs targeting DUBs and also conjugating enzymes will not only permit the discovery of DUB/E3 ligase activity and substrates, but also allow the testing of small molecule inhibitors as part of a drug discovery pipeline towards applications in the clinic. ABPs have already led to the development of highly specific DUB inhibitors that have promising anti-tumour effects in vivo.
In summary, ubiquitomics has significantly evolved in the last decade and allowed us to better understand the biological meaning of the ubiquitin modification. However, there are still big challenges for the field. Promising technological developments and dedicated databases will help us to untangle the complexity of ubiquitination and to discover novel ways of targeting the ubiquitination machinery in disease.
Author Contributions
G.V., conceptualisation, review of the literature and original draft preparation; R.K., visualisation; and A.P.-F. and B.M.K., conceptualisation and supervision. All the authors contributed to the final draft and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
B.M.K. was supported by an EPSRC grant (grant number: EP/N034295/1) and by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (CIFMS), China (grant number: 2018-I2M-2–002). G.V. and A.P.-F were supported by Pfizer Inc. funding awarded to B.M.K.
Acknowledgments
We would like to thank other members of the Kessler lab and the TDI Discovery Proteomics Facility (DPF) for their helpful discussions and feedback on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, S.; Haendler, B. The Ubiquitin System in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Kliza, K.; Husnjak, K. Resolving the Complexity of Ubiquitin Networks. Front. Mol. Biosci. 2020, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, E.; Palaniyappan, N.; Tooth, D.; Layfield, R. Methods for the purification of ubiquitinated proteins. Proteomics 2007, 7, 1016–1022. [Google Scholar] [CrossRef]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Beaudette, P.; Popp, O.; Dittmar, G. Proteomic techniques to probe the ubiquitin landscape. Proteomics 2016, 16, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, J.M.R.; Sylvestersen, K.B.; Bekker-Jensen, S.; Szklarczyk, D.; Poulsen, J.W.; Horn, H.; Jensen, L.J.; Mailand, N.; Nielsen, M.L. Mass Spectrometric Analysis of Lysine Ubiquitylation Reveals Promiscuity at Site Level. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lectez, B.; Migotti, R.; Lee, S.Y.; Ramirez, J.; Beraza, N.; Mansfield, B.; Sutherland, J.D.; Martinez-Chantar, M.L.; Dittmar, G.; Mayor, U. Ubiquitin profiling in liver using a transgenic mouse with biotinylated ubiquitin. J. Proteome Res. 2014, 13, 3016–3026. [Google Scholar] [CrossRef]
- Meierhofer, D.; Wang, X.; Huang, L.; Kaiser, P. Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J. Proteome Res. 2008, 7, 4566–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimov, V.; Olsen, L.C.; Hansen, S.V.; Barrio-Hernandez, I.; Puglia, M.; Jensen, S.S.; Solov’Yov, I.A.; Kratchmarova, I.; Blagoev, B. StUbEx PLUS—A Modified Stable Tagged Ubiquitin Exchange System for Peptide Level Purification and In-Depth Mapping of Ubiquitination Sites. J. Proteome Res. 2018, 17, 296–304. [Google Scholar] [CrossRef]
- Stes, E.; Laga, M.; Walton, A.; Samyn, N.; Timmerman, E.; De Smet, I.; Goormachtig, S.; Gevaert, K. A COFRADIC protocol to study protein ubiquitination. J. Proteome Res. 2014, 13, 3107–3113. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 2010, 28, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined preparation and use of anti-diglycine remnant (K-ϵ-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Schölz, C.; Kelstrup, C.D.; Young, C.; Nielsen, M.L.; Olsen, J.V.; Brakebusch, C.; Choudhary, C. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol. Cell. Proteom. 2012, 11, 1578–1585. [Google Scholar] [CrossRef] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and n-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Hansen, F.M.; Tanzer, M.C.; Brüning, F.; Bludau, I.; Schulman, B.A.; Robles, M.S.; Karayel, O.; Mann, M. Data-independent acquisition method for ubiquitinome analysis reveals regulation of circadian biology. bioRxiv 2020. [Google Scholar] [CrossRef]
- Steger, M.; Ihmor, P.; Backman, M.; Müller, S.; Daub, H. Deep ubiquitination site profiling by single-shot data-independent acquisition mass spectrometry. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.E.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef]
- Rose, C.M.; Isasa, M.; Ordureau, A.; Prado, M.A.; Beausoleil, S.A.; Jedrychowski, M.P.; Finley, D.J.; Harper, J.W.; Gygi, S.P. Highly Multiplexed Quantitative Mass Spectrometry Analysis of Ubiquitylomes. Cell Syst. 2016, 3, 395–403.e4. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Mani, D.C.; Satpathy, S.; Fereshetian, S.; Gasser, J.A.; Svinkina, T.; Olive, M.E.; Ebert, B.L.; Mertins, P.; Carr, S.A. Rapid and deep-scale ubiquitylation profiling for biology and translational research. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Elia, A.E.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Ordureau, A.; Paulo, J.A.; Zhang, J.; An, H.; Swatek, K.N.; Cannon, J.R.; Wan, Q.; Komander, D.; Harper, J.W. Global Landscape and Dynamics of Parkin and USP30-Dependent Ubiquitylomes in iNeurons during Mitophagic Signaling. Mol. Cell 2020, 77, 1124–1142.e10. [Google Scholar] [CrossRef]
- Theurillat, J.P.P.; Udeshi, N.D.; Errington, W.J.; Svinkina, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Chapman, J.D.; Goodlett, D.R.; Masselon, C.D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 2014, 33, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 min. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Back, S.; Gorman, A.W.; Vogel, C.; Silva, G.M. Site-Specific K63 Ubiquitinomics Provides Insights into Translation Regulation under Stress. J. Proteome Res. 2019, 18, 309–318. [Google Scholar] [CrossRef]
- Minguez, P.; Parca, L.; Diella, F.; Mende, D.R.; Kumar, R.; Helmer-Citterich, M.; Gavin, A.C.; Van Noort, V.; Bork, P. Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 2012, 8, 599. [Google Scholar] [CrossRef]
- Fouad, S.; Wells, O.S.; Hill, M.A.; D’Angiolella, V. Cullin Ring Ubiquitin Ligases (CRLs) in Cancer: Responses to Ionizing Radiation (IR) Treatment. Front. Physiol. 2019, 10, 1144. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genom. 2017, 44, 243–250. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications – writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Saeki, Y.; Sakamoto, K.; Ohtake, K.; Nishikawa, H.; Tsuchiya, H.; Ohta, T.; Tanaka, K.; Kanno, J. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep. 2015, 16, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhou, T.; He, B.; Yu, H.; Guo, X.; Song, X.; Sha, J. mUbiSiDa: A comprehensive database for protein ubiquitination sites in mammals. PLoS ONE 2014, 9, e85744. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Liu, Z.; Wang, Y.; Cheng, H.; Yang, Q.; Guo, A.; Ren, J.; Xue, Y. UUCD: A family-based database of ubiquitin and ubiquitin-like conjugation. Nucleic Acids Res. 2013, 41, D445–D451. [Google Scholar] [CrossRef] [Green Version]
- Kennelly, P.J.; Krebs, E.G. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 1991, 266, 15555–15558. [Google Scholar]
- Gavel, Y.; Heijne, G.V. Sequence differences between glycosylated and non-glycosylated asn-x-thr/ser acceptor sites: Implications for protein engineering. Protein Eng. Des. Sel. 1990, 3, 433–442. [Google Scholar] [CrossRef]
- Jadhav, T.; Wooten, M.W. Defining an embedded code for protein Ubiquitination. J. Proteom. Bioinform. 2009, 2, 316–333. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhou, Y.; Zhang, Z.; Song, J. Towards more accurate prediction of ubiquitination sites: A comprehensive review of current methods, tools and features. Brief. Bioinform. 2014, 16, 640–657. [Google Scholar] [CrossRef] [Green Version]
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins Struct. Funct. Bioinform. 2010, 78, 365–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Qiu, J.D.; Shi, S.P.; Suo, S.B.; Huang, S.Y.; Liang, R.P. Incorporating key position and amino acid residue features to identify general and species-specific Ubiquitin conjugation sites. Bioinformatics 2013, 29, 1614–1622. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Yang, Y.; Wang, X.; Wang, H.; Xu, Y. DeepUbi: A deep learning framework for prediction of ubiquitination sites in proteins. BMC Bioinform. 2019, 20, 86. [Google Scholar] [CrossRef] [PubMed]
- Weissman, A.M.; Shabek, N.; Ciechanover, A. The predator becomes the prey: Regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 2011, 12, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Iconomou, M.; Saunders, D.N. Systematic approaches to identify E3 ligase Substrates. Biochem. J. 2016, 473, 4083–4101. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef]
- Verdine, G.L.; Walensky, L.D. The challenge of drugging undruggable targets in cancer: Lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 2007, 13, 7264–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, D.; Anderton, B.; Goga, A. Taking on Challenging Targets: Making MYC Druggable. Am. Soc. Clin. Oncol. Educ. B 2014, 34, e497–e502. [Google Scholar] [CrossRef] [Green Version]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, T.; Li, Z.; Sun, K.; Fu, Y.; Cheng, T.; Guo, J.; Yu, B.; Shi, X.; Liu, H. Discovery of [1,2,3]triazolo[4,5-d] pyrimidine derivatives as highly potent, selective, and cellularly active USP28 inhibitors. Acta Pharm. Sin. B 2020, 10, 1476–1491. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z.; Zhang, L.; Yang, Z.; Chen, X.; Luo, J.; Zhou, Z.; Mei, X.; Yu, X.; Shao, Z.; et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–77. [Google Scholar] [CrossRef]
- Pinto-Fernandez, A.; Kessler, B.M. DUBbing cancer: Deubiquitylating enzymes involved in epigenetics, DNA damage and the cell cycle as therapeutic targets. Front. Genet. 2016, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Steklov, M.; Pandolfi, S.; Baietti, M.F.; Batiuk, A.; Carai, P.; Najm, P.; Zhang, M.; Jang, H.; Renzi, F.; Cai, Y.; et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.W.; Nagel, J.; Hoving, S.; Gerrits, B.; Bauer, A.; Thomas, J.R.; Kirschner, M.W.; Schirle, M.; Luchansky, S.J. Quantitative Lys-ϵ-Gly-Gly (diGly) proteomics coupled with inducible RNAi reveals ubiquitin-mediated proteolysis of DNA damage-inducible transcript 4 (DDIT4) by the E3 Ligase HUWE1. J. Biol. Chem. 2014, 289, 28942–28955. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Saeki, Y.; Murakami, A.; Kawawaki, J.; Tsuchiya, H.; Yoshihara, H.; Shindo, M.; Tanaka, K. A comprehensive method for detecting ubiquitinated substrates using TR-TUBE. Proc. Natl. Acad. Sci. USA 2015, 112, 4630–4635. [Google Scholar] [CrossRef] [Green Version]
- Potu, H.; Peterson, L.F.; Kandarpa, M.; Pal, A.; Sun, H.; Durham, A.; Harms, P.W.; Hollenhorst, P.C.; Eskiocak, U.; Talpaz, M.; et al. Usp9x regulates Ets-1 ubiquitination and stability to control NRAS expression and tumorigenicity in melanoma. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, J.J.; Vasilevskaya, I.A.; Neupane, N.P.; Shafi, A.A.; McNair, C.; Dylgjeri, E.; Mandigo, A.C.; Schiewer, M.J.; Schrecengost, R.S.; Gallagher, P.; et al. USP22 functions as an oncogenic driver in prostate cancer by regulating cell proliferation and DNA repair. Cancer Res. 2020, 80, 430–443. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Phu, L.; Rose, C.M.; Tea, J.S.; Wall, C.E.; Verschueren, E.; Cheung, T.K.; Kirkpatrick, D.S.; Bingol, B. Dynamic Regulation of Mitochondrial Import by the Ubiquitin System. Mol. Cell 2020, 77, 1107–1123.e10. [Google Scholar] [CrossRef] [PubMed]
- Rusilowicz-Jones, E.V.; Jardine, J.; Kallinos, A.; Pinto-Fernandez, A.; Guenther, F.; Giurrandino, M.; Barone, F.G.; McCarron, K.; Burke, C.J.; Murad, A.; et al. USP30 sets a trigger threshold for PINK1-PARKIN amplification of mitochondrial ubiquitylation. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- Sapmaz, A.; Berlin, I.; Bos, E.; Wijdeven, R.H.; Janssen, H.; Konietzny, R.; Akkermans, J.J.; Erson-Bensan, A.E.; Koning, R.I.; Kessler, B.M.; et al. USP32 regulates late endosomal transport and recycling through deubiquitylation of Rab7. Nat. Commun. 2019, 10, 1454. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.A.; Satpathy, S.; Beli, P.; Choudhary, C. SPATA 2 links CYLD to the TNF-α receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 2016, 35, 1868–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unsworth, A.J.; Bombik, I.; Pinto-Fernandez, A.; McGouran, J.F.; Konietzny, R.; Zahedi, R.P.; Watson, S.P.; Kessler, B.M.; Pears, C.J. Human Platelet Protein Ubiquitylation and Changes following GPVI Activation. Thromb. Haemost. 2019, 119, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.; Mace, Y.; Drouet, F.; Bony, E.; Boidot, R.; Draoui, N.; Lobysheva, I.; Corbet, C.; Polet, F.; Martherus, R.; et al. A new ER-specific photosensitizer unravels 1O2-driven protein oxidation and inhibition of deubiquitinases as a generic mechanism for cancer PDT. Oncogene 2016, 35, 3976–3985. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Mani, D.R.; Eisenhaure, T.; Mertins, P.; Jaffe, J.D.; Clauser, K.R.; Hacohen, N.; Carr, S.A. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol. Cell. Proteom. 2012, 11, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Aravamudhan, S.; Nolte, H.; Türk, C.; Hölper, S.; Mü Ller, S.; Gü Nther, S.; Blaauw, B.; Braun, T.; Krüger, M. Dynamic changes in the mouse skeletal muscle proteome during denervation-induced atrophy. Dis. Model. Mech. 2017, 10, 881–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.; Charles, P.D.; He, L.; Mowlds, P.; Kessler, B.M.; Fischer, R. Expanding Proteome Coverage with CHarge Ordered Parallel Ion aNalysis (CHOPIN) Combined with Broad Specificity Proteolysis. J. Proteome Res. 2017, 16, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, L.; Bezstarosti, K.; Sap, K.A.; Dekkers, D.H.; Rijkers, E.; Mientjes, E.; Elgersma, Y.; Demmers, J.A. Improvement of ubiquitylation site detection by Orbitrap mass spectrometry. J. Proteom. 2018, 172, 49–56. [Google Scholar] [CrossRef]
- Ordureau, A.; Paulo, J.A.; Zhang, W.; Ahfeldt, T.; Zhang, J.; Cohn, E.F.; Hou, Z.; Heo, J.M.; Rubin, L.L.; Sidhu, S.S.; et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol. Cell 2018, 70, 211–227.e8. [Google Scholar] [CrossRef] [Green Version]
- Sobott, F.; Watt, S.J.; Smith, J.; Edelmann, M.J.; Kramer, H.B.; Kessler, B.M. Comparison of CID Versus ETD Based MS/MS Fragmentation for the Analysis of Protein Ubiquitination. J. Am. Soc. Mass Spectrom. 2009, 20, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Porras-Yakushi, T.R.; Sweredoski, M.J.; Hess, S. ETD Outperforms CID and HCD in the Analysis of the Ubiquitylated Proteome. J. Am. Soc. Mass Spectrom. 2015, 26, 1580–1587. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Geladaki, A.; Kočevar Britovšek, N.; Breckels, L.M.; Smith, T.S.; Vennard, O.L.; Mulvey, C.M.; Crook, O.M.; Gatto, L.; Lilley, K.S. Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gillotin, S.; Davies, J.D.; Philpott, A. Subcellular localisation modulates ubiquitylation and degradation of Ascl1. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelsall, I.R.; Zhang, J.; Knebel, A.; Arthur, S.J.; Cohen, P. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl. Acad. Sci. USA 2019, 116, 13293–13298. [Google Scholar] [CrossRef] [Green Version]
- Pao, K.C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; Van Aalten, D.M.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 2018, 556, 381–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Liu, X.; Xia, T.; Tekcham, D.S.; Wang, W.; Chen, H.; Li, T.; Lu, C.; Ning, Z.; Liu, X.; et al. A Multidimensional Characterization of E3 Ubiquitin Ligase and Substrate Interaction Network. iScience 2019, 16, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Needham, E.J.; Parker, B.L.; Burykin, T.; James, D.E.; Humphrey, S.J. Illuminating the dark phosphoproteome. Sci. Signal. 2019, 12, eaau8645. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Xu, P.; Qin, J. Ubiquitinated proteome: Ready for global? Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.L.; Vermeulen, M.; Bonaldi, T.; Cox, J.; Moroder, L.; Mann, M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 2008, 5, 459–460. [Google Scholar] [CrossRef]
- Sanman, L.E.; Bogyo, M. Activity-Based Profiling of Proteases. Annu. Rev. Biochem. 2014, 83, 249–273. [Google Scholar] [CrossRef] [Green Version]
- Borodovsky, A.; Kessler, B.M.; Casagrande, R.; Overkleeft, H.S.; Wilkinson, K.D.; Ploegh, H.L. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001, 20, 5187–5196. [Google Scholar] [CrossRef] [Green Version]
- Hewings, D.S.; Flygare, J.A.; Bogyo, M.; Wertz, I.E. Activity-based probes for the ubiquitin conjugation- deconjugation machinery: New chemistries, new tools, and new insights. FEBS J. 2017, 284, 1555–1576. [Google Scholar] [CrossRef] [Green Version]
- Byrne, R.; Mund, T.; Licchesi, J.D. Activity-Based Probes for HECT E3 Ubiquitin Ligases. ChemBioChem 2017, 18, 1415–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, S.; Fletcher, A.J.; Branigan, E.; Hay, R.T.; Virdee, S. Photocrosslinking Activity-Based Probes for Ubiquitin RING E3 Ligases. Cell Chem. Biol. 2020, 27, 74–82.e6. [Google Scholar] [CrossRef] [Green Version]
- Ekkebus, R.; Van Kasteren, S.I.; Kulathu, Y.; Scholten, A.; Berlin, I.; Geurink, P.P.; De Jong, A.; Goerdayal, S.; Neefjes, J.; Heck, A.J.; et al. On terminal alkynes that can react with active-site cysteine nucleophiles in proteases. J. Am. Chem. Soc. 2013, 135, 2867–2870. [Google Scholar] [CrossRef] [PubMed]
- Hameed, D.S.; Sapmaz, A.; Burggraaff, L.; Amore, A.; Slingerland, C.J.; van Westen, G.J.; Ovaa, H. Development of Ubiquitin-Based Probe for Metalloprotease Deubiquitinases. Angew. Chem. Int. Ed. 2019, 58, 14477–14482. [Google Scholar] [CrossRef] [Green Version]
- Pinto-Fernández, A.; Davis, S.; Schofield, A.B.; Scott, H.C.; Zhang, P.; Salah, E.; Mathea, S.; Charles, P.D.; Damianou, A.; Bond, G.; et al. Comprehensive Landscape of Active Deubiquitinating Enzymes Profiled by Advanced Chemoproteomics. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Mulder, M.P.; Witting, K.; Berlin, I.; Pruneda, J.N.; Wu, K.P.; Chang, J.G.; Merkx, R.; Bialas, J.; Groettrup, M.; Vertegaal, A.C.; et al. A cascading activity-based probe sequentially targets E1-E2-E3 ubiquitin enzymes. Nat. Chem. Biol. 2016, 12, 523–530. [Google Scholar] [CrossRef]
- Hewings, D.S.; Heideker, J.; Ma, T.P.; Ahyoung, A.P.; El Oualid, F.; Amore, A.; Costakes, G.T.; Kirchhofer, D.; Brasher, B.; Pillow, T.; et al. Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Kwasna, D.; Abdul Rehman, S.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I.; et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol. Cell 2018, 70, 150–164.e6. [Google Scholar] [CrossRef] [Green Version]
- Artavanis-Tsakonas, K.; Misaghi, S.; Comeaux, C.A.; Catic, A.; Spooner, E.; Duraisingh, M.T.; Ploegh, H.L. Identification by functional proteomics of a deubiquitinating/deNeddylating enzyme in Plasmodium falciparum. Mol. Microbiol. 2006, 61, 1187–1195. [Google Scholar] [CrossRef]
- Damianou, A.; Burge, R.J.; Catta-Preta, C.M.; Geoghegan, V.; Nievas, Y.R.; Newling, K.; Brown, E.; Burchmore, R.; Rodenko, B.; Mottram, J.C. Essential roles for deubiquitination in Leishmania life cycle progression. PLoS Pathog. 2020, 16, e1008455. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and Cellular Roles of Ubiquitin-Specific Deubiquitinating Enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummari, E.; Alugubelly, N.; Hsu, C.Y.; Dong, B.; Nanduri, B.; Edelmann, M.J. Activity-based proteomic profiling of deubiquitinating enzymes in salmonella-infected macrophages leads to identification of putative function of UCH-L5 in inflammasome regulation. PLoS ONE 2015, 10, e0135531. [Google Scholar] [CrossRef]
- Zhang, Z.; Fang, X.; Wu, X.; Ling, L.; Chu, F.; Li, J.; Wang, S.; Zang, J.; Zhang, B.; Ye, S.; et al. Acetylation- Dependent Deubiquitinase OTUD3 Controls MAVS Activation in Innate Antiviral Immunity. Mol. Cell 2020, 79, 304–319.e7. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; González-Prieto, R.; Zhang, M.; Geurink, P.P.; Kooij, R.; Iyengar, P.V.; van Dinther, M.; Bos, E.; Zhang, X.; Le Dévédec, S.E.; et al. Deubiquitinase activity profiling identifies UCHL1 as a candidate oncoprotein that promotes TGFβ-induced breast cancer metastasis. Clin. Cancer Res. 2020, 26, 1460–1473. [Google Scholar] [CrossRef] [Green Version]
- McGouran, J.F.; Gaertner, S.R.; Altun, M.; Kramer, H.B.; Kessler, B.M. Deubiquitinating enzyme specificity for ubiquitin chain topology profiled by di-ubiquitin activity probes. Chem. Biol. 2013, 20, 1447–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stangl, A.; Elliott, P.R.; Pinto-Fernandez, A.; Bonham, S.; Harrison, L.; Schaub, A.; Kutzner, K.; Keusekotten, K.; Pfluger, P.T.; El Oualid, F.; et al. Regulation of the endosomal SNX27-retromer by OTULIN. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Ritorto, M.S.; Ewan, R.; Perez-Oliva, A.B.; Knebel, A.; Buhrlage, S.J.; Wightman, M.; Kelly, S.M.; Wood, N.T.; Virdee, S.; Gray, N.S.; et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 2014, 5, 4763. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Pérez Berrocal, D.A.; Witting, K.F.; Ovaa, H.; Mulder, M.P. Hybrid Chains: A Collaboration of Ubiquitin and Ubiquitin-Like Modifiers Introducing Cross-Functionality to the Ubiquitin Code. Front. Chem. 2020, 7, 931. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Smits, A.H.; van Tilburg, G.B.; Jansen, P.W.; Makowski, M.M.; Ovaa, H.; Vermeulen, M. An Interaction Landscape of Ubiquitin Signaling. Mol. Cell 2017, 65, 941–955.e8. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Lutz, J.; Höllmüller, E.; Scheffner, M.; Marx, A.; Stengel, F. Identification of Proteins Interacting with Ubiquitin Chains. Angew. Chem. Int. Ed. 2017, 56, 15764–15768. [Google Scholar] [CrossRef]
- Radley, E.H.; Long, J.; Gough, K.C.; Layfield, R. The ‘dark matter’ of ubiquitin-mediated processes: Opportunities and challenges in the identification of ubiquitin-binding domains. Biochem. Soc. Trans. 2019, 47, 1949–1962. [Google Scholar] [CrossRef]
- Herhaus, L.; Dikic, I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015, 16, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Cui, J.; Yao, Q.; Li, S.; Ding, X.; Lu, Q.; Mao, H.; Liu, L.; Zheng, N.; Chen, S.; Shao, F. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science 2010, 329, 1215–1218. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.Q. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009, 10, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattern, M.; Sutherland, J.; Kadimisetty, K.; Barrio, R.; Rodriguez, M.S. Using Ubiquitin Binders to Decipher the Ubiquitin Code. Trends Biochem. Sci. 2019, 44, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN Restricts Met1-Linked Ubiquitination to Control Innate Immune Signaling. Mol. Cell 2013, 50, 818–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, G.M.; Finley, D.; Vogel, C. K63 polyubiquitination is a new modulator of the oxidative stress response. Nat. Struct. Mol. Biol. 2015, 22, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Phu, L.; Izrael-Tomasevic, A.; Matsumoto, M.L.; Bustos, D.; Dynek, J.N.; Fedorova, A.V.; Bakalarski, C.E.; Arnott, D.; Deshayes, K.; Dixit, V.M.; et al. Improved quantitative mass spectrometry methods for characterizing complex ubiquitin signals. Mol. Cell. Proteom. 2011, 10, M110.003756. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, H.; Burana, D.; Ohtake, F.; Arai, N.; Kaiho, A.; Komada, M.; Tanaka, K.; Saeki, Y. Ub-ProT reveals global length and composition of protein ubiquitylation in cells. Nat. Commun. 2018, 9, 524. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [CrossRef] [Green Version]
- Boname, J.M.; Thomas, M.; Stagg, H.R.; Xu, P.; Peng, J.; Lehner, P.J. Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic 2010, 11, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Goto, E.; Yamanaka, Y.; Ishikawa, A.; Aoki-Kawasumi, M.; Mito-Yoshida, M.; Ohmura-Hoshino, M.; Matsuki, Y.; Kajikawa, M.; Hirano, H.; Ishido, S. Contribution of lysine 11-linked ubiquitination to MIR2-mediated major histocompatibility complex class I internalization. J. Biol. Chem. 2010, 285, 35311–35319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.J.; Rape, M. Enhanced protein degradation by branched ubiquitin chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hospenthal, M.K.; Mevissen, T.E.; Komander, D. Deubiquitinase-based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest). Nat. Protoc. 2015, 10, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Peng, J. Characterization of polyubiquitin chain structure by middle-down mass spectrometry. Anal. Chem. 2008, 80, 3438–3444. [Google Scholar] [CrossRef] [Green Version]
- Valkevich, E.M.; Sanchez, N.A.; Ge, Y.; Strieter, E.R. Middle-Down mass spectrometry enables characterization of branched ubiquitin chains. Biochemistry 2014, 53, 4979–4989. [Google Scholar] [CrossRef]
- Crowe, S.O.; Rana, A.S.; Deol, K.K.; Ge, Y.; Strieter, E.R. Ubiquitin Chain Enrichment Middle-Down Mass Spectrometry Enables Characterization of Branched Ubiquitin Chains in Cellulo. Anal. Chem. 2017, 89, 4428–4434. [Google Scholar] [CrossRef] [Green Version]
- Swatek, K.N.; Usher, J.L.; Kueck, A.F.; Gladkova, C.; Mevissen, T.E.T.; Pruneda, J.N.; Skern, T.; Komander, D. Insights into ubiquitin chain architecture using Ub-clipping. Nature 2019, 572, 533–537. [Google Scholar] [CrossRef]
- Mathis, B.; Lai, Y.; Qu, C.; Janicki, J.; Cui, T. CYLD-Mediated Signaling and Diseases. Curr. Drug Targets 2014, 16, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, M.E.; Schot, R.; Buta, S.; Oudesluijs, G.; Tinschert, S.; Speer, S.D.; Li, Z.; van Unen, L.; Heijsman, D.; Goldmann, T.; et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TOR CH syndrome. J. Exp. Med. 2016, 213, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef]
- Cowey, C.L.; Rathmell, W.K. VHL gene mutations in renal cell carcinoma: Role as a biomarker of disease outcome and drug efficacy. Curr. Oncol. Rep. 2009, 11, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25, S32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thery, F.; Wu, N.C.; Luhmann, E.K.; Dussurget, O.; Foecke, M.; Bredow, C.; Jiménez-Fernández, D.; Leandro, K.; Beling, A.; et al. The in vivo ISGylome links ISG15 to metabolic pathways and autophagy upon Listeria monocytogenes infection. Nat. Commun. 2019, 10, 5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The scope of the cellular ubiquitome. (A) The number of ubiquitinated proteins and sites reported by key studies in the last 20 years [10,12,14,15,17,19,20,21,22,23]. (B–D) Insight from the PhosphositePlus database [24] on ubiquitinated proteins: (B) the most frequently ubiquitinated protein domains; (C) the overlap of studies reporting ubiquitination sites; and (D) the frequency of the number of ubiquitination sites per protein.
Figure 1.
The scope of the cellular ubiquitome. (A) The number of ubiquitinated proteins and sites reported by key studies in the last 20 years [10,12,14,15,17,19,20,21,22,23]. (B–D) Insight from the PhosphositePlus database [24] on ubiquitinated proteins: (B) the most frequently ubiquitinated protein domains; (C) the overlap of studies reporting ubiquitination sites; and (D) the frequency of the number of ubiquitination sites per protein.

Figure 2.
Activity based profiling for DUBs. (A) A schematic for an activity-based probe profiling experiment coupled with LC-MS/MS. (B) Types of ABPP experiment: (i) catalogue of DUBs to show active DUBs in cells and to discover new DUBs in mammals and other species; (ii) specific probes allow pulldown of DUBs to study their interactomes; (iii) inhibitor profiling can show specificity of DUB inhibitors; and (iv) DUB ABPs can show how DUB activity is upregulated in certain conditions, giving clues to the function of that DUB.
Figure 2.
Activity based profiling for DUBs. (A) A schematic for an activity-based probe profiling experiment coupled with LC-MS/MS. (B) Types of ABPP experiment: (i) catalogue of DUBs to show active DUBs in cells and to discover new DUBs in mammals and other species; (ii) specific probes allow pulldown of DUBs to study their interactomes; (iii) inhibitor profiling can show specificity of DUB inhibitors; and (iv) DUB ABPs can show how DUB activity is upregulated in certain conditions, giving clues to the function of that DUB.

Figure 3.
MS advances in deciphering ubiquitin chain topology and the ubiquitin code. (A) Ubiquitin itself can be modified by a plethora of PTMs which are amenable to detection by MS-methods. (B) Novel interactors of ubiquitin chains have been identified through incubation of synthetic diUb chains with cell lysate. Pulldown of these short chains followed by proteomic analysis in the eluate enables detection of novel interactors for different ubiquitin chain types. (C) Typical ubiquitin chain structures have been suggested by Ub-Prot, a technique allowing analysis of the length of different ubiquitin chain types. (D) Quantitation of ubiquitin chain branch points has been enabled by Ub-Clipping. Lb cleaves ubiquitin between the 74th and 75th amino acid. For ubiquitin monomers in a chain, this cleavage leaves the characteristic GG remnant from a distal ubiquitin (furthest from substrate) conjugated to a proximal ubiquitin. The number of GG remnants can be detected by intact MS suggesting the degree of branching.
Figure 3.
MS advances in deciphering ubiquitin chain topology and the ubiquitin code. (A) Ubiquitin itself can be modified by a plethora of PTMs which are amenable to detection by MS-methods. (B) Novel interactors of ubiquitin chains have been identified through incubation of synthetic diUb chains with cell lysate. Pulldown of these short chains followed by proteomic analysis in the eluate enables detection of novel interactors for different ubiquitin chain types. (C) Typical ubiquitin chain structures have been suggested by Ub-Prot, a technique allowing analysis of the length of different ubiquitin chain types. (D) Quantitation of ubiquitin chain branch points has been enabled by Ub-Clipping. Lb cleaves ubiquitin between the 74th and 75th amino acid. For ubiquitin monomers in a chain, this cleavage leaves the characteristic GG remnant from a distal ubiquitin (furthest from substrate) conjugated to a proximal ubiquitin. The number of GG remnants can be detected by intact MS suggesting the degree of branching.


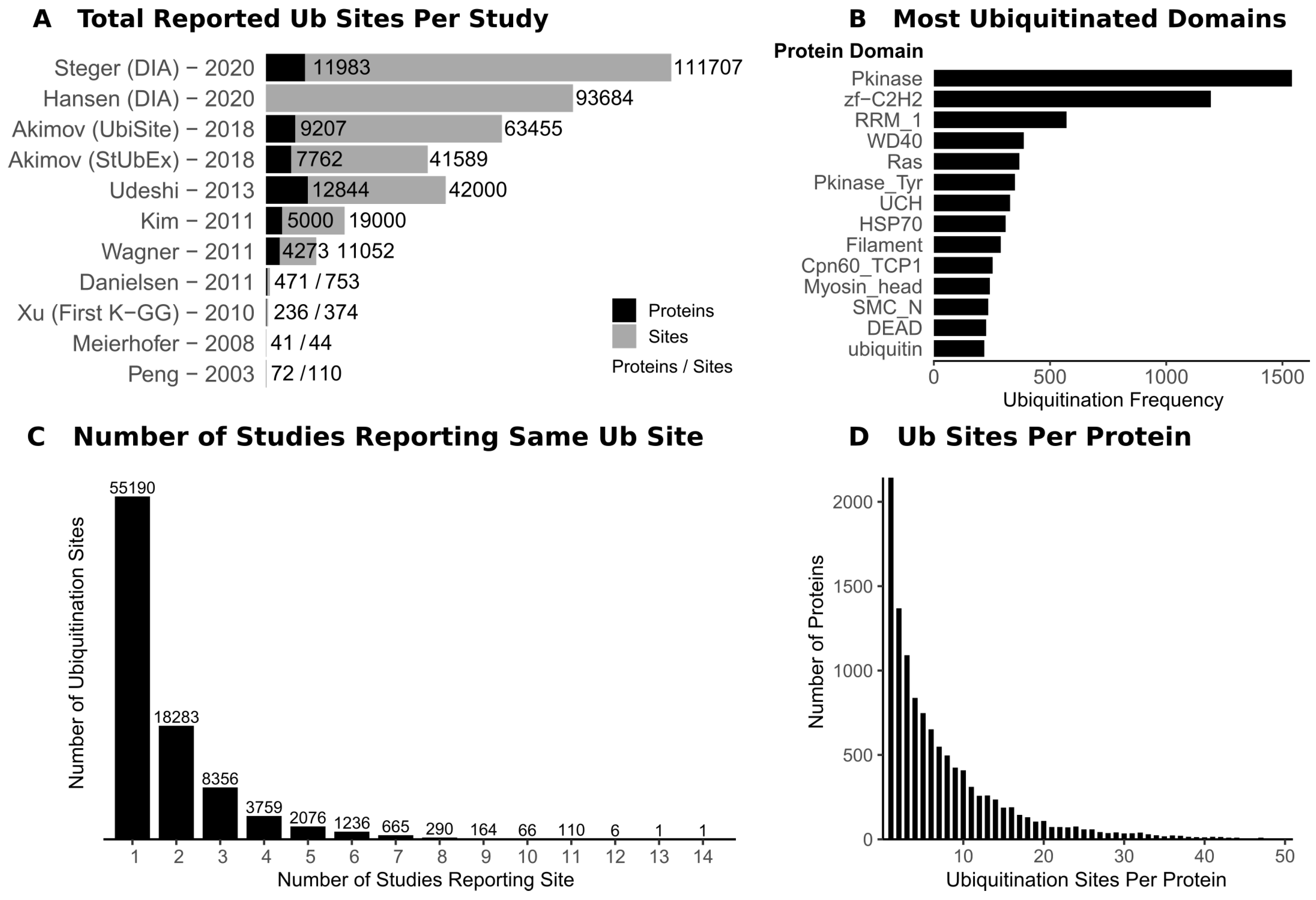{kind=link}
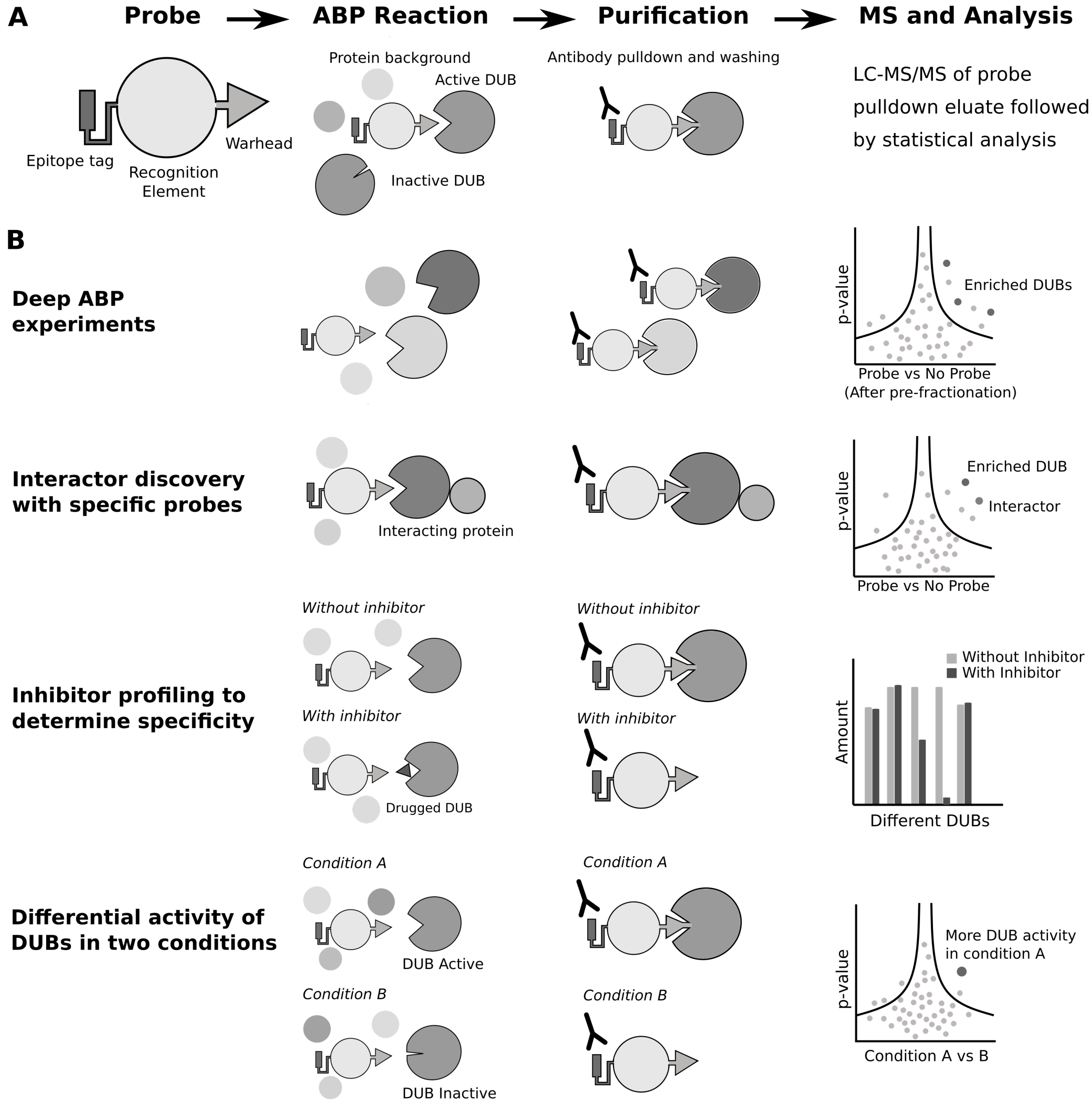{kind=link}
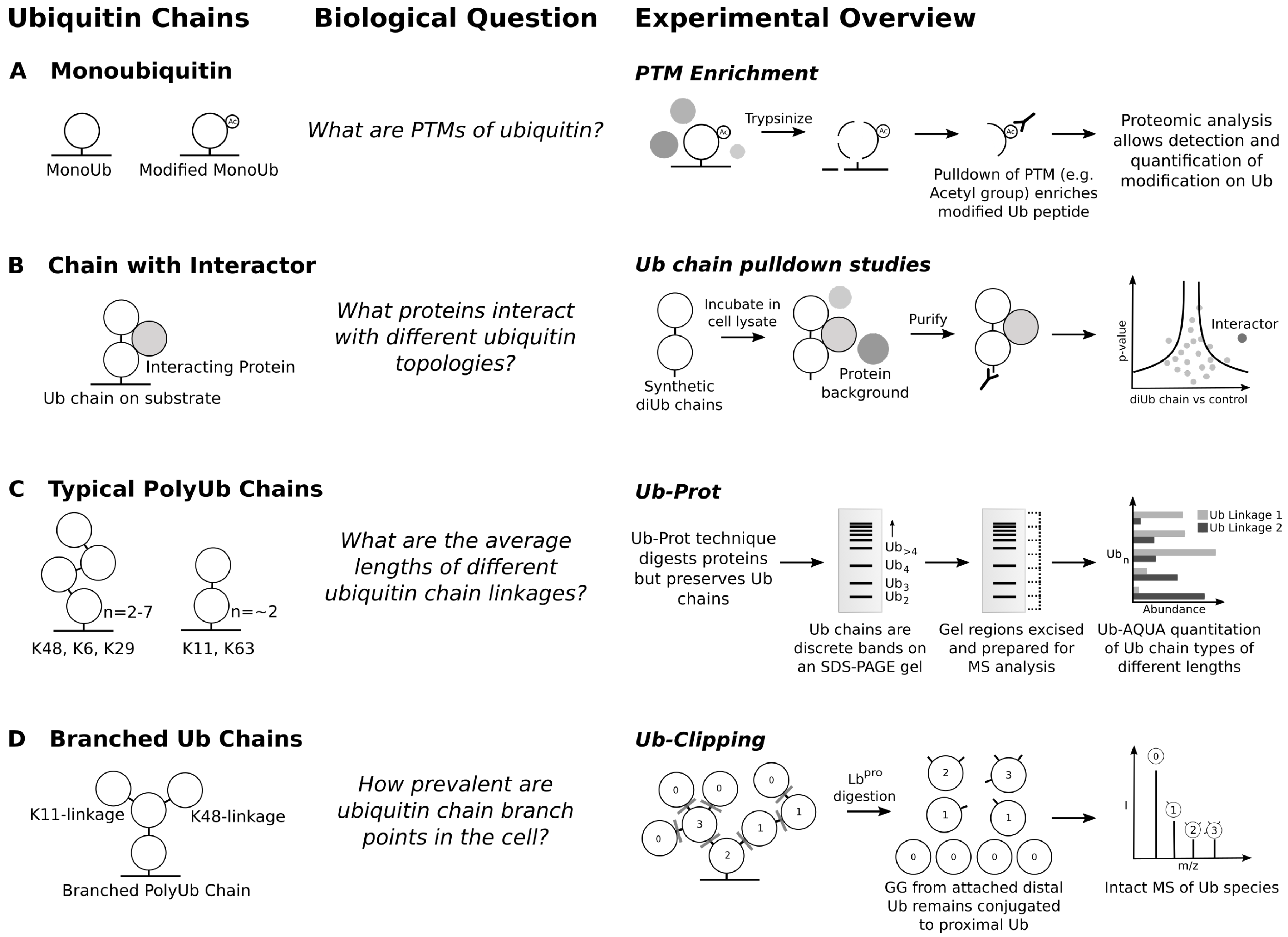{kind=link}
Table 1.
Types of ubiquitomics experiment.
| Aim | Sample Requirement | Enrichment Method | MS Factors and Analysis | Example |
|---|---|---|---|---|
| Deep ubiquitome | Up to 50 mg cell culture | UbiSite | Prefractionation to increase depth | [21] |
| Multiple PTMs | 1–20 mg cell culture | UbiSite or K-GG with additional PTM pulldowns | Multi-Omic data analysis | [31] |
| Multiple conditions | 0.5–20 mg cell culture | K-GG or TUBE | Use of SILAC or TMT— in-solution or on-bead | [19,29] |
| Chain type specific | 1–200 mg cell culture/yeast | TUBE, possible to combine with K-GG | - | [38] |
| Low abundance modifications | <1 mg lysate | K-GG | Use of DIA to increase MS sensitivity | [22,23] |
Table 2.
List of resources for ubiquitination site identification.
| Databases | Information | Reference |
|---|---|---|
| PhosphositePlus Database | Most comprehensive database for protein ubiquitination including most recent studies | [24] |
| Protein Lysine Modification Database (PLMD) | Contains information on lysine ubiquitination and on other lysine modifications. Potential for investigating PTM crosstalk | [41] |
| Mammalian Ubiquitination Site Database (mUbiSiDa) | A database of ubiquitination sites assembled in 2013 | [44] |
| Ubiquitin and Ubiquitin-like conjugation Database (UUCD) | A database of actual and predicted ubiquitin and Ubl associated machinery in several species | [45] |
Table 3.
Examples of ubiquitin site profiling studies to investigate substrates of the ubiquitin machinery and for the discovery of ubiquitinated proteins following specific stimuli.
Table 3.
Examples of ubiquitin site profiling studies to investigate substrates of the ubiquitin machinery and for the discovery of ubiquitinated proteins following specific stimuli.
| Ubiquitin Modifying Enzyme | Study Details | Reference |
|---|---|---|
| Cullin Ring Ligases | K-GG, CRL inhibition | [18] |
| SPOP | K-GG, SILAC, mutant and overexpression | [33] |
| Parkin | K-GG, inactive mutant | [28] |
| K-GG, inactive mutant | [32] | |
| LZTR1 | K-GG, knockout | [63] |
| HUWE1 | K-GG, knockdown | [64] |
| Skp2 | TUBE, overexpression | [65] |
| USP7 | K-GG, DIA-MS, inhibitor | [23] |
| USP9X | K-GG, knockdown | [66] |
| USP22 | K-GG, knockdown and overexpression | [67] |
| USP30 | K-GG, knockdown | [68] |
| K-GG, knockout | [32] | |
| K-GG, inhibitor | [69] | |
| K-GG, knockout and inhibitor | [70] | |
| USP32 | TUBE, knockdown | [71] |
| Stimulus | ||
| UV-induced DNA damage | K-GG, SILAC | [27] |
| UV- and radiation-induced DNA damage | K-GG, SILAC | [31] |
| TNF signalling | K-GG, SILAC | [72] |
| Cell cycle synchronisation | K-GG, DIA-MS | [23] |
| Lenalidomide treatment | K-GG, UbiFast/TMT | [29] |
| CRP-XL signalling | K-GG | [73] |
| Photosensitiser treatment | K-GG | [74] |
| Proteasome inhibition | K-GG, SILAC | [18] |
| K-GG, SILAC | [75] | |
| UbiSite | [21] | |
| Muscle atrophy | K-GG, time course examining mouse muscle ubiquitome following atrophy | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vere, G.; Kealy, R.; Kessler, B.M.; Pinto-Fernandez, A. Ubiquitomics: An Overview and Future. Biomolecules 2020, 10, 1453. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101453
AMA Style
Vere G, Kealy R, Kessler BM, Pinto-Fernandez A. Ubiquitomics: An Overview and Future. Biomolecules. 2020; 10(10):1453. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101453
Chicago/Turabian StyleVere, George, Rachel Kealy, Benedikt M. Kessler, and Adan Pinto-Fernandez. 2020. "Ubiquitomics: An Overview and Future" Biomolecules 10, no. 10: 1453. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101453
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.