2. Materials and Methods
2.1. Chemicals
Melting points were determined on a Kofler hot-stage apparatus and are uncorrected. The NMR spectra were recorded on Bruker Aspect 3000 (1H: 360 MHz, 13C: 90 MHz) and Bruker Avance II 400 (1H: 400 MHz; 13C: 100 MHz) spectrometers using TMS as internal standard. Chemical shifts were reported as ppm and 3JH,H coupling constants in Hz. Chiral HPLC separation of rac-20a-g, rac-23a-g, rac-3a-g, and rac-4a-g were performed on a JASCO HPLC system with Chiralpak-IA column (5 μm, 150 × 4.6 mm, hexane/2-propanol 80:20, 90:10 eluent, respectively, 1 mL min−1 flow rate) or Chiralpak-IC column (5 μm, 250 × 4.6 mm, hexane/2-propanol 70:30 eluent, respectively, 1 mL min−1 flow rate) and HPLC-ECD spectra were recorded in stopped-flow mode on a JASCO J-810 electronic circular dichroism spectropolarimeter equipped with a 10 mm HPLC flow cell. ECD ellipticity (ϕ) values were not corrected for concentration. For an HPLC-ECD spectrum, three consecutive scans were recorded and averaged with 2 nm bandwidth, 1 s response, and standard sensitivity. The HPLC-ECD spectrum of the eluent recorded in the same way was used as background. The concentration of the injected sample was set so that the HT value did not exceed 500 V in the HT channel down to 230 nm. IR spectra were recorded on a JASCO FT/IR-4100 spectrometer and absorption bands are presented as wavenumber in cm−1. Electrospay quadrupole time-of-flight HRMS measurements were performed with a MicroTOF-Q type QqTOF MS or maXis II UHR ESI- QTOF MS instrument equipped with an ESI source from Bruker (Bruker Daltoniks, Bremen, Germany).
2.2. General Procedure for the Synthesis of Tosyl Oxime Analogues (5a-g)
Oxime derivative 16a-g (12.54 mmol) and Et3N (2.11 mL, 15.04 mmol) were dissolved in anhydrous CH2Cl2 (50 mL) under inert atmosphere. At room temperature, p-toluenesulfonyl chloride (15.04 mmol) was added to the solution. The mixture was refluxed for 3 h. Extraction with water, drying over MgSO4 and concentration under reduced pressure afforded the crude product as orange oil. The oil was triturated with cold hexane, which resulted in the pure product.
N-{[(4-methylphenyl)sulfonyl]oxy}-2-phenyl-2,3-dihydro-4H-chromen-4-imine (5a). White crystals, yield 87%, mp 142–143 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.43 (s, 3H, CH3), 2.75 (dd, J = 17.2, 12.4 Hz, 1H, 3-Ha), 3.44 (dd, J = 17.2, 2.8 Hz, 1H, 3-Hb), 5.01 (dd, J = 12.4, 2.8 Hz, 1H, 2-H), 6.94 (m, 2H, 6-H, 8-H), 7.34 (m, 8H, 7-H, 2′-H, 3′-H, 4′-H, 5′-H, 6′-H, 3″-H, 5″-H) 7.81 (dd, J = 8.0, 1.6 Hz, 1H, 5-H), 7.92 (d, 2H, 2″-H, 6″-H); 13C-NMR (100 MHz, CDCl3): δ: 21.8 (C-CH3), 31.9 (C-3), 76.8 (C-2), 115.6 (C-4a), 118.3 (C-8), 121.8 (C-6), 125.1 (C-5), 126.2 (C-2′, C-6′), 128.9 (C-3′, C-4′, C-5′), 129.1 (C-3″, C-5″), 129.7 (C-2″, C-6″), 132.6 (C-1″), 133.5 (C-7), 138.7 (C-1′), 145.3 (C-4″), 157.2 (C-4), 157.9 (C-8a); HRMS (ESI) calcd. for C22H19NaNO4S [M+Na]+ 416.093; found 416.093.
N-{[(4-methylphenyl)sulfonyl]oxy}-2-(4-methoxyphenyl)-2,3-dihydro-4H-chromen-4-imine (5b). Off-white crystals, yield 93%, mp 165–167 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.44 (s, 3H, CH3), 2.78 (dd, J = 17.6, 12.8 Hz, 1H, 3-Ha), 3.42 (dd, J = 17.6, 2.8 Hz, 1H, 3-Hb), 3.81 (s, 3H, OCH3), 4.97 (dd, J = 12.8, 2.8 Hz, 1H, 2-H), 6.91 (m, 4 H, 6-H, 8-H, 3′-H, 5′-H), 7.32 (m, 5H, 7-H, 2′-H, 6′-H, 3″-H, 5″-H), 7.81 (dd, J = 8.0, 1.2 Hz, 1H, 5-H), 7.93 (d, 2H, 2″-H, 6″-H); 13C-NMR (100 MHz, CDCl3) δ: 21.8 (C-CH3), 31.6 (C-3), 55.4 (C-OCH3), 76.5 (C-2), 114.2 (C-3′, C-5′) 115.5 (C-4a), 118.3 (C-8), 121.7 (C-6), 125.1 (C-5), 127.7 (C-2′, C-6′), 129.1 (C-3″, C-5″), 129.7 (C-2″, C-6″), 130.7 (C-1′), 132.6 (C-1″), 133.5 (C-7), 145.3 (C-4″), 157.5 (C-4), 158.0 (C-8a), 160.0 (C-4′); HRMS (ESI) calcd. for C23H21NaNO5S [M+Na]+ 446.104; found 446.105.
N-{[(4-methylphenyl)sulfonyl]oxy}-2-(3,4-dimethoxyphenyl)-2,3-dihydro-4H-chromen-4-imine (5c). Off-white crystals, yield 92%, mp 148–150 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.44 (s, 3H, CH3), 2.82 (dd, J = 17.6, 12.4 Hz, 1H, 3-Ha), 3.43 (d, J = 17.6 Hz, 1H, 3-Hb), 3.89 (d, 6H, 2 × OCH3), 4.99 (d, J = 12.4 Hz, 1H, 2-H), 6.87 (d, J = 7.6 Hz, 1H, 8-H), 6.94 (m, 4 H, 6-H, 2′-H, 5′-H, 6′-H), 7.35 (m, 3H, 7-H, 3″-H, 5″-H), 7.80 (d, J = 8.4 Hz, 1H, 5-H), 7.93 (d, 2H, 2″-H, 6″-H); 13C-NMR (100 MHz, CDCl3): δ = 21.7 (C-CH3), 31.7 (C-3), 56.0 (2 × C-OCH3), 76.7 (C-2), 109.4 (C-5′), 111.2 (C-2′), 115.5 (C-4a), 118.3 (C-8), 118.9 (C-6′), 121.8 (C-6), 125.0 (C-5), 129.0 (C-3″, C-5″), 129.7 (C-2″, C-6″), 131.1 (C-1″), 132.5 (C-1′), 133.4 (C-7), 145.3 (C-4″), 149.3 (C-4′), 149.5 (C-3′), 157.3 (C-4), 157.8 (C-8a); HRMS (ESI) calcd. for C24H23NaNO6S [M+Na]+ 476.114; found 476.113.
N-{[(4-methylphenyl)sulfonyl]oxy}-2-(3,5-dimethoxyphenyl)-2,3-dihydro-4H-chromen-4-imine (5d). Off-white crystals, yield: 89%, mp 142–144 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.44 (s, 3H, CH3), 2.76 (d, J = 17.6, 12.8 Hz, 1H, 3-Ha), 3.44 (dd, J = 17.6, 3.2 Hz, 1H, 3-Hb), 3.8 (s, 6H, 2xOCH3), 4.96 (dd, J = 12.8, 3.2 Hz, 1H, 2-H), 6.44 (t, J = 2.4 Hz, 1H, 4′-H), 6.55 (d, 2H, 2′-H, 6′-H), 6.94 (m, 2H, 6-H, 8-H), 7.32 (7-H, 3″-H, 5″-H), 7.80 (dd, J = 8.4, 1.6 Hz, 1H, 5-H), 7.92 (d, 2H, 2″-H, 6″-H); 13C-NMR (100 MHz, CDCl3) δ: 21.8 (C-CH3), 32.0 (C-3), 55.5 (2xC-OCH3), 76.8 (C-2), 100.6 (C-4′), 104.2 (C-2′, C-6′), 115.6 (C-4a), 118.4 (C-8), 121.9 (C-6), 125.1 (C-5), 129.1 (C-3″, C-5″), 129.7 (C-2″, C-6″), 132.6 (C-1″), 133.5 (C-7), 141.1 (C-1′), 145.3 (C-4″), 157.2 (C-4), 157.7 (C-8a), 161.2 (C-3′, C-5′); HRMS (ESI) calcd. for C24H23NO6S [M + H]+ 454.132; found 454.131.
N-{[(4-methylphenyl)sulfonyl]oxy}-2-(3,4,5-trimethoxyphenyl)-2,3-dihydro-4H-chromen-4-imine (5e). Off-white crystals, yield: 84%, mp 157–159 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.45 (s, 3H, CH3), 2.81 (dd, J = 17.6, 12.4 Hz, 1H, 3-Ha), 3.45 (dd, J = 17.6, 3.2 Hz, 1H, 3-Hb), 3.85 (m, 9H, 3 × OCH3), 4.98 (dd, J = 12.8, 2.8 Hz, 1H, 2-H), 6.65 (s, 2H, 2′-H, 6′-H), 6.95 (m, 2H, 6-H, 8-H), 7.34 (m, 3H, 7-H, 3″-H, 5″-H), 7.81 (dd, J = 8.4, 1.6 Hz, 1H, 5-H), 7.92 (d, 2H, 2″-H, 6″-H); 13C-NMR (100 MHz, CDCl3) δ: 21.8 (C-CH3), 32.0 (C-3), 56.3 (2xC-OCH3), 60.9 (C-OCH3), 77.0 (C-2), 103.3 (C-2′, C-6′), 115.6 (C-4a), 118.3 (C-8), 121.9 (C-6), 125.1 (C-5), 129.1 (C-3″, C-5″), 129.7 (C-2″, C-6″), 132.5 (C-1″), 133.5 (C-7), 134.3 (C-1′), 138.3 (C-4′), 145.3 (C-4″), 153.6 (C-3′, C-5′), 157.1 (C-4), 157.7 (C-8a); HRMS (ESI) calcd. for C25H25NO7S [M + H]+ 484.143; found 484.141.
N-{[(4-methylphenyl)sulfonyl]oxy}-2-(naphthalene-1-yl)-2,3-dihydro-4H-chromen-4-imine (5f). Off-white crystals, yield 80%, mp 136–138 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.40 (s, 3H, CH3), 2.93 (dd, J = 17.6, 12.8 Hz, 1H, 3-Ha), 3.61 (dd, J = 17.6, 3.2 Hz, 1H, 3-Hb), 5.69 (dd, J = 12.8, 2.8 Hz, 1H, 2-H), 6.95 (m, 2H, 6-H, 8-H), 7.31 (m, 3H, 7-H, 3″-H, 5″-H), 7.44 (m, 3H, 2′-H, 3′-H, 7′-H), 7.62 (d, J = 6.8 Hz, 1H, 6′-H), 7.82 (m, 5H, 5-H, 4′-H, 5′-H, 8′-H, 2″-H, 6″-H); 13C-NMR (100 MHz, CDCl3) δ: 21.8 (C-CH3), 31.1 (C-3), 74.2 (C-2), 115.8 (C-4a), 118.4 (C-8), 122.0 (C-6), 122.9 (C-8′), 124.0 (C-5), 125.2 (C-2′), 125.4 (C-3′), 126.0 (C-7′), 126.8 (C-6′), 129.0 (C-3″, C-5″), 129.2 (C-5′), 129.5 (C-4′), 129.7 (C-2″, C-6″), 130.3 (C-8a’), 132.5 (C-1″), 133.5 (C-7), 133.9 (C-4a’), 134.0 (C-2′), 145.3 (C-4″), 157.5 (C-4), 158.1 (C-8a); HRMS (ESI) calcd. for C26H21NaNO4S [M+Na]+ 466.109; found 466.108.
N-{[(4-methylphenyl)sulfonyl]oxy}-2-(naphthalene-2-yl)-2,3-dihydro-4H-chromen-4-imine (5g). Off-white crystals, yield: 83%, mp 207–209 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.45 (s, 3H, CH3), 2.90 (dd, J = 17.6, 12.4 Hz, 1H, 3-Ha), 3.55 (dd, J = 17.6, 3.2 Hz, 1H, 3-Hb), 5.22 (dd, J = 12.4, 3.2 Hz, 1H, 2-H), 6.96 (m, 2H, 6-H, 8-H), 7.35 (m, 3H, 7-H, 3″-H, 5″-H), 7.50 (m, 3H, 3′-H, 6′-H, 7′-H), 7.85 (m, 7H, 5-H, 1′-H, 4′-H, 5′-H, 8′-H, 2″-H, 6″-H); 13C-NMR (100 MHz, CDCl3) δ: 21.9 (C-CH3), 31.8 (C-3), 76.9 (C-2), 115.7 (C-4a), 118.4 (C-8), 121.9 (C-6), 123.7 (C-5), 125.2 (C-1′), 125.5 (C-8′), 126.7 (C-3′, C-7′), 127.9 (C-6′), 128.3 (C-5′), 128.9 (C-4′), 129.2 (C-3″, C-5″), 129.8 (C-2″, C-6″), 132.6 (C-8a’), 133.3 (C-1″), 133.5 (C-4a’), 133.6 (C-7), 136.1 (C-2′), 145.4 (C-4″), 157.3 (C-4), 157.9 (C-8a); HRMS (ESI) calcd. for C26H21NaNO4S [M+Na]+ 466.109; found 466.108.
2.3. General Procedure for Neber Rearrangement (rac-cis-1a-g, rac-trans-1a-g, 17a-g)
Tosyl oxime derivatives 5a-g (8.895 mmol) were dissolved in anhydrous toluene (50 mL) under inert atmosphere and then 10.9 mL NaOEt (940 mg Na in 50 mL EtOH) was added dropwise to the solution. Stirring for 1 day at room temperature afforded an orange suspension. The suspension was filtered through cellite and washed with EtOH. Concentration of the filtrate under vacuum provided the crude product as an orange oil. Then it was dissolved in CH2Cl2 and 3 N HCl solution (3 mL) was added to it. After 2 h stirring at room temperature, an orange suspension was obtained. Filtration and washing with acetone provided the cis product as white powder. Then the filtrate was thoroughly concentrated under reduced pressure and trituration with acetone afforded the trans product as off-white powder. The residue filtrate was purified after concentration by column chromatography using toluene/ethyl acetate 4:1 as eluent. The benzoxazole derivates 17a-g were obtained by this procedure.
(±)-(2R*,3R*)-3-amino-2-phenyl-2,3-dihydro-4H-chroman-4-one hydrochloride (
rac-
trans-
1a) [
5]: Off-white solid, yield 30%, mp 197–199 °C;
1H-NMR (400 MHz, DMSO-
d6) δ: 5.01 (d,
J = 12.4 Hz, 1H, 3-H), 5.77 (d,
J = 12.4 Hz, 1H, 2-H), 7.13 (d,
J = 8 Hz, 1H, 8-H), 7.20 (m, 1H, 6-H), 7.50 (m, 3H, 3′-H, 4′-H, 5′-H), 7.68 (m, 3H, 7-H, 2′-H, 6′-H), 7.87 (dd,
J = 8.0, 1.6 Hz, 1H, 5-H), 8.72 (bs, 3H, NH
3);
13C-NMR (100 MHz, DMSO-
d6) δ: 55.7 (C-3), 80.2 (C-2), 118.0 (C-8), 118.5 (C-4a), 122.5 (C-6), 126.9 (C-5), 128.6 (C-2′, C-6′), 128.8 (C-3′, C-5′), 129.9 (C-4′), 134.3 (C-1′), 137.6 (C-7), 160.8 (C-8a), 187.6 (C-4); HRMS (ESI) calcd. for C
15H
13NO
2 [M + H]
+ 240.102; found 240.101.
(±)-(2R*,3R*)-3-amino-2-(4-methoxyphenyl)-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-trans-1b): Off-white solid, yield 32%, mp 206–209 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.80 (s, 3H, OCH3), 5.03 (d, J = 12.4 Hz, 1H, 3-H), 5.83 (d, J = 12.4 Hz, 1H, 2-H), 7.03 (d, 2H, 3′-H, 5′-H), 7.10 (d, J = 8.0 Hz, 1H, 8-H), 7.18 (t, J = 7.6 Hz, 1H, 6-H), 7.64 (m, 3H, 7-H, 2′-H, 6′-H), 7.85 (d, J = 7.6 Hz, 1H, 5-H), 8.83 (s, 3H, NH3); 13C-NMR (100 MHz, DMSO-d6) δ: 55.4 (C-OCH3), 55.9 (C-3), 80.0 (C-2), 114.2 (C-3′, C-5′), 118.0 (C-8), 118.6 (C-4a), 122.4 (C-6), 126.5 (C-1′), 126.9 (C-5), 130.2 (C-2′, C-6′), 137.5 (C-7), 160.4 (C-4′), 160.9 (C-8a), 187.8 (C-4); HRMS (ESI) calcd. for C16H15NO3 [M + H]+ 270.113; found 270.111.
(±)-(2R*,3R*)-3-amino-2-(3,4-dimethoxyphenyl)-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-trans-1c): Off-white solid, yield 39%, mp 187–189 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.80 (s, 6H, 2 × OCH3), 5.08 (d, J = 12.6 Hz, 1H, 3-H), 5.71 (d, J = 12.6 Hz, 1H, 2-H), 7.03 (d, J = 9.2 Hz, 1H, 8-H), 7.12 (m, 3H, 6-H, 5′-H, 6′-H), 7.38 (s, 1H, 2′-H), 7.67 (t, J = 8.0 Hz, 1H, 7-H), 7.86 (d, J = 8.0 Hz, 1H, 5-H), 8.71 (bs, 3H, NH3); 13C-NMR (100 MHz, DMSO-d6) δ: 55.6 (2 × C-OCH3), 55.9 (C-3), 80.3 (C-2), 111.7 (C-2′), 111.9 (C-5′), 118.0 (C-8), 118.5 (C-4a), 121.6 (C-6′), 122.3 (C-6), 126.5 (C-1′), 126.8 (C-5), 137.4 (C-7), 148.9 (C-4′), 150.0 (C-3′), 160.8 (C-8a), 187.7 (C-4); HRMS (ESI) calcd. for C17H17NO4 [M + H]+ 300.123; found 300.122.
(±)-(2R*,3R*)-3-amino-2-(3,5-dimethoxyphenyl)-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-trans-1d): Off-white solid, yield: 46%, mp 187–189 °C; 1H-NMR (360 MHz, DMSO-d6) δ: 3.79 (s, 6H, 2 × OCH3), 5.04 (d, J = 12.6 Hz, 1H, 3-H), 5.68 (d, J = 12.6 Hz, 1H, 2-H), 6.60 (t, J = 1.8 Hz, 1H, 4′-H), 6.88 (d, 2 H, 2′-H, 6′-H), 7.17 (d, J = 8.3 Hz, 1H, 8-H), 7.20 (t, J = 7.9 Hz, 1H, 6-H), 7.69 (m, 1H, 7-H), 7.87 (dd, J = 7.9, 1.4 Hz, 1H, 5-H), 8.69 (bs, 3H, NH3); 13C-NMR (90 MHz, DMSO-d6) δ: 55.8 (2 × C-OCH3), 56.2 (C-3), 80.7 (C-2), 102.0 (C-4′), 107.0 (C-2′, C-6′), 118.5 (C-8), 118.9 (C-4a), 123.0 (C-6), 127.3 (C-5), 136.7 (C-1′), 138.1 (C-7), 161.2 (C-8a, C-3′, C-5′), 188.1 (C-4); HRMS (ESI) calcd. for C17H17NO4 [M + H]+ 300.123; found 300.124.
(±)-(2R*,3R*)-3-amino-2-(3,4,5-trimethoxyphenyl)-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-trans-1e): Off-white solid, yield 48%, mp 188–190 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.71 (s, 3H, OCH3), 3.82 (s, 6H, 2 × OCH3), 5.09 (d, J = 12.4 Hz, 1H, 3-H), 5.65 (d, J = 12.4 Hz, 1H, 2-H), 7.05 (s, 2H, 2′-H, 6′-H), 7.15 (d, J = 8.4 Hz, 1H, 8-H), 7.20 (m, 1H, 6-H), 7.69 (m, 1H, 7-H), 7.87 (d, J = 8.0, 1.6 Hz, 1H, 5-H), 8.63 (bs, 3H, NH3); 13C-NMR (100 MHz, DMSO-d6) δ: 55.9 (C-3), 56.0 (2 × C-OCH3), 60.0 (C-OCH3), 80.5 (C-2), 106.1 (C-2′, C-6′), 118.1 (C-8), 118.5 (C-4a), 122.5 (C-6), 126.9 (C-5), 129.6 (C-1′), 137.6 (C-7), 138.5 (C-4′), 153.1 (C-3′, C-5′), 160.8 (C-8a), 187.8 (C-4); HRMS (ESI) calcd. for C18H19NO5 [M + H]+ 330.134; found 330.133.
(±)-(2R*,3R*)-3-amino-2-naphthalen-1-yl-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-trans-1f): Off-white solid, yield 64%, mp 212–215 °C; 1H-NMR (360 MHz, DMSO-d6) δ: 5.40 (d, J = 12.6 Hz, 1H, 3-H), 6.53 (d, J = 12.6 Hz, 1H, 2-H), 7.11 (d, J = 8.3 Hz, 1H, 8-H), 7.23 (t, J = 7.9 Hz, 1H, 6-H), 7.56 (m, 3H, 2′-H, 3′-H, 7′-H), 7.68 (t, J = 7.9 Hz, 1H, 7-H), 7.94 (m, 2H, 5-H, 6′-H), 8.04 (m, 2H, 4′-H, 5′-H), 8.49 (d, J = 7.9 Hz, 1H, 8′-H), 8.79 (bs, 3H, NH3); 13C-NMR (90 MHz, DMSO-d6) δ: 55.2 (C-3), 78.8 (C-2), 118.5 (C-8), 119.3 (C-4a), 123.0 (C-6), 124.6 (C-8′), 125.9 (C-2′), 126.5 (C-3′), 126.5 (C-5), 127.1 (C-7′), 127.5 (C-6′), 129.3 (C-5′), 130.1 (C-8a’), 131.1 (C-4′), 131.7 (C-1′), 134.4 (C-4a’), 137.9 (C-7), 161.3 (C-8a), 188.2 (C-4); HRMS (ESI) calcd. for C19H15NO2 [M + H]+ 290.118; found 290.118.
(±)-(2R*,3R*)-3-amino-2-naphthalen-2-yl-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-trans-1g): Off-white solid, yield 48%, mp 201–203 °C; 1H-NMR (360 MHz, DMSO-d6) δ: 5.16 (d, J = 12.6 Hz, 1H, 3-H), 5.96 (d, J = 12.6 Hz, 1H, 2-H), 7.16 (d, J = 8.3 Hz, 1H, 8-H), 7.22 (m, 1H, 6-H), 7.60 (m, 2H, 3′-H, 7′-H), 7.70 (m, 1H, 7-H), 7.84 (d, J = 8.3 Hz, 1H, 5-H), 7.90 (d, J = 8.3 Hz, 1H, 6′-H), 7.97 (m, 2H, 4′-H, 5′-H), 8.06 (d, J = 8.6 Hz, 1H, 8′-H), 8.21 (s, 1H, 1′-H), 8.74 (bs, 3H, NH3); 13C-NMR (90 MHz, DMSO-d6) δ: 55.8 (C-3), 80.4 (C-2), 118.0 (C-6), 118.6 (C-4a), 122.5 (C-8), 125.1 (C-5), 126.5 (C-1′), 126.9 (C-8′), 127.7 (C-6′, C-7′), 128.2 (C-3′), 128.7 (C-5′), 128.8 (C-4′), 131.8 (C-2′), 132.6 (C-8a’), 133.7 (C-4a’), 137.5 (C-7), 160.8 (C-8a), 187.4 (C-4).
(±)-(2R*,3S*)-3-amino-2-phenyl-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-cis-1a): White solid, yield 30%, mp 202–204 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 5.09 (d, J = 5.6 Hz, 1H, 3-H), 6.23 (d, J = 5.6 Hz, 1H, 2-H), 7.15 (m, 2H, 6-H, 8-H), 7.38 (m, 5 H, 2′-H, 3′-H, 4′-H, 5′-H, 6′-H), 7.68 (m, 1H, 7-H), 7.82 (dd, J = 7.6, 1.6 Hz, 1H, 5-H), 9.09 (bs, 3H, NH3); 13C-NMR (100 MHz, DMSO-d6): δ = 54.8 (C-3), 78.6 (C-2), 118.4 (C-8), 119.6 (C-4a), 122.4 (C-6), 126.7 (C-5), 127.7 (C-2′, C-6′), 129.2 (C-3′, C-5′), 129.6 (C-4′), 133.7 (C-1′), 138.0 (C-7), 160.2 (C-8a), 187.2 (C-4); HRMS (ESI) calcd. for C15H13NO2 [M + H]+ 240.102; found 240.101.
(±)-(2R*,3S*)-3-amino-2-(4-methoxyphenyl)-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-cis-1b): white solid, yield 14%. mp 186–188 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.72 (s, 3H, OCH3), 5.11 (d, J = 6.0 Hz, 1H, 3-H), 6.18 (d, J = 6.0 Hz, 1H, 2-H), 6.90 (d, 2H, 3′-H, 5′-H), 7.11 (d, J = 8.4 Hz, 1H, 8-H), 7.14 (t, J = 7.6 Hz, 1H, 6-H), 7.28 (d, 2H, 2′-H, 6′-H), 7.65 (m, 1H, 7-H), 7.80 (d, J = 7.6 Hz, 1H, 5-H), 9.08 (bs, 3H, NH3); 13C-NMR (100 MHz, DMSO-d6) δ: 54.5 (C-3), 55.0 (C-OCH3), 78.0 (C-2), 114.1 (C-3′, C-5′), 118.0 (C-8), 119.2 (C-4a), 121.8 (C-6), 125.1 (C-1′), 126.1 (C-5), 128.9 (C-2′, C-6′), 137.5 (C-7), 159.6 (C-4′), 159.8 (C-8a), 187.1 (C-4); HRMS (ESI) calcd. for C16H15NO3 [M + H]+ 270.113; found 270.111.
(±)-(2R*,3S*)-3-amino-2-(3,4-dimethoxyphenyl)-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-cis-1c): White solid, yield 23%, mp 191–195°C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.70 (d, 6H, 2 × OCH3), 5.09 (d, J = 5.6 Hz, 1H, 3-H), 6.16 (d, J = 5.6 Hz, 1H, 2-H), 6.76 (dd, J = 8.4, 2.0 Hz, 1H, 8-H), 6.89 (d, J = 8.4 Hz, 1H, 5′-H), 7.14 (m, 3H, 6-H, 2′-H, 6′-H), 7.68 (m, 1H, 7-H), 7.80 (dd, J = 8.0, 1.6 Hz, 1H, 5-H), 9.07 (bs, 3H, NH3); 13C-NMR (100 MHz, DMSO-d6) δ: 54.8 (C-3), 55.4 (C-OCH3), 55.5 (C-OCH3), 78.2 (C-2), 111.8 (C-2′, C-5′), 118.1 (C-8), 119.2 (C-6′), 119.4 (C-4a), 122.1 (C-6), 125.5 (C-1′), 126.3 (C-5), 137.7 (C-7), 148.6 (C-4′), 149.4 (C-3′), 160.1 (C-8a), 187.2 (C-4); HRMS (ESI) calcd. for C17H17NO4 [M + H]+ 300.123; found 300.122.
(±)-(2R*,3S*)-3-amino-2-(3,5-dimethoxyphenyl)-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-cis-1d): White solid, yield 20%, mp 195–197 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.68 (s, 6H, 2 × OCH3), 5.05 (d, J = 5.6 Hz, 1H, 3-H), 6.15 (d, J = 5.6 Hz, 1H, 2-H), 6.49 (t, J = 2.4 Hz, 1H, 4′-H), 6.56 (d, 2H, 2′-H, 6′-H), 7.16 (m, 2H, 6-H, 8-H), 7.68 (m, 1H, 7-H), 7.79 (dd, J = 8.0, 1.6 Hz, 1H, 5-H), 9.08 (bs, 3H, NH3); 13C-NMR (100 MHz, DMSO-d6) δ: 54.6 (C-3), 55.3 (2 × C-OCH3), 78.2 (C-2), 100.5 (C-4′), 105.5 (C-2′, C-6′), 118.1 (C-8), 119.4 (C-4a), 122.2 (C-6), 126.4 (C-5), 135.5 (C-1′), 137.8 (C-7), 160.1 (C-8a), 160.6 (C-3′, C-5′), 186.8 (C-4); HRMS (ESI) calcd. for C17H17NO4 [M + H]+ 300.123; found 300.124.
(±)-(2R*,3S*)-3-amino-(3,4,5-trimethoxyphenyl)-2,3-dihydro-4H-chroman-4-one hydrochloride (rac-cis-1e): Off-white solid, yield 17%, mp 194–196 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.64 (d, 9H, 3 × OCH3), 5.01 (d, J = 5.2 Hz, 1H, 3-H), 6.14 (d, J = 5.2 Hz, 1H, 2-H), 6.75 (s, 2H, 2′-H, 6′-H), 7.17 (m, 2H, 6-H, 8-H), 7.70 (t, J = 7.6 Hz, 1H, 7-H), 7.82 (d, J = 8.0 Hz, 1H, 5-H), 9.08 (bs, 3H, NH3); 13C-NMR (100 MHz, DMSO-d6) δ: 54.8 (C-3), 55.8 (2 × C-OCH3), 59.9 (C-OCH3), 78.4 (C-2), 104.8 (C-2′, C-6′), 118.2 (C-8), 119.4 (C-4a), 122.3 (C-6), 126.4 (C-5), 128.8 (C-1′), 137.8 (C-7), 137.9 (C-4′), 153.0 (C-3′, C-5′), 160.2 (C-8a), 187.0 (C-4); HRMS (ESI) calcd. for C18H19NO5 [M + H]+ 330.134; found 330.133.
2. -[(E)-2-phenylethenyl]-1,3-benzoxazole (17a) [
18]: Pale yellow crystals, yield 15%, mp 62–64 °C;
1H-NMR (400 MHz, CDCl
3) δ: 7.05 (d,
J = 16.4 Hz, 1H, 2′-H), 7.30 (m, 2H, 4-H, 7-H), 7.36 (m, 3H, 5′-H, 6′-H, 7′-H), 7.50 (m, 1H, 5-H), 7.57 (d, 2H, 4′-H, 8′-H), 7.69 (m, 1H, 6-H), 7.76 (d,
J = 16.4 Hz, 1H, 1′-H);
13C-NMR (100 MHz, CDCl
3) δ: 110.4 (C-2′), 114.0 (C-5), 119.9 (C-6), 124.6 (C-4), 125.3 (C-7), 127.6 (C-5′, C-7′), 129.0 (C-4′, C-8′), 129.9 (C-6′), 135.2 (C-3′), 139.6 (C-1′), 142.2 (C-3a), 150.5 (C-7a), 162.9 (C-2); HRMS (ESI) calcd. for C
15H
11NO [M + H]
+ 222.092; found 222.089.
2. -[(E)-2-(4-methoxyphenyl)ethenyl]-1,3-benzoxazole (17b) [
19]: White chrystals, yield 20%, mp 128–130 °C;
1H-NMR (400 MHz, CDCl
3) δ: 3.79 (s, 3H, OCH
3), 6.88 (m, 3H, 2′-H, 5′-H, 7′-H), 7.27 (m, 2H, 4-H, 7-H), 7.46 (m, 3H, 5-H, 4′-H, 8′-H), 7.67 (m, 2H, 1’-H, 6-H);
13C-NMR (100 MHz, CDCl
3) δ: 55.4 (C-OCH
3), 110.2 (C-5), 111.5 (C-2′), 114.4 (C-5′, C-7′), 119.7 (C-6), 124.4 (C-4), 124.9 (C-7), 127.9 (C-3′), 129.1 (C-4′, C-8′), 139.1 (C-1′), 142.3 (C-3a), 150.4 (C-7a), 161.0 (C-6′), 163.2 (C-2); HRMS (ESI) calcd. for C
16H
13NO
2 [M + H]
+ 252.102; found 252.103.
2. -[(E)-2-(3,4-dimethoxyphenyl)ethenyl]-1,3-benzoxazole (17c): Pale yellow crystals, yield: 16%, mp 119–120 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.90 (d, 6H, 2 × OCH3), 6.86 (d, J = 8.0 Hz, 1H, 7′-H), 6.91 (d, J = 16.4 Hz, 1H, 2′-H), 7.11 (s, 1H, 4′-H), 7.13 (d, J = 8.0 Hz, 1H, 8′-H), 7.29 (m, 2H, 4-H, 7-H), 7.48 (m, 1H, 5-H), 7.68 (m, 2H, 6-H, 1′-H); 13C-NMR (100 MHz, CDCl3) δ: 55.9 (C-OCH3), 56.0 (C-OCH3), 109.3 (C-4′), 110.2 (C-5), 111.2 (C-7′), 111.8 (C-2′), 119.7 (C-6), 121.9 (C-8′), 124.5 (C-4), 125.0 (C-7), 128.3 (C-3′), 139.3 (C-1′), 142.3 (C-3a), 149.4 (C-6′), 150.4 (C-5′), 150.8 (C-7a), 163.2 (C-2); HRMS (ESI) calcd. for C17H15NO3 [M + H]+ 282.113; found 282.112.
2. -[(E)-2-(3,4,5-trimethoxyphenyl)ethenyl]-1,3-benzoxazole (17e): Pale yellow crystals, yield 14%, mp. 147–149 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.91 (s, 9 H, 3 × OCH3), 6.82 (s, 2H, 4′-H, 8′-H), 6.96 (d, J = 16.0 Hz, 1H, 2′-H), 7.33 (m, 2H, 4-H, 7-H), 7.50 (m, 1H, 5-H), 7.68 (m, 2H, 6-H, 1′-H); 13C-NMR (100 MHz, CDCl3) δ: 56.2 (2 × C-OCH3), 61.1 (C-OCH3), 104.7 (C-4′, C-8′), 110.3 (C-5), 113.3 (C-2′), 119.9 (C-6), 124.6 (C-4), 125.2 (C-7), 130.8 (C-3′), 139.4 (C-1′), 139.8 (C-6′), 142.3 (C-3a), 150.5 (C-7a), 153.6 (C-5′, C-7′), 162.8 (C-2); HRMS (ESI) calcd. for C18H17NO4 [M + H]+ 312.123; found 312.125.
2. -[(E)-2-(naphthalen-1-yl)ethenyl]-1,3-benzoxazole (17f): Pale yellow crystals, yield: 10%, mp 124–126 °C; 1H-NMR (400 MHz, CDCl3) δ: 7.15 (d, J = 16.0 Hz, 1H, 2′-H), 7.34 (m, 2H, 4-H, 7-H), 7.50 (m, 4 H, 5-H, 4′-H, 5′-H, 9′-H), 7.74 (m, 1H, 6-H), 7.84 (m, 3H, 6′-H, 7′-H, 8′-H), 8.28 (d, J = 8.4 Hz, 1H, 10′-H), 8.59 (d, J = 16.0 Hz, 1H, 1′-H); 1H-NMR (400 MHz, CDCl3) δ: 110.5 (C-5), 116.5 (C-2′), 120.1 (C-6), 123.5 (C-10′), 124.6 (C-4), 124.7 (C-4′), 125.4 (C-7), 125.7 (C-5′), 126.3 (C-9′), 126.9 (C-8′), 128.9 (C-7′), 130.2 (C-6′), 131.4 (C-6a’), 132.6 (C-10a’), 133.9 (C-3′), 136.4 (C-1′), 142.4 (C-3a), 150.6 (C-7a), 162.9 (C-2); HRMS (ESI) calcd. for C19H13NO [M + H]+ 272.107; found 272.106.
2. -[(E)-2-(naphthlaen-2-yl)ethenyl]-1,3-benzoxazole (17g) [
18]: White crystals, yield 11%, mp 129–131 °C;
1H-NMR (400 MHz, CDCl
3) δ: 7.11 (d,
J = 16.4 Hz, 1H, 2′-H), 7.28 (m, 2H, 4-H, 7-H), 7.45 (m, 3H, 5-H, 4′-H, 8′-H), 7.68 (m, 2H, 6-H, 7′-H), 7.77 (m, 3H, 5′-H, 6′-H, 9′-H), 7.90 (m, 2H, 10′-H, 1′-H);
1H-NMR (400 MHz, CDCl
3) δ: 110.4 (C-5), 114.1 (C-2′), 120.0 (C-6), 123.2 (C-10′), 124.6 (C-4), 125.3 (C-7), 126.8 (C-9′), 127.1 (C-4′), 127.9 (C-8′), 128.5 (C-7′), 128.8 (C-6′), 129.2 (C-5′), 132.7 (C-5a’), 133.5 (C-9a’), 134.0 (C-3′), 139.5 (C-1′), 142.3 (C-3a), 150.5 (C-7a), 162.9 (C-2); HRMS (ESI) calcd. for C
19H
13NO [M + H]
+ 272.107; found 272.107.
2.4. General Procedure for the Synthesis of 2-Chloroacetamide Derivatives (rac-trans-18a-g)
The hydrochloride salt of 3-aminoflavanone derivatives rac-cis-1a-g or rac-trans-1a-g (1.452 mmol) was suspended in anhydrous THF under inert atmosphere. After addition of Et3N (510 µL, 3.630 mmol), the reaction was stirred for 5 min at room temperature or at 0 °C. Then chloroacetyl chloride (139 µL, 1.742 mmol) was added dropwise to the suspension and it was stirred further for 15 min. The reaction was quenched with water and then it was extracted with CH2Cl2. The combined organic phases were dried over MgSO4 and concentrated under reduced pressure. Trituration with cold Et2O afforded the pure product.
(±)-2-chloro-N-[(2R*,3R*)-2-phenyl-4-oxochroman-3-yl]acetamide (rac-trans-18a): White crystals, yield 77%, mp 214–216 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.82 (d, J = 15.6 Hz, 1H, CH2-Ha), 3.95 (d, J = 15.6 Hz, 1H, CH2-Hb), 5.08 (dd, J = 12.0, 8.4 Hz, 1H, 3-H), 5.39 (d, J = 12.0 Hz, 1H, 2-H), 6.79 (d, J = 8.0 Hz, 1H, NH), 7.06 (d, J = 8.4 Hz, 1H, 8-H), 7.09 (t, J = 7.6 Hz, 1H, 6-H), 7.41 (m, 3H, 3′-H, 4′-H, 5′-H), 7.50 (m, 3H, 7-H, 2′-H, 6′-H), 7.93 (dd, J = 7.6, 1.2 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 42.4 (C-CH2), 58.4 (C-3), 83.2 (C-2), 118.2 (C-8), 120.0 (C-4a), 122.4 (C-6), 127.7 (C-2′, C-6′), 127.8 (C-5), 128.7 (C-3′, C-5′), 129.6 (C-4′), 135.7 (C-1′), 136.9 (C-7), 161.4 (C-8a), 166.2 (amide carbonyl), 189.9 (C-4); HRMS (ESI) calcd. for C17H14ClNaNO3 [M+Na]+ 338.056; found 338.056.
(±)-2-chloro-N-[(2R*,3R*)-2-(4-methoxyphenyl)-4-oxochroman-3-yl]acetamide (rac-trans-18b): White crystals, yield 71%, mp 179–180 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.77 (s, 3H, OCH3), 3.99 (m, 2H, CH2), 5.00 (dd, J = 12.4, 8.4 Hz, 1H, 3-H), 5.57 (d, J = 12.4 Hz, 1H, 2-H), 6.95 (d, 2H, 3′-H, 5′-H), 7.07 (d, J = 8.0 Hz, 1H, 8-H), 7.13 (m, 1H, 6-H), 7.42 (d, 2H, 2′-H, 6′-H), 7.61 (m, 1H, 7-H), 7.81 (dd, J = 7.6, 1.6 Hz, 1H, 5-H), 8.51 (d, J = 8.4 Hz, 1H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 42.3 (C-CH2), 55.3 (C-OCH3), 57.8 (C-3), 81.2 (C-2), 113.8 (C-3′, C-5′), 118.2 (C-8), 120.1 (C-4a), 122.1 (C-6), 127.1 (C-5), 128.9 (C-1′), 129.4 (C-2′, C-6′), 136.7 (C-7), 159.8 (C-4′), 161.1 (C-8a), 166.2 (amide carbonyl), 190.0 (C-4); HRMS (ESI) calcd. for C18H16ClNaNO4 [M+Na]+ 368.066; found 368.067.
(±)-2-chloro-N-[(2R*,3R*)-2-(3,4-dimethoxyphenyl)-4-oxochroman-3-yl]acetamide (rac-trans-18c): White crystals, yield 80%, mp 183–185 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.77 (s, 6H, 2xOCH3), 3.96 (m, 2H, CH2), 5.03 (dd, J = 12.4, 8.4 Hz, 1H, 3-H), 5.56 (d, J = 12.4 Hz, 1H, 2-H), 6.95 (d, J = 8.4 Hz, 1H, 5′-H), 7.00 (dd, J = 8.4, 1.6 Hz, 1H, 6′-H), 7.08 (d, J = 8.0 Hz, 1H, 8-H), 7.12 (m, 2H, 6-H, 2′-H), 7.60 (m, 1H, 7-H), 7.82 (dd, J = 7.6, 1.6 Hz, 1H, 5-H), 8.52 (d, J = 8.4 Hz, 1H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 42.2 (C-CH2), 55.5 (2xC-OCH3), 57.6 (C-3), 81.2 (C-2), 109.5 (C-2′), 111.2 (C-5′), 118.0 (C-8), 119.9 (C-4a), 120.7 (C-6′), 121.9 (C-6), 126.9 (C-5), 129.0 (C-1′), 136.4 (C-7), 148.4 (C-4′), 149.2 (C-3′), 160.8 (C-8a), 166.0 (amide carbonyl), 189.8 (C-4); HRMS (ESI) calcd. for C19H18ClNaNO5 [M+Na]+ 398.077; found 398.078.
(±)-2-chloro-N-[(2R*,3R*)-2-(3,5-dimethoxyphenyl)-4-oxochroman-3-yl]acetamide (rac-trans-18d): White crystals, yield 80%, mp 206–208 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.74 (s, 6 H, 2 × OCH3), 4.01 (s, 2H, CH2), 4.96 (dd, J = 12.0, 8.4 Hz, 1H, 3-H), 5.57 (d, J = 12.0 Hz, 1H, 2-H), 6.49 (t, J = 2.4 Hz, 1H, 4′-H), 6.68 (d, 2H, 2′-H, 6′-H), 7.09 (d, J = 8.0 Hz, 1H, 8-H), 7.13 (t, J = 7.6 Hz, 1H, 6-H), 7.61 (m, 1H, 7-H), 7.80 (dd, J = 7.6, 1.6 Hz, 5-H), 8.67 (d, J = 8.4 Hz, 1H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 42.1 (C-CH2), 55.3 (2 × C-OCH3), 57.6 (C-3), 81.0 (C-2), 100.7 (C-4′), 105.8 (C-2′, C-6′), 118.0 (C-8), 119.8 (C-4a), 122.0 (C-6), 126.9 (C-5), 136.5 (C-7), 138.8 (C-1′), 160.2 (C-3′, C-5′), 160.7 (C-8a), 166.1 (amide carbonyl), 189.5 (C-4); HRMS (ESI) calcd. for C19H18ClNaNO5 [M+Na]+ 398.077; found 398.077.
(±)-2-chloro-N-[(2R*,3R*)-2-(3,4,5-trimethoxyphenyl)-4-oxochroman-3-yl]acetamide (rac-trans-18e): White crystals, yield 74%, mp 140–142 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.83 (m, 9H, 3 × OCH3), 3.84 (m, 2H, CH2), 5.03 (dd, J = 12.4, 8.8 Hz, 1H, 3-H), 5.41 (d, J = 12.4 Hz, 1H, 2-H), 6.74 (s, 2H, 2′-H, 6′-H), 6.99 (m, 2H, 6-H, 8-H), 7.29 (d, J = 8.8 Hz, 1H, NH), 7.47 (m, 1H, 7-H), 7.80 (d, J = 7.6, 1.2 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 41.9 (C-CH2), 55.8 (2 × C-OCH3), 57.8 (C-3), 60.4 (C-OCH3), 82.2 (C-2), 104.4 (C-2′, C-6′), 117.7 (C-8), 119.5 (C-4a), 121.8 (C-6), 127.1 (C-5), 131.0 (C-1′), 136.2 (C-7), 138.1 (C-4′), 152.8 (C-3′, C-5′), 160.7 (C-8a), 166.3 (amide carbonyl), 189.5 (C-4); HRMS (ESI) calcd. for C20H20ClNaNO6 [M+Na]+ 428.088; found 428.089.
(±)-2-chloro-N-[(2R*,3R*)-2-(naphthalen-1-yl)-4-oxochroman-3-yl]acetamide (rac-trans-18f): White crystals, yield 79%, mp 252–254 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.80 (q, 2H, CH2), 5.33 (t, J = 10.8 Hz, 1H, 3-H), 6.48 (d, J = 12.0 Hz, 1H, 2-H), 7.09 (d, J = 8.0 Hz, 1H, 8-H), 7.18 (t, J = 7.6 Hz, 1H, 6-H), 7.55 (m, 4 H, 7-H, 2′-H, 3′-H, 7′-H), 7.82 (s, 1H, 6′-H), 7.90 (d, J = 7.6 Hz, 1H, 5-H), 7.98 (m, 2H, 4′-H, 5′-H), 8.29 (s, 1H, 8′-H), 8.57 (d, J = 8.0 Hz, 1H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 42.0 (C-CH2), 57.3 (C-3), 78.1 (C-2), 118.0 (C-8), 120.1 (C-4a), 122.1 (C-6), 125.2 (C-5), 125.8 (C-2′, C-3′), 126.5 (C-7′), 127.0 (C-6′), 128.7 (C-4′), 129.5 (C-5′), 131.2 (C-4a’), 132.4 (C-8a’), 133.3 (C-1′), 136.5 (C-7), 160.9 (C-8a), 166.1 (amide carbonyl), 189.6 (C-4); HRMS (ESI) calcd. for C21H16ClNaNO3 [M+Na]+ 388.072; found 388.073.
(±)-2-chloro-N-[(2R*,3R*)-2-(naphthalen-2-yl)-4-oxochroman-3-yl]acetamide (rac-trans-18g). White crystals, yield, 82%, mp 226–228 °C; 1H-NMR (360 MHz, DMSO-d6) δ: 3.91 (q, 2H, CH2), 5.11 (dd, J = 12.2, 8.3 Hz, 1H, 3-H), 5.82 (d, J = 12.2 Hz, 1H, 2-H), 7,12 (m, 2H, 6-H, 8-H), 7.54 (m, 2H, 3′-H, 7′-H), 7.62 (m, 1H, 7-H), 7.69 (dd, J = 8.6, 1.4 Hz, 1H, 5-H), 7.85 (dd, J = 7.6, 1.4 Hz, 1H, 6′-H), 7.94 (m, 4 H, 1′-H, 4′-H, 5′-H, 8′-H), 8.59 (d, J = 8.3 Hz, 1H, NH); 13C-NMR (90 MHz, DMSO-d6) δ: 42.0 (C-CH2), 57.7 (C-3), 81.3 (C-2), 118.0 (C-8), 120.0 (C-4a), 122.0 (C-6), 124.9 (C-5), 126.3 (C-1′), 126.5 (C-8′), 126.9 (C-3′), 127.2 (C-7′), 127.5 (C-6′), 127.8 (C-5′), 128.0 (C-4′), 132.4 (C-8a’), 133.1 (C-4a’), 134.2 (C-2′), 136.4 (C-7), 160.7 (C-8a), 166.0 (amide carbonyl), 189.4 (C-4); HRMS (ESI) calcd. for C21H16ClNaNO3 [M+Na]+ 388.072; found 388.073.
2.5. General Procedure for the Synthesis of Flavan-4-ol Derivatives rac-19a-g and rac-22a-g
To the solution of the chloroacetamide derivatives (0.958 mmol) in MeOH (10 mL), NaBH4 (1.150 mmol) was added and reaction mixture was stirred at room temperature. The reaction was completed in 10 min. The pH was adjusted to about 5 with 10% HCl solution and the mixture was concentrated and the residue was extracted with ethyl acetate and water. The combined organic phase was dried over MgSO4 and the solvent was evaporated in vacuum. Column chromatography with CHCl3 as eluent provided the pure product.
(±)-2-chloro-N-[(2R*,3S*,4R*)-4-Hydroxy-2-phenyl-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-19a): White crystals, yield 90%, mp 157–159 °C; 1H-NMR (400 MHz, acetone-d6) δ: 3.76 (d, J = 14.0 Hz, 1H, CH2-Ha), 3.86 (d, J = 14.0 Hz, 1H, CH2-Hb), 4.31 (q, J = 9.2 Hz, 1H, 3-H), 4.87 (d, J = 6.4 Hz, 1H, OH), 5.11 (dd, J = 9.2, 6.4 Hz, 1H, 4-H), 5.22 (d, J = 10.4 Hz, 1H, 2-H), 6.80 (dd, J = 8.0, 0.8 Hz, 1H, 8-H), 6.96 (m, 1H, 6-H), 7.17 (m, 1H, 7-H), 7.32 (m, 3H, 3′-H, 4′-H, 5′-H), 7.47 (m, 2H, 2′-H, 6′-H), 7.54 (d, J = 8.0 Hz, 1H, 5-H), 7.61 (d, J = 8.8 Hz, NH); 13C-NMR (100 MHz, acetone-d6) δ: 43.2 (C-CH2), 56.6 (C-3), 69.6 (C-4), 80.5 (C-2), 116.7 (C-8), 121.7 (C-6), 126.6 (C-4a), 128.7 (C-2′, C-6′), 128.8 (C-3′, C-5′), 128.9 (C-4′), 129.1 (C-5), 129.5 (C-7), 138.8 (C-1′), 154.9 (C-8a), 166.6 (amide carbonyl); HRMS (ESI) calcd. for C17H16ClNaNO3 [M+Na]+ 340.071; found 340.073.
(±)-2-chloro-N-[(2R*,3S*,4R*)-4-Hydroxy-2-(4-methoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-19b): White crystals, yield 92%, mp 163–164 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.74 (bs, 1H, OH), 3.81 (m, 4 H, CH2-Ha, OCH3), 3.99 (d, J = 15.6 Hz, 1H, CH2-Hb), 4.27 (m, 1H, 3-H), 4.99 (d, 2H, 2-H, 4-H), 6.51 (d, J = 6.4 Hz, 1H, NH), 6.88 (d, J = 7.2 Hz, 1H, 8-H), 6.92 (d, 2H, 3′-H, 5′-H), 7.02 (m, 1H, 6-H), 7.21 (m, 1H, 7-H), 7.35 (d, 2H, 2′-H, 6′-H), 7.54 (d, J = 7.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, DMSO-d6) δ: 42.9 (C-CH2), 55.8 (C-OCH3), 57.5 (C-3), 70.8 (C-4), 79.4 (C-2), 114.8 (C-3′, C-5′), 116.9 (C-8), 122.3 (C-6), 124.4 (C-4a), 128.6 (C-1′), 128.7 (C-5), 129.3 (C-2′, C-6′), 129.9 (C-7), 154.0 (C-8a), 160.8 (C-4′), 167.5 (amide carbonyl); HRMS (ESI) calcd. for C18H18NaNO4 [M+Na]+ 370.109; found 370.110.
(±)-2-chloro-N-[(2R*,3S*,4R*)-4-Hydroxy-2-(3,4-dimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-19c): White crystals, yield 98%, mp 156–158 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.78 (d, J = 15.2 Hz, 1H, CH2-Ha), 3.87 (d, 6H, 2 × OCH3), 3.92 (m, 2H, CH2-Hb, OH), 4.26 (q, J = 9.2 Hz, 1H, 3-H), 4.96 (m, 2H, 2-H, 4-H), 6.56 (d, J = 6.8 Hz, 1H, NH), 6.85 (m, 2H, 2′-H, 5′-H), 6.95 (m, 3H, 8-H, 2′-H, 6′-H), 7.00 (t, J = 7.6 Hz, 1H, 6-H), 7.19 (t, J = 8.0 Hz, 1H, 7-H), 7.51 (d, J = 7.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 42.6 (C-CH2), 56.0 (C-OCH3), 56.1 (C-OCH3), 56.9 (C-3), 70.3 (C-4), 79.3 (C-2), 110.1 (C-5′), 111.1 (C-2′), 116.6 (C-6′), 120.5 (C-8), 121.9 (C-6), 124.2 (C-4a), 128.3 (C-5), 128.7 (C-1′), 129.5 (C-7), 149.5 (C-4′), 149.9 (C-3′), 153.6 (C-8a), 167.2 (amide carbonyl); HRMS (ESI) calcd. for C19H20ClNaNO5 [M+Na]+ 400.092; found 400.094.
(±)-2-chloro-N-[(2R*,3S*,4R*)-4-Hydroxy-2-(3,5-dimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-19d). White crystals, yield 94%, mp 160–162 °C; 1H-NMR (400 MHz, acetone-d6) δ: 3.78 (d, 6H, 2xOCH3), 3.83 (d, J = 14.0 Hz, 1H, CH2-Ha), 3.92 (d, J = 14.0 Hz, 1H, CH2-Hb), 4.31 (q, J = 9.2 Hz, 1H, 3-H), 4.90 (d, J = 6.0 Hz, 1H, OH), 5.11 (dd, J = 9.2, 6.0 Hz, 1H, 4-H), 5.17 (d, J = 10.4 Hz, 1H, 2-H), 6.44 (t, J = 2.4 Hz, 1H, 4′-H), 6.69 (d, 2H, 2′-H, 6′-H), 6.81 (d, J = 8.4 Hz, 1H, 8-H), 6.96 (m, 1H, 6-H), 7.17 (m, 1H, 7-H), 7.54 (d, J = 7.6 Hz, 1H, 5-H), 7.64 (d, J = 9.2 Hz, 1H, NH); 13C-NMR (101 MHz, acetone-d6) δ: 44.2 (C-CH2), 56.6 (2xC-OCH3), 57.4 (C-3), 70.6 (C-4), 81.4 (C-2), 102.2 (C-4′), 107.5 (C-2′, C-6′), 117.7 (C-8), 122.7 (C-6), 127.5 (C-4a), 129.8 (C-5), 130.4 (C-7), 141.9 (C-1′), 155.8 (C-8a), 162.4 (C-3′, C-5′), 167.7 (amide carbonyl); HRMS (ESI) calcd. for C19H19NaNO5 [M+Na]+ 364.116; found 364.118; HRMS (ESI) calcd. for C19H20ClNaNO5 [M+Na]+ 400.092; found 400.094.
(±)-2-chloro-N-[(2R*,3S*,4R*)-4-Hydroxy-2-(3,4,5-trimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-19e): White crystals, yield 92%, mp 155–157 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.79 (m, 10 H, CH2-Ha, 3xOCH3), 3.92 (d, J = 15.2 Hz, 1H, CH2-Hb), 4.06 (bs, 1H, OH), 4.23 (q, J = 8.8 Hz, 1H, 3-H), 4.96 (m, 2H, 2-H, 4-H), 6.64 (m, 3H, NH, 2′-H, 6′-H), 6.88 (d, J = 8.4 Hz, 1H, 8-H), 7.00 (t, J = 7.6 Hz, 1H, 6-H), 7.20 (t, J = 7.2 Hz, 1H, 7-H), 7.50 (d, J = 7.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 42.2 (C-CH2), 55.9 (2xC-OCH3), 56.6 (C-3), 60.5 (C-OCH3), 69.6 (C-4), 79.2 (C-2), 104.2 (C-2′, C-6′), 116.1 (C-8), 121.6 (C-6), 123.8 (C-4a), 127.8 (C-5), 129.1 (C-7), 131.5 (C-1′), 138.2 (C-4′), 153.1 (C-8a), 153.2 (C-3′, C-5′), 166.8 (amide carbonyl); HRMS (ESI) calcd. for C20H22ClNaNO6 [M+Na]+ 430.103; found 430.104.
(±)-2-chloro-N-[(2R*,3S*,4R*)-4-Hydroxy-2-(naphthalen-1-yl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-19f): White crystals, yield 88%, mp 247–249 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.64 (m, 2H, CH2), 4.49 (q, J = 9.6 Hz, 1H, 3-H), 5.00 (t, J = 8.0 Hz, 1H, 4-H), 5.77 (d, J = 6.8 Hz, 1H, OH), 5.89 (d, J = 10.4 Hz, 1H, 2-H), 6.80 (d, J = 8.0 Hz, 1H, 8-H), 7.00 (t, J = 7.6 Hz, 1H, 6-H), 7.18 (t, J = 7.2 Hz, 1H, 7-H), 7.48 (m, 4 H, 2′-H, 3′-H, 6′-H, 7′-H), 7.65 (d, J = 7.2 Hz, 1H, 5-H), 7.90 (m, 2H, 4′-H, 5′-H), 8.21 (d, J = 9.2 Hz, 1H, NH), 8.28 (d, J = 9.2 Hz, 1H, 8′-H); 13C-NMR (100 MHz, DMSO-d6) δ: 42.4 (C-CH2), 53.8 (C-3), 67.9 (C-4), 76.2 (C-2), 115.7 (C-8), 120.9 (C-6), 124.0 (C-8′), 125.1 (C-2′, C-3′), 125.5 (C-5), 126.0 (C-7′), 126.2 (C-4a), 128.1 (C-6′), 128.5 (C-7, C-5′), 128.8 (C-4′), 131.4 (C-1′), 133.2 (C-4a’), 133.3 (C-8a’), 153.5 (C-8a), 165.3 (amide carbonyl); HRMS (ESI) calcd. for C21H18ClNaNO3 [M+Na]+ 390.087; found 390.088.
(±)-2-chloro-N-[(2R*,3S*,4R*)-4-Hydroxy-2-(naphthalen-2-yl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-19g): White crystals, yield 89%, mp 210–212 °C; 1H-NMR (400 MHz, acetone-d6) δ: 3.69 (d, J = 14.0 Hz, 1H, CH2-Ha), 3.83 (d, J = 14.0 Hz, 1H, CH2-Hb), 4.41 (q, J = 9.2 Hz, 1H, 3-H), 4.89 (d, J = 6.4 Hz, 1H, OH), 5.20 (dd, J = 9.2, 6.8 Hz, 1H, 4-H), 5.43 (d, J = 10.4 Hz, 1H, 2-H), 6.85 (d, J = 8.4 Hz, 1H, 8-H), 6.99 (m, 1H, 6-H), 7.20 (m, 1H, 7-H), 7.50 (m, 2H, 3′-H, 7′-H), 7.58 (d, J = 7.6 Hz, 1H, 5-H), 7.64 (m, 2H, 6′-H, NH), 7.88 (m, 3H, 4′-H, 5′-H, 8′-H), 7.98 (s, 1H, 1′-H); 13C-NMR (100 MHz, acetone-d6) δ: 43.2 (C-CH2), 56.7 (C-3), 69.6 (C-4), 80.6 (C-2), 116.8 (C-8), 121.8 (C-6), 126.1 (C-8′), 126.7 (C-4a), 126.9 (C-1′), 127.0 (C-3′), 128.2 (C-5), 128.5 (C-7′), 128.6 (C-6′), 128.9 (C-4′, C-5′), 129.5 (C-7), 133.9 (C-4a’), 134.4 (C-8a’), 136.4 (C-2′), 154.9 (C-8a), 166.6 (amide carbonyl); HRMS (ESI) calcd. for C21H18ClNaNO3 [M+Na]+ 390.087; found 390.087.
(±)-2-chloro-N-[(2R*,3R*,4R*)-4-Hydroxy-2-phenyl-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-22a): White crystals, overall yield of acylation and reduction 62%, mp 142–144 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.70 (d, J = 15.2 Hz, 1H, CH2-Ha), 3.84 (d, J = 15.2 Hz, 1H, CH2-Hb), 3.90 (d, J = 5.2 Hz, 1H, OH), 4.72 (dd, J = 9.2, 5.2 Hz, 1H, 3-H), 5.27 (t, J = 5.2 Hz, 1H, 4-H), 5.34 (s, 1H, 2-H), 6.80 (d, J = 9.2 Hz, 1H, NH), 6.98 (d, J = 8.0 Hz, 1H, 8-H), 7.02 (t, J = 7.2 Hz, 1H, 6-H), 7.22 (t, J = 7.2 Hz, 1H, 7-H), 7.32 (m, 5H, 2′-H, 3′-H, 4′-H, 5′-H, 6′-H), 7.54 (d, J = 8.0 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 42.6 (C-CH2), 51.8 (C-3), 67.4 (C-4), 77.3 (C-2), 116.6 (C-8), 122.4 (C-6), 122.9 (C-4a), 125.8 (C-2′, C-6′), 128.3 (C-5), 128.4 (C-4′), 128.6 (C-3′, C-5′), 129.5 (C-7), 136.9 (C-1′), 153.2 (C-8a), 168.1 (amide carbonyl); HRMS (ESI) calcd. for C17H16ClNaNO3 [M+Na]+ 340.071; found 340.073.
(±)-2-chloro-N-[(2R*,3R*,4R*)-4-Hydroxy-2-(4-methoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-22b): White crystals, overall yield of acylation and reduction 54%, mp 179–181 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.18 (bs, 1H, OH), 3.82 (s, 3H, OCH3), 3.83 (d, J = 15.2 Hz, 1H, CH2-Ha), 3.95 (d, J = 15.2 Hz, 1H, CH2-Hb), 4.72 (m, 1H, 3-H), 5.31 (bs, 1H, 4-H), 5.34 (s, 1H, 2-H), 6.83 (d, J = 8.8 Hz, 1H, NH), 6.91 (m, 2H, 3′-H, 5′-H), 6.98 (dd, J = 8.4, 1.2 Hz, 1H, 8-H), 7.05 (m, 1H, 6-H), 7.26 (m, 1H, 7-H), 7.36 (d, 2H, 2′-H, 6′-H), 7.56 (d, J = 7.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 42.6 (C-CH2), 52.1 (C-3), 55.4 (C-OCH3), 68.0 (C-4), 76.9 (C-2), 114.2 (C-3′, C-5′), 116.7 (C-8), 122.5 (C-6), 123.0 (C-4a), 127.1 (C-2′, C-6′), 128.4 (C-5), 128.8 (C-1′), 129.6 (C-7), 153.3 (C-8a), 159.7 (C-4′), 168.4 (amide carbonyl); HRMS (ESI) calcd. for C18H18NaNO4 [M+Na]+ 370.109; found 370.110.
(±)-2-chloro-N-[(2R*,3R*,4R*)-4-Hydroxy-2-(3,4-dimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-22c): White crystals, overall yield of acylation and reduction 70%, mp 124–126 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.57 (d, J = 5.2 Hz, 1H, OH), 3.81 (d, J = 15.2 Hz, 1H, CH2-Ha), 3.88 (d, 6H, 2xOCH3), 3.94 (d, J = 15.2 Hz, 1H, CH2-Hb), 4.71 (m, 1H, 3-H), 5.29 (m, 2H, 2-H, 4-H), 6.86 (m, 2H, NH, 5′-H), 6.98 (m, 3H, 8-H, 2′-H, 6′-H), 7.05 (m, 1H, 6-H), 7.24 (m, 1H, 7-H), 7.55 (d, J = 7.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 42.7 (C-CH2), 52.0 (C-3), 56.0 (C-OCH3), 56.1 (C-OCH3), 67.7 (C-4), 77.0 (C-2), 108.9 (C-5′), 111.2 (C-2′), 116.7 (C-6′), 118.2 (C-8), 122.5 (C-6), 123.0 (C-4a), 128.3 (C-5), 129.3 (C-1′), 129.5 (C-7), 149.0 (C-4′), 149.1 (C-3′), 153.2 (C-8a), 168.1 (amide carbonyl); HRMS (ESI) calcd. for C19H20ClNaNO5 [M+Na]+ 400.092; found 400.094.
(±)-2-chloro-N-[(2R*,3R*,4R*)-4-Hydroxy-2-(3,5-dimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-22d): White crystals, overall yield of acylation and reduction 59%, mp 156–158 °C; 1H-NMR (360 MHz, CDCl3) δ: 3.61 (d, J = 5.0 Hz, 1H, OH), 3.78 (m, 8 H, CH2, 2xOCH3), 4.72 (dd, J = 7.9, 5.0 Hz, 1H, 3-H), 5.30 (s, 2H, 2-H, 4-H), 6.42 (t, J = 2.4 Hz, 1H, 4′-H), 6.60 (d, 2H, 2′-H, 6′-H), 6.87 (d, J = 9.0 Hz, 1H, NH), 6.99 (d, J = 7.9 Hz, 1H, 8-H), 7.04 (t, J = 7.6 Hz, 1H, 6-H), 7.24 (m, 1H, 7-H), 7.55 (d, J = 7.6 Hz, 1H, 5-H); 13C-NMR (90 MHz, CDCl3) δ: 42.7 (C-CH2), 52.2 (C-3), 55.5 (2xC-OCH3), 67.9 (C-4), 77.1 (C-2), 100.4 (C-4′), 103.9 (C-2′, C-6′), 116.6 (C-8), 122.5 (C-6), 123.1 (C-4a), 128.4 (C-5), 129.5 (C-7), 139.2 (C-1′), 153.1 (C-8a), 161.2 (C-3′, C-5′), 168.3 (amide carbonyl); HRMS (ESI) calcd. for C19H20ClNaNO5 [M+Na]+ 400.092; found 400.094.
(±)-2-chloro-N-[(2R*,3R*,4R*)-4-Hydroxy-2-(3,4,5-trimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-22e): White crystals, overall yield of acylation and reduction 66%, mp 76–78 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.77 (m, 12H, CH2, OH, 3 × OCH3), 4.70 (dd, J = 8.8, 4.8 Hz, 1H, 3-H), 5.27 (s, 2H, 2-H, 4-H), 6.66 (s, 2H, 2′-H, 6′-H), 6.84 (d, J = 9.2 Hz, 1H, NH), 6.98 (d, J = 8.4 Hz, 1H, 8-H), 7.02 (t, J = 7.6 Hz, 1H, 6-H), 7.22 (t, J = 7.6 Hz, 1H, 7-H), 7.52 (d, J = 7.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 42.5 (C-CH2), 51.6 (C-3), 56.1 (2 × C-OCH3), 60.7 (C-OCH3), 67.3 (C-4), 77.1 (C-2), 102.8 (C-2′, C-6′), 116.4 (C-8), 122.3 (C-6), 122.8 (C-4a), 128.1 (C-5), 129.3 (C-7), 132.2 (C-1′), 137.7 (C-4′), 152.9 (C-8a), 153.2 (C-3′, C-5′), 167.9 (amide carbonyl); HRMS (ESI) calcd. for C20H22ClNaNO6 [M+Na]+ 430.103; found 430.104.
(±)-2-chloro-N-[(2R*,3R*,4R*)-4-Hydroxy-2-(naphthalen-2-yl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-22g): White crystals, overall yield of acylation and reduction 68%, mp 69–71 °C; 1H-NMR (360 MHz, acetone-d6) δ: 3.82 (q, 2H, CH2), 4.59 (d, J = 7.9 Hz, 1H, OH), 4.94 (m, 1H, 3-H), 5.40 (t, J = 5.4 Hz, 1H, 4-H), 5.71 (s, 1H, 2-H), 6.95 (d, J = 8.3 Hz, 1H, 8-H), 7.01 (m, 1H, 6-H), 7.21 (m, 2H, 7-H, NH), 7.48 (m, 2H, 6′-H, 7′-H), 7.58 (d, J = 7.6 Hz, 1H, 5-H), 7.69 (dd, J = 8.6, 1.4 Hz, 1H, 3′-H), 7.9 (m, 3H, 4′-H, 5′-H, 8′-H), 8.09 (s, 1H, 1′-H); 13C-NMR (90 MHz, acetone-d6) δ: 43.3 (C-CH2), 52.0 (C-3), 67.3 (C-4), 78.8 (C-2), 117.0 (C-8), 122.3 (C-6), 125.2 (C-8′), 125.3 (C-4a), 126.0 (C-1′), 126.8 (C-3′), 126.9 (C-5), 128.5 (C-6′, C-7′), 128.9 (C-4′), 129.1 (C-5′), 129.6 (C-7), 134.0 (C-4a’, C-8a’), 136.6 (C-2′), 154.6 (C-8a), 167.4 (amide carbonyl); HRMS (ESI) calcd. for C21H18ClNaNO3 [M+Na]+ 390.087; found 390.087.
2.6. General Procedure for the Synthesis of 1,4-oxazin-3-one Derivatives rac-20a-g and rac-23a-e, g
The flavan-4-ol derivative rac-19g or rac-22g (0.629 mmol) was dissolved in anhydrous THF (10 mL) under inert atmosphere. To the stirred solution, 60% dispersion of NaH (0.755 mmol) was added at room temperature. The reaction was quenched after 15 min with the addition of water. The pH was adjusted to about 5 with 10% HCl solution and then the mixture was extracted with CH2Cl2. The organic phases dried over MgSO4 and concentrated under reduced pressure. Column chromatography using CHCl3 as eluent provided the pure product.
(±)-(4aS*,5R*,10bR*)-5-phenyl-4,4a,5,10b-tetrahydrochromeno [4,3-b][1,4]oxazin-3(2H)-one (rac-20a): White crystals, yield 88%, mp 255–257 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.84 (t, J = 10.0 Hz, 1H, 4a-H), 4.40 (q, 2H, 2-H), 4.83 (d, J = 9.2 Hz, 1H, 10b-H), 4.98 (d, J = 10.4 Hz, 1H, 5-H), 5.45 (bs, 1H, NH), 6.90 (d, J = 8.0 Hz, 1H, 7-H), 7.02 (t, J = 7.6 Hz, 1H, 9-H), 7.24 (t, J = 7.6 Hz, 1H, 8-H), 7.44 (m, 6 H, 10-H, 2′-H, 3′-H, 4′-H, 5′-H, 6′-H); 13C-NMR (100 MHz, CDCl3) δ: 55.6 (C-4a), 68.4 (C-2), 73.8 (C-10b), 79.4 (C-5), 116.5 (C-7), 120.1 (C-10a), 121.6 (C-9), 125.6 (C-10), 127.7 (C-2′, C-6′), 129.6 (C-3′, C-5′), 130.0 (C-8), 130.2 (C-4′), 134.7 (C-1′), 153.3 (C-6a), 168.9 (C-3); HRMS (ESI) calcd. for C17H15NO3 [M + H]+ 282.113; found 282.115.
(4aS,5R,10bR)-20a: tR = 4.52 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 282sh (−3.79), 274 (−3.54), 228 (−11.27), 217 (4.28).
(4aR,5S,10bS)-20a: tR = 5.30 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 282sh (4.70), 274 (5.26), 228 (14.69), 217 (−5.67).
(±)-(4aS*,5SR*,10bR*)-5-(4-methoxyphenyl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-20b): White crystals, yield 89%, mp 224–226 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.79 (s, 3H, OCH3), 3.90 (t, J = 10.0 Hz, 1H, 4-H), 4.33 (s, 2H, 2-H), 4.95 (d, J = 9.2 Hz, 1H, 10b-H), 5.11 (d, J = 10.0 Hz, 1H, 5-H), 6.80 (d, J = 8.0 Hz, 1H, 7-H), 6.98 (m, 4 H, 9-H, 3′-H, 5′-H, NH), 7.20 (t, J = 8.0 Hz, 1H, 8-H), 7.37 (d, J = 7.6 Hz, 1H, 10-H), 7.41 (d, 2H, 2′-H, 6′-H); 13C-NMR (100 MHz, DMSO-d6) δ: 53.9 (C-OCH3), 55.2 (C-4a), 67.8 (C-2), 72.7 (C-10b), 78.6 (C-5), 114.2 (C-3′, C-5′), 115.7 (C-7), 120.7 (C-9), 120.9 (C-10a), 125.2 (C-10), 127.3 (C-1′), 129.3 (C-8), 130.0 (C-2′, C-6′), 153.2 (C-6a), 160.1 (C-4′), 168.3 (C-3); HRMS (ESI) calcd. for C18H17NO4 [M + H]+ 312.123; found 312.124.
(4aS,5R,10bR)-20b: tR = 5.82 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 273 (−3.94), 233 (7.54), 215 (−1.82).
(4aR,5S,10bS)-20b: tR = 6.85 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 273 (3.70), 233 (−9.20), 215 (2.98).
(±)-(4aS*,5R*,10bR*)-5-(3,4-dimethoxyphenyl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-20c): White crystals, yield 90%, mp 255–257 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.78 (d, 6H, 2 × OCH3), 3.97 (t, J = 9.6 Hz, 1H, 4a-H), 4.33 (s, 2H, 2-Ha, 2-Hb), 4.95 (d, J = 9.2 Hz, 1H, 10b-H), 5.10 (d, J = 10.4 Hz, 1H, 5-H), 6.81 (d, J = 8.0 Hz, 1H, 7-H), 6.97 (m, 4 H, 9-H, 2′-H, 5′-H, 6′-H), 7.21 (t, J = 8.0 Hz, 1H, 8-H), 7.37 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, DMSO-d6) δ: 53.8 (C-4a), 55.5 (2 × C-OCH3), 67.7 (C-2), 72.6 (C-10b), 78.9 (C-5), 111.8 (C-5′), 111.8 (C-2′), 115.7 (C-6′), 120.5 (C-7), 120.9 (C-10a), 121.3 (C-9), 125.1 (C-10), 127.4 (C-1′), 129.2 (C-8), 149.1 (C-4′), 150.2 (C-3′), 153.2 (C-6a), 168.1 (C-3); HRMS (ESI) calcd. for C19H19NaNO5 [M+Na]+ 364.116; found 364.113.
(4aR,5S,10bS)-20c: tR = 7.86 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 287 (−0.89), 282sh (1.66), 274 (1.92), 235 (6.17), 224sh (4.71).
(4aS,5R,10bR)-20c: tR = 9.50 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}:287 (0.35), 282sh (−1.61), 274 (−1.93), 235 (−6.62), 224sh (−5.44).
(±)-(4aS*,5R*,10bR*)-5-(3,5-dimethoxyphenyl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-20d): White crystals, yield 80%, mp 189–190 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.77 (m, 7H, 4a-H, 2 × OCH3), 4.38 (d, J = 17.2 Hz, 1H, 2-Ha), 4.45 (d, J = 17.2 Hz, 1H, 2-Hb), 4.78 (d, J = 9.2 Hz, 1H, 10b-H), 4.88 (d, J = 10.4 Hz, 1H, 5-H), 5.59 (s, 1H, NH), 6.50 (t, J = 2.4 Hz, 1H, 4′-H), 6.56 (d, 2H, 2′-H, 6′-H), 6.91 (d, J = 8.0 Hz, 1H, 7-H), 7.01 (t, J = 7.6 Hz, 1H, 9-H), 7.23 (m, 1H, 8-H), 7.44 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, CDCl3) δ: 55.1 (C-4a), 55.2 (2 × C-OCH3), 67.9 (C-2), 73.3 (C-10b), 79.0 (C-5), 101.2 (C-4′), 105.1 (C-2′, C-6′), 116.1 (C-7), 119.8 (C-10a), 121.2 (C-9), 125.3 (C-10), 129.6 (C-8), 136.5 (C-1′), 152.9 (C-6a), 161.3 (C-3′, C-5′), 168.5 (C-3); HRMS (ESI) calcd. for C19H19NaNO5 [M+Na]+ 364.116; found 364.113.
(4aR,5S,10bS)-20d: tR = 6.72 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (4.08), 240sh (−1.16), 233 (−3.03), 222 (1.82), 213 (−10.39).
(4aS,5R,10bR)-20d: tR = 7.07 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}:283 (−4.52), 240sh (1.28), 233 (2.95), 222 (−3.55), 213 (6.44).
(±)-(4aS*,5R*,10bR*)-5-(3,4,5-trimethoxyphenyl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-20e): White crystals, yield 83%, mp 146–148 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.84 (m, 10 H, 4a-H, 3 × OCH3), 4.41 (d, J = 16.8 Hz, 1H, 2-Ha), 4.48 (d, J = 16.8 Hz, 1H, 2-Hb), 4.82 (d, J = 9.2 Hz, 1H, 10b-H), 4.91 (d, J = 10.4 Hz, 1H, 5-H), 5.63 (s, 1H, NH), 6.65 (s, 2H, 2′-H, 6′-H), 6.92 (d, J = 8.4 Hz, 1H, 7-H), 7.03 (t, J = 7.2 Hz, 1H, 9-H), 7.26 (t, J = 7.2 Hz, 1H, 8-H), 7.46 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (101 MHz, CDCl3) δ: 55.2 (C-4a), 56.0 (2 × C-OCH3), 60.6 (C-OCH3), 68.0 (C-2), 73.4 (C-10b), 79.3 (C-5), 104.2 (C-2′, C-6′), 116.2 (C-7), 119.8 (C-10a), 121.3 (C-9), 125.3 (C-10), 129.6 (C-1′), 129.6 (C-8), 138.9 (C-4′), 152.9 (C-6a), 153.8 (C-3′, C-5′), 168.7 (C-3); HRMS (ESI) calcd. for C20H21NaNO6 [M+Na]+ 394.126; found 394.124.
(4aR,5S,10bS)-20e: tR = 10.18 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 282 (1.51), 275sh (1.37), 245 (−1.52), 233sh (1.49), 225 (2.54), 216 (−0.36).
(4aS,5R,10bR)-20e: tR = 15.71 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}:282 (−1.44), 275sh (−1.31), 245 (0.65), 233sh (−1.82), 225 (−2.09), 216 (0.52).
(±)-(4aS*,5R*,10bR*)-5-(naphthalen-1-yl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-20f): White crystals, yield 83%, mp 254–255 °C; 1H-NMR (360 MHz, DMSO-d6) δ: 4.20 (t, J = 9.0 Hz, 1H, 4a-H), 4.37 (s, 2H, 2-H), 5.19 (d, J = 8.3 Hz, 1H, 10b-H), 6.12 (bs, 1H, 5-H), 6.83 (d, J = 7.9 Hz, 1H, 7-H), 7.01 (t, J = 7.2 Hz, 1H, 9-H), 7.24 (m, 2H, 8-H, NH), 7.44 (d, J = 7.2 Hz, 1H, 10-H), 7.59 (m, 3H, 2′-H, 3′-H, 7′-H), 7.77 (m, 1H, 6′-H), 8.01 (m, 2H, 4′-H, 5′-H), 8.28 (m, 1H, 8′-H); 13C-NMR (91 MHz, DMSO-d6) δ: 53.8 (C-4a), 67.8 (C-2), 72.7 (C-10b), 82.4 (C-5), 115.7 (C-7), 120.7 (C-9), 121.2 (C-10a), 123.3 (C-8′), 125.2 (C-10), 125.7 (C-2′, C-3′), 126.3 (C-6′, C-7′), 128.8 (C-5′), 129.3 (C-4′), 129.6 (C-8), 131.0 (C-4a’), 131.9 (C-8a’), 133.7 (C-1′), 153.2 (C-6a), 168.3 (C-3); HRMS (ESI) calcd. for C21H17NaNO3 [M+Na]+ 354.110; found 354.112.
(4aS,5R,10bR)-20f: tR = 5.71 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 293 (−1.64), 280 (−1.36), 270 (−1.79), 225 (34.72), 211 (−8.76).
(4aR,5S,10bS)-20f: tR = 7.06 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 293 (1.13), 280 (1.01), 270 (0.98), 225 (−34.55), 211 (13.28).
(±)-(4aS*,5R*,10bR*)-5-(naphthalen-2-yl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-20g): White crystals, yield 91%, mp 273–275 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 4.04 (t, J = 10.0 Hz, 1H, 4a-H), 4.31 (m, 2H, 2-H), 5.03 (d, J = 9.6 Hz, 1H, 10b-H), 5.35 (d, J = 10.4 Hz, 1H, 5-H), 6.85 (d, J = 8.0 Hz, 1H, 7-H), 6.99 (m, 1H, 9-H), 7.23 (m, 1H, 8-H), 7.37 (s, 1H, NH), 7.41 (d, J = 7.6 Hz, 1H, 10-H), 7.56 (m, 2H, 6′-H, 7′-H), 7.62 (dd, J = 8.4, 1.2 Hz, 1H, 3′-H), 7.96 (m, 3H, 4′-H, 5′-H, 8′-H), 8.04 (s, 1H, 1′-H); 13C-NMR (100 MHz, DMSO-d6) δ: 54.0 (C-4a), 67.8 (C-2), 72.6 (C-10b), 79.3 (C-5), 115.7 (C-7), 120.8 (C-8), 121.0 (C-10a), 125.2 (C-1′, C-8′), 126.2 (C-3′), 126.5 (C-7′), 127.6 (C-9), 128.2 (C-6′), 128.6 (C-5′), 128.8 (C-4′), 129.3 (C-10), 132.9 (C-4a’, C-8a’), 133.5 (C-2′), 153.2 (C-6a), 168.3 (C-3); HRMS (ESI) calcd. for C21H17NaNO3 [M+Na]+ 354.110; found 354.111.
(4aS,5R,10bR)-20g: tR = 21.46 min on Chiralpak IA column (hexane/2-propanol 90:10), HPLC-ECD {λ [nm] (ϕ)}: 283 (−1.06), 240sh (1.27), 228 (9.56), 216 (−3.76).
(4aR,5S,10bS)-20g: tR = 22.63 min on Chiralpak IA column (hexane/2-propanol 90:10), HPLC-ECD {λ [nm] (ϕ)}: 283 (1.35), 240sh (−0.81), 228 (−10.02), 216 (3.61).
(±)-(4aR*,5R*,10bR*)-5-phenyl-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-23a): White crystals, yield 93%, mp 151–153 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.83 (d, J = 16.8 Hz, 1H, 2-Ha), 4.02 (d, J = 16.8 Hz, 1H, 2-Hb) 4.20 (d, J = 5.6 Hz, 1H, 4a-H), 5.29 (s, 1H, 5-H), 5.46 (d, J = 5.6 Hz, 1H, 10b-H), 5.70 (bs, 1H, NH), 6.98 (d, J = 8.0 Hz, 1H, 7-H), 7.06 (m, 1H, 9-H), 7.28 (m, 1H, 8-H), 7.40 (m, 1H, 4′-H), 7.45 (m, 5H, 5-H, 2′-H, 3′-H, 5′-H, 6′-H); 13C-NMR (100 MHz, CDCl3) δ: 52.4 (C-4a), 61.9 (C-2), 67.7 (C-10b), 76.4 (C-5), 117.5 (C-10a), 117.8 (C-7), 122.8 (C-9), 126.0 (C-2′, C-6′), 128.2 (C-5), 129.3 (C-4′), 129.7 (C-3′, C-5′), 130.5 (C-8), 135.8 (C-1′), 155.4 (C-6a), 169.1 (C-3); HRMS (ESI) calcd. for C17H15NO3 [M + H]+ 282.113; found 282.115.
(4aS,5S,10bS)-23a: tR = 6.38 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283sh (10.79), 276 (11.06), 237 (3.05), 229 (−13.14), 214 (32.68).
(4aR,5R,10bR)-23a: tR = 7.92 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283sh (−7.27), 276 (−7.62), 237 (−1.40), 229 (11.40), 214 (−25.80).
(±)-(4aR*,5R*,10bR*)-5-(4-methoxyphenyl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-23b): White crystals, yield 74%, mp 167–169 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.79 (m, 4 H, OCH3, 2-Ha), 3.98 (d, J = 16.8 Hz, 1H, 2-Hb), 4.12 (d, J = 5.6 Hz, 1H, 4a-H), 5.20 (s, 1H, 10b-H), 5.40 (d, J = 5.6 Hz, 1H, 5-H), 5.81 (s, 1H, NH), 6.92 (d, 2H, 3′-H, 5′-H), 6.96 (d, J = 8.4 Hz, 1H, 7-H), 7.04 (t, J = 7.6 Hz, 1H, 9-H), 7.24 (t, J = 7.2 Hz, 1H, 8-H), 7.38 (d, 2H, 2′-H, 6′-H), 7.43 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, CDCl3) δ: 51.9 (C-4a), 55.4 (C-OCH3), 61.5 (C-2), 67.2 (C-10b), 75.7 (C-5), 114.6 (C-3′, C-5′), 117.1 (C-10a), 117.3 (C-7), 122.3 (C-9), 126.8 (C-2′, C-6′), 127.2 (C-1′), 127.9 (C-10), 130.0 (C-8), 155.1 (C-6a), 159.8 (C-4′), 168.6 (C-3); HRMS (ESI) calcd. for C18H17NO4 [M + H]+ 312.123; found 312.124.
(4aR,5R,10bR)-23b: tR = 8.78 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (−7.85), 275 (−7.80), 231sh (−18.86), 225 (−19.61).
(4aS,5S,10bS)-23b: tR = 9.75 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (8.34), 275 (7.98), 231sh (18.54), 225 (19.96).
(±)-(4aR*,5R*,10bR*)-5-(3,4-dimethoxyphenyl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-23c): White crystals, yield 69%, mp 139–140 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.83 (d, J = 17.2 Hz, 1H, 2-Ha), 3.92 (d, 6H, 2 × OCH3), 4.04 (d, J = 17.2 Hz, 1H, 2-Hb), 4.18 (d, J = 5.2 Hz, 1H, 4a-H), 5.24 (s, 1H, 10b-H), 5.45 (d, J = 5.2 Hz, 1H, 5-H), 5.73 (s, 1H, NH), 6.94 (m, 3H, 7-H, 2′-H, 5′-H), 7.05 (m, 2H, 9-H, 6′-H), 7.27 (m, 1H, 8-H), 7.46 (d, J = 7.2 Hz, 1H, 10-H); 13C-NMR (100 MHz, CDCl3) δ: 52.2 (C-4a), 56.2 (2 × C-OCH3), 61.6 (C-2), 67.3 (C-10b), 75.9 (C-5), 108.6 (C-5′), 111.8 (C-2′), 117.2 (C-10a), 117.5 (C-6′), 117.9 (C-7), 122.5 (C-9), 127.8 (C-1′), 127.9 (C-10), 130.2 (C-8), 149.4 (C-4′), 149.8 (C-3′), 155.1 (C-6a), 168.7 (C-3); HRMS (ESI) calcd. for C19H19NaNO5 [M+Na]+ 364.116; found 364.113.
(4aR,5R,10bR)-23c: tR = 12.43 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (−10.06), 276sh (−9.22), 235 (−10.58), 224sh (−2.55), 211 (−18.11).
(4aS,5S,10bS)-23c: tR = 15.97 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (10.17), 276sh (9.61), 235 (11.92), 224sh (4.81), 211 (11.93).
(±)-(4aR*,5R*,10bR*)-5-(3,5-dimethoxyphenyl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-23d): White crystals, yield 96%, mp 136–138 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.83 (m, 7H, 2-Ha, 2 × OCH3), 4.02 (d, J = 16.8 Hz, 1H, 2-Hb), 4.18 (d, J = 5.6 Hz, 1H, 4a-H), 5.20 (s, 1H, 10b-H), 5.44 (d, J = 5.6 Hz, 1H, 5-H), 5.73 (s, 1H, NH), 6.47 (s, 1H, 4′-H), 6.64 (s, 2H, 2′-H, 6′-H), 6.98 (d, J = 8.0 Hz, 1H, 7-H), 7.05 (t, J = 7.6 Hz, 1H, 9-H), 7.26 (t, J = 8.0 Hz, 1H, 8-H), 7.44 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, CDCl3) δ: 52.2 (C-4a), 55.6 (2 × C-OCH3), 61.6 (C-2), 67.3 (C-10b), 76.0 (C-5), 100.4 (C-4′), 103.7 (C-2′, C-6′), 117.2 (C-10a), 117.5 (C-7), 122.5 (C-8), 127.9 (C-9), 130.2 (C-10), 137.8 (C-1′), 154.9 (C-6a), 161.7 (C-3′, C-5′), 168.6 (C-3); HRMS (ESI) calcd. for C19H19NaNO5 [M+Na]+ 364.116; found 364.113.
(4aS,5S,10bS)-23d: tR = 10.35 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (2.62), 276sh (2.71), 241 (0.83), 231 (−3.03), 222sh (4.32), 210 (15.46).
(4aR,5R,10bR)-23d: tR = 11.98 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (−3.52), 276sh (−3.47), 241 (−1.00), 231 (2.81), 222sh (−7.12), 210 (−27.95).
(±)-(4aR*,5R*,10bR*)-5-(3,4,5-trimethoxyphenyl)-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-23e): White crystals, yield 89%, mp 148–150 °C; 1H-NMR (400 MHz, CDCl3) δ: 3.83 (m, 10 H, 2-Ha, 3 × OCH3), 4.04 (d, J = 16.8 Hz, 1H, 2-Hb), 4.19 (d, J = 5.6 Hz, 1H, 4a-H), 5.22 (s, 1H, 10b-H), 5.45 (d, J = 5.6 Hz, 1H, 5-H), 5.73 (s, 1H, NH), 6.71 (s, 2H, 2′-H, 6′-H), 7.00 (d, J = 8.0 Hz, 1H, 7-H), 7.07 (t, J = 7.6 Hz, 1H, 9-H), 7.27 (t, J = 8.0 Hz, 1H, 8-H), 7.45 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, CDCl3) δ: 52.1 (C-4a), 56.2 (2 × C-OCH3), 60.8 (C-OCH3), 61.4 (C-2), 67.1 (C-10b), 75.8 (C-5), 102.3 (C-2′, C-6′), 117.0 (C-10a), 117.3 (C-7), 122.4 (C-9), 127.7 (C-10), 130.0 (C-8), 130.8 (C-1′), 138.0 (C-4′), 153.8 (C-3′, C5′), 154.7 (C-6a), 168.5 (C-3); HRMS (ESI) calcd. for C20H21NaNO6 [M+Na]+ 394.126; found 394.124.
(4aR,5R,10bR)-23e: tR = 12.23 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (−4.07), 276sh (−4.33), 240 (−6.43), 228 (1.76), 212 (−21.15).
(4aS,5S,10bS)-23e: tR = 16.85 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (3.63), 276sh (3.23), 240 (5.25), 228 (−0.84), 212 (11.85).
(±)-(4aR*,5R*,10bR*)-5-naphthalen-2-yl-4,4a,5,10b-tetrahydrochromeno[4,3-b][1,4]oxazin-3(2H)-one (rac-23g): White crystals, yield 79%, mp 193–195 °C; 1H-NMR (360 MHz, CDCl3) δ: 3.83 (d, J = 16.9 Hz, 1H, 2-Ha), 4.01 (d, J = 16.9 Hz, 1H, 2-Hb), 4.27 (d, J = 5.4 Hz, 1H, 4a-H), 5.41 (s, 1H, 5-H), 5.49 (d, J = 5.4 Hz, 1H, 10b-H), 5.69 (s, 1H, NH), 7.05 (m, 2H, 7-H, 9-H), 7.28 (t, J = 7.2 Hz,1H, 8-H), 7.44 (m, 4 H, 3′-H, 6′-H, 7′-H, 10-H), 7.86 (m, 3H, 4′-H, 5′-H, 8′-H), 8.08 (s, 1H, 1′-H); 13C-NMR (90 MHz, CDCl3) δ: 51.8 (C-4a), 61.6 (C-2), 67.4 (C-10b), 76.2 (C-5), 117.2 (C-10a), 117.5 (C-7), 122.5 (C-8), 122.8 (C-1′), 125.2 (C-8′), 126.9 (C-3′), 127.0 (C-7′), 127.9 (C-9), 128.0 (C-6′), 128.3 (C-5′), 129.3 (C-4′), 130.2 (C-10), 132.6 (C-4a’), 133.2 (C-8a’), 133.3 (C-2′), 155.1 (C-6a), 168.6 (C-3); HRMS (ESI) calcd. for C21H17NaNO3 [M+Na]+ 354.110; found 354.111.
(4aR,5R,10bR)-23g: tR = 18.43 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (−4.33), 234sh (−2.97), 219 (−30.92), 203 (21.46).
(4aS,5S,10bS)-23e: tR = 19.30 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 283 (3.93), 234sh (2.35), 219 (28.64), 203 (−21.27).
2.7. General Procedure for the Synthesis of Condensed Morpholinee Derivatives [rac-(4aR*,5S*,10aS*)-2a-g, rac-(4aR*,5S*,10aR*)-2a-e, g, rac-(4aR*,5R*,10aR*)-2a-e, g]
Under inert atmosphere, the condensed 1,4-oxazinone derivatives rac-20a-g or rac-23a-e, g (0.359 mmol) were dissolved in anhydrous dioxane (5 mL) and after heating the reaction mixture to 90 °C, 2 M LiAlH4 solution in THF (216 µL) was added. The reaction was quenched after 15 min with the addition of ethyl acetate and water. The organic phase was collected and dried over MgSO4, then it was concentrated under reduced pressure. Procedure A: The product was obtained as the hydrochloride salt after stirring for 2 h at room temperature in a mixture of ethyl acetate (5 mL) and 3 N HCl solution (124 µL). Procedure B: The product was isolated as the amine base after column chromatography using CHCl3 as eluent.
(±)-(4aR*,5S*,10bS*)-5-phenyl-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine hydrochloride [rac-(4aR*,5S*,10aS*)-2a]: White solid, yield 72%, mp > 300 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.15 (bs, 1H, 3-Ha), 3.23 (d, J = 12.4 Hz, 1H, 3-Hb), 3.84 (m, 1H, 4a-H), 4.17 (d, 2H, 2-Ha, 2-Hb), 5.30 (d, J = 9.6 Hz, 1H, 10b-H), 5.51 (d, J = 10.0 Hz, 1H, 5-H), 6.87 (d, J = 8.4 Hz, 1H, 7-H), 7.02 (t, J = 7.2 Hz, 1H, 9-H), 7.26 (t, J = 7.2 Hz, 1H, 8-H), 7.39 (d, J = 7.6 Hz, 1H, 10-H), 7.49 (m, 3H, 3′-H, 4′-H, 5′-H), 7.63 (m, 2H, 2′-H, 6′-H), 8.38 (bs, 1H, NH2-Ha), 11.29 (bs, 1H, NH2-Hb); 13C-NMR (100 MHz, DMSO-d6) δ: 43.9 (C-3), 55.5 (C-4a), 63.4 (C-2), 71.9 (C-10b), 76.4 (C-5), 116.0 (C-7), 120.4 (C-10a), 121.2 (C-9), 125.6 (C-10), 128.5 (C-2′, C-6′), 128.9 (C-3′, C-5′), 129.6 (C-8), 129.7 (C-4′), 134.2 (C-1′), 152.7 (C-6a); HRMS (ESI) calcd. for C17H17NO2 [M + H]+ 268.1332; found 268.1137.
(±)-(4aR*,5S*,10bR*)-5-phenyl-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aR*)-2a]. Colorless oil, yield 60%, 1H-NMR (400 MHz, CDCl3) δ: 2.26 (s, 1H, NH), 2.90 (m, 3H, 2-Ha, 3-Ha, 3-Hb), 3.56 (d, J = 11.6 Hz, 1H, 2-Hb), 4.36 (s, 1H, 5-H), 4.89 (d, J = 7.6 Hz, 1H, 4a-H), 5.41 (d, J = 7.6 Hz, 1H, 10b-H), 6.92 (m, 2H, 7-H, 9-H), 7.26 (m, 5H, 3′-H, 4′-H, 5′-H, 8-H, 10-H), 7.48 (d, 2′-H, 6′-H); 13C-NMR (100 MHz, CDCl3) δ: 50.5 (C-3), 61.2 (C-5), 68.3 (C-2), 78.9 (C-10b), 90.1 (C-4a), 110.4 (C-7), 121.4 (C-9), 124.8 (C-10a), 126.4 (C-4′), 126.9 (C-2′, C-6′), 127.1 (C-10), 128.5 (C-3′, C-5′), 130.9 (C-8), 143.4 (C-1′), 160.1 (C-10a); HRMS (ESI) calcd. for C17H17NO2 [M + H]+ 268.1332; found 268.1132.
(±)-(4aR*,5R*,10bR*)-5-phenyl-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5R*,10aR*) -2a]: White crystals, yield 20%, mp 134–136 °C; 1H-NMR (400 MHz, acetone-d6) δ: 2.64 (d, J = 12.0 Hz, 1H, 3-Ha), 2.75 (m, 1H, 3-Hb), 3.36 (4 H, 2-Ha, 2-Hb, 4a-H, NH), 5.13 (d, J = 4.0 Hz, 1H, 10b-H), 5.29 (s, 1H, 5-H), 6.89 (dd, J = 8.4, 1.2 Hz, 1H, 7-H), 6.97 (m, 1H, 9-H), 7.19 (m, 1H, 8-H), 7.23 (m, 1H, 4′-H), 7.39 (m, 3H, 3′-H, 5′-H, 10-H), 7.55 (d, 2H, 2′-H, 6′-H); 13C-NMR (100 MHz, acetone-d6) δ: 46.3 (C-3), 54.7 (C-4a), 61.2 (C-2), 70.5 (C-10b), 79.2 (C-5), 117.0 (C-7), 121.8 (C-9), 122.4 (C-10a), 127.1 (C-2′, C-6′), 128.3 (C-10), 128.5 (C-4′), 129.0 (C-3′, C-5′), 129.2 (C-8), 139.4 (C-1′), 156.2 (C-6a); HRMS (ESI) calcd. for C17H17NO2 [M + H]+ 268.1332; found 268.1137.
(±)-(4aR*,5S*,10bS*)-5-(4-methoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine hydrochloride [rac-(4aR*,5S*,10aS*)-2b]: White crystals, yield 70%, mp 245–247 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.15 (bs, 1H, 3-Ha), 3.22 (d, J = 12.8 Hz, 1H, 3-Hb), 3.81 (m, 4 H, 4a-H, OCH3), 4.16 (d, 2H), 5.28 (d, J = 10.0 Hz, 1H, 10b-H), 5.45 (d, J = 10.4 Hz, 1H, 5-H), 6.85 (d, J = 8.4 Hz, 1H, 7-H), 7.00 (m, 3H, 3′-H, 5′-H, 9-H), 7.24 (m, 1H, 8-H), 7.38 (d, J = 7.6 Hz, 1H, 10-H), 7.54 (d, 2H, 2′-H, 6′-H), 8.33 (bs, 1H, NH2-Ha), 11.25 (d, J = 5.6 Hz, 1H, NH2-Hb); 13C-NMR (100 MHz, DMSO-d6) δ: 43.9 (C-3), 55.3 (C-OCH3), 55.6 (C-4a), 63.4 (C-2), 72.0 (C-10b), 76.0 (C-5), 114.3 (C-3′, C-5′), 116.0 (C-7), 120.4 (C-10a), 121.1 (C-9), 125.6 (C-10), 126.1 (C-1′), 129.5 (C-8), 130.0 (C-2′, C-6′), 152.8 (C-6a), 160.3 (C-4′); HRMS (ESI) calcd. for C18H19NO3 [M + H]+ 298.1438; found 298.1439.
(±)-(4aR*,5S*,10bR*)-5-(4-methoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aR*)-2b]: Colorless oil, yield: 41%, 1H-NMR (400 MHz, CDCl3) δ: 2.90 (m, 4 H, 2-Ha, 3-Ha, 3-Hb, NH), 3.56 (d, J = 11.6 Hz, 1H, 2-Hb), 3.80 (s, 3H, OCH3), 4.34 (s, 1H, 5-H), 4.88 (d, J = 7.6 Hz, 1H, 4a-H), 5.41 (d, J = 7.6 Hz, 1H, 10b-H), 6.90 (d, 2H, 3′-H, 5′-H), 6.94 (d, J = 8.4 Hz, 1H, 7-H), 6.97 (m, 1H, 9-H), 7.28 (m, 1H, 8-H), 7.39 (d, J = 7.2 Hz, 1H, 10-H), 7.43 (d, 2H, 2′-H, 6′-H); 13C-NMR (100 MHz, CDCl3) δ: 50.6 (C-3), 55.4 (C-OCH3), 60.9 (C-5), 68.1 (C-2), 79.0 (C-10b), 90.2 (C-4a), 110.6 (C-7), 114.1 (C-3′, C-5′), 121.5 (C-9), 124.9 (C-10a), 126.5 (C-10), 128.2 (C-2′, C-6′), 131.1 (C-8), 135.5 (C-1′), 158.8 (C-4′), 160.3 (C-6a); HRMS (ESI) calcd. for C18H19NO3 [M + H]+ 298.1438; found 298.1440.
(±)-(4aR*,5R*,10bR*)-5-(4-methoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5R*,10aR*)-2b]: White crystals, yield 13%, mp 153–155 °C; 1H-NMR (400 MHz, aceton-d6) δ: 2.66 (d, J = 12.0 Hz, 1H, 3-Ha), 2.76 (m, 1H, 3-Hb), 3.33 (m, 4 H, 2-Ha, 2-Hb, 4a-H, NH), 3.81 (s, 3H, OCH3), 5.10 (d, J = 4.8 Hz, 1H, 10b-H), 5.23 (s, 1H, 5-H), 6.86 (dd, J = 8.4, 1.2 Hz, 1H, 7-H), 6.96 (m, 3H, 3′-H, 5′-H, 9-H), 7.18 (m, 1H, 8-H), 7.42 (d, J = 7.6 Hz, 1H, 10-H), 7.46 (d, 2H, 2′-H, 6′-H); 13C-NMR (100 MHz, acetone-d6) δ: 46.3 (C-3), 54.8 (C-4a), 55.5 (C-OCH3), 61.2 (C-2), 70.5 (C-10b), 78.9 (C-5), 114.4 (C-3′, C-5′), 116.9 (C-7), 121.6 (C-9), 122.4 (C-10a), 128.3 (C-2′, C-6′), 129.2 (C-8), 131.3 (C-1′), 156.4 (C-6a), 160.2 (C-4′); HRMS (ESI) calcd. for C18H19NO3 [M + H]+ 298.1438; found 298.1439.
(±)-(4aR*,5S*,10bS*)-5-(3,4-dimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aS*)-2c]: White crystals, yield 55%, mp 178–180 °C; 1H-NMR (360 MHz, CDCl3) δ: 1.71 (bs, 1H, NH), 2.89 (m, 2H, 3-Ha, 3-Hb, 4a-H), 3.86 (m, 7H, 2-Ha, 2xOCH3), 4.04 (dd, J = 11.2, 2.2 Hz, 1H, 2-Hb), 4.56 (d, J = 9.0 Hz, 1H, 10b-H), 4.90 (d, J = 9.7 Hz, 1H, 5-H), 6.87 (m, 2H, 2′-H, 5′-H), 6.97 (m, 3H, 6′-H, 7-H, 9-H), 7.21 (m, 1H, 8-H), 7.43 (d, J = 7.9 Hz, 1H, 10-H); 13C-NMR (90 MHz, CDCl3) δ: 46.4 (C-3), 56.1 (2xC-OCH3), 59.1 (C-4a), 67.5 (C-2), 76.1 (C-10b), 80.3 (C-5), 110.0 (C-5′), 111.3 (C-2′), 116.2 (C-6′), 120.2 (C-7), 121.0 (C-9), 122.3 (C-10a), 125.5 (C-10), 129.0 (C-8), 129.2 (C-1′), 149.5 (C-3′), 149.7 (C-4′), 153.6 (C-6a); HRMS (ESI) calcd. for C19H21NO4 [M + H]+ 328.1543; found 328.1543.
(±)-(4aR*,5S*,10bR*)-5-(3,4-dimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aR*)-2c]): White crystals, yield 34%, mp 124–126 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.31 (bs, 1H, NH), 2.90 (m, 3H, 2-Ha, 3-Ha, 3-Hb), 3.57 (d, J = 12.4 Hz, 1H, 2-Hb), 3.87 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 4.32 (s, 1H, 5-H), 4.89 (d, J = 7.6 Hz, 1H, 4a-H), 5.42 (d, J = 7.6 Hz, 1H, 10b-H), 6.85 (d, J = 8.4 Hz, 1H, 5′-H), 6.94 (d, J = 8.0 Hz, 1H, 7-H), 6.98 (m, 1H, 9-H), 7.03 (dd, J = 8.0, 2.0 Hz, 1H, 6′-H), 7.09 (d, J = 1.6 Hz, 1H, 2′-H), 7.28 (m, 1H, 8-H), 7.40 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, CDCl3) δ: 50.6 (C-3), 56.0 (2xC-OCH3), 61.2 (C-5), 68.1 (C-2), 79.0 (C-10b), 90.3 (C-4a), 110.5 (C-7), 110.6 (C-5′), 111.3 (C-2′), 119.1 (C-6′), 121.5 (C-9), 124.9 (C-10a), 126.5 (C-10), 131.0 (C-8), 136.2 (C-1′), 148.2 (C-4′), 149.1 (C-3′), 160.2 (C-6a); HRMS (ESI) calcd. for C19H21NO4 [M + H]+ 328.1543; found 328.1544.
(±)-(4aR*,5R*,10bR*)-5-(3,4-dimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5R*,10aR*)-2c]: White crystals, yield 16%, mp 139–140 °C; 1H-NMR (400 MHz, acetone-d6) δ: 2.66 (d, J = 12.0 Hz, 1H, 3-Ha), 2.77 (m, 1H, 3-Hb), 3.35 (m, 4 H, 2-Ha, 2-Hb, 4a-H, NH), 3.82 (d, 6H, 2 × OCH3), 5.10 (d, J = 4.0 Hz, 1H, 10b-H), 5.23 (s, 1H, 5-H), 6.86 (dd, J = 8.4, 1.2 Hz, 1H, 7-H), 6.96 (m, 2H, 6′-H, 9-H), 7.06 (m, 1H, 5′-H), 7.16 (d, J = 2.0 Hz, 1H, 2′-H), 7.18 (m, 1H, 8-H), 7.42 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, acetone-d6) δ: 45.5 (C-3), 54.1 (C-OCH3), 55.2 (C-4a), 55.3 (C-OCH3), 60.3 (C-2), 69.7 (C-10b), 78.1 (C-5), 110.3 (C-5′), 111.7 (C-2′), 116.1 (C-6′), 118.4 (C-7), 120.7 (C-9), 121.5 (C-10a), 127.3 (C-10), 128.3 (C-8), 130.9 (C-1′), 149.0 (C-4′), 149.3 (C-3′) 155.4 (C-6a); HRMS (ESI) calcd. for C19H21NO4 [M + H]+ 328.1543; found 328.1543.
(±)-(4aR*,5S*,10bS*)-5-(3,5-dimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine hydrochloride [rac-(4aR*,5S*,10aS*)-2d]: White crystals, yield 84%, mp 238–241 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.12 (m, 1H, 3-Ha), 3.24 (d, J = 12.8 Hz, 1H, 3-Hb), 3.75 (m, 7H, 4a-H, 2xOCH3), 4.08 (m, 1H, 2-Ha), 4.17 (dd, J = 12.4, 4.0 Hz, 1H, 2-Hb), 5.20 (d, J = 9.6 Hz, 1H, 10b-H), 5.38 (d, J = 10.4 Hz, 1H, 5-H), 6.58 (t, J = 2.0 Hz, 1H, 4′-H), 6.60 (d, 2H, 2′-H, 6′-H), 6.89 (d, J = 8.4 Hz, 1H, 7-H), 7.01 (m, 1H, 9-H), 7.26 (m, 1H, 8-H), 7.38 (d, J = 7.6 Hz, 1H, 10-H), 8.33 (bs, 1H, NH2-Ha), 10.93 (bs, 1H, NH2-Hb); 13C-NMR (100 MHz, DMSO-d6) δ: 43.9 (C-3), 55.3 (2 × C-OCH3), 55.6 (C-4a), 63.4 (C-2), 71.9 (C-10b), 76.2 (C-5), 101.3 (C-4′), 106.2 (C-2′, C-6′), 116.1 (C-7), 120.5 (C-10a), 121.3 (C-9), 125.6 (C-10), 129.6 (C-8), 136.3 (C-1′), 152.6 (C-6a), 160.7 (C-3′, C-5′); HRMS (ESI) calcd. for C19H21NO4 [M + H]+ 328.1543; found 328.1544.
(±)-(4aR*,5S*,10bR*)-5-(3,5-dimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aR*)-2d]. Colorless oil, yield 48%, 1H-NMR (400 MHz, CDCl3) δ: 2.80 (m, 4 H, 2-Ha, 3-Ha, 3-Hb, NH), 3.46 (d, J = 12.0 Hz, 1H, 2-Hb), 3.71 (s, 6H, 2 × OCH3), 4.19 (s, 1H, 5-H), 4.78 (d, J = 7.6 Hz, 1H, 4a-H), 5.31 (d, J = 7.6 Hz, 1H, 10b-H), 6.27 (t, J = 2.0 Hz, 1H, 4′-H), 6.57 (d, 2H, 2′-H, 6′-H), 6.82 (d, J = 8.4 Hz, 1H, 7-H), 6.87 (t, J = 7.2 Hz, 1H, 9-H), 7.18 (m, 1H, 8-H), 7.29 (d, J = 7.2 Hz, 1H, 10-H); 13C-NMR (100 MHz, CDCl3) δ: 50.5 (C-3), 55.5 (2 × C-OCH3), 61.5 (C-5), 68.2 (C-2), 79.0 (C-10b), 90.2 (C-4a), 99.2 (C-4′), 105.2 (C-2′, C-6′), 110.6 (C-7), 121.5 (C-9), 124.8 (C-10a), 126.5 (C-10), 131.1 (C-8), 145.7 (C-1′), 160.2 (C-6a), 161.0 (C-3′, C-5′); HRMS (ESI) calcd. for C19H21NO4 [M + H]+ 328.1543; found 328.1543.
(±)-(4aR*,5R*,10bR*)-5-(3,5-dimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5R*,10aR*)-2d]: White crystals, yield 5%, mp 145–147 °C; 1H-NMR (400 MHz, acetone-d6) δ: 2.68 (d, J = 12.0 Hz, 1H, 3-Ha), 2.79 (m, 2H, 3-Hb, NH), 3.38 (m, 3H, 2-Ha, 2-Hb, 4a-H), 3.81 (s, 6H, 2OCH3), 5.13 (d, J = 4.0 Hz, 1H, 10b-H), 5.25 (s, 1H, 5-H), 6.45 (t, J = 2.4 Hz, 1H, 4′-H), 6.74 (d, 2H, 2′-H, 6′-H), 6.88 (d, J = 8.4 Hz, 1H, 7-H), 6.97 (t, J = 7.6 Hz, 1H, 9-H), 7.19 (m, 1H, 8-H), 7.42 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, acetone-d6) δ: 46.3 (C-3), 54.9 (C-4a), 55.6 (2 × C-OCH3), 61.1 (C-2), 70.4 (C-10b), 79.0 (C-5), 100.2 (C-4′), 105.0 (C-2′, C-6′), 117.0 (C-7), 121.7 (C-9), 122.3 (C-10a), 128.2 (C-10), 129.3 (C-8), 141.7 (C-1′), 156.1 (C-6′), 161.9 (C-3′, C-5′); HRMS (ESI) calcd. for C19H21NO4 [M + H]+ 328.1543; found 328.1543.
(±)-(4aR*,5S*,10bS*)-5-(3,4,5-trimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aS*)-2e]: Colorless oil, yield 46%, 1H-NMR (360 MHz, CDCl3) δ: 1.72 (bs, 1H, NH), 2.91 (m, 3H, 3-Ha, 3-Hb, 4a-H), 3.87 (m, 10 H, 2-Ha, 3 × OCH3), 4.06 (dd, J = 11.2, 1.8 Hz, 1H, 2-Hb), 4.56 (d, J = 9.4 Hz, 1H, 10b-H), 4.88 (d, J = 10.1Hz, 1H, 5-H), 6.70 (s, 2H, 2′-H, 6′-H), 6.89 (dd, J = 8.3, 1.1Hz, 1H, 7-H), 6.98 (m, 1H, 9-H), 7.20 (m, 1H, 8-H), 7.44 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (90 MHz, CDCl3) δ: 46.4 (C-3), 56.3 (2 × C-OCH3), 59.2 (C-4a), 60.9 (C-OCH3), 67.5 (C-2), 76.1 (C-10b), 80.6 (C-5), 104.2 (C-2′, C-6′), 116.2 (C-7), 121.1 (C-9), 122.2 (C-10a), 125.5 (C-10), 129.1 (C-8), 132.5 (C-1′), 138.4 (C-4′), 153.5 (C-6a), 153.7 (C-3′, C-5′); HRMS (ESI) calcd. for C20H23NO5 [M + H]+ 358.1649; found 358.1648.
(±)-(4aR*,5S*,10bR*)-5-(3,4,5-trimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aR*)-2e]: Colorless oil, yield 33%, 1H-NMR (400 MHz, CDCl3) δ: 2.13 (bs, 1H, NH), 2.91 (m, 3H, 2-Ha, 3-Ha, 3-Hb), 3.58 (d, J = 12.0 Hz, 1H, 2-Hb), 3.83 (s, 3H, OCH3), 3.91 (s, 6 H, 2 × OCH3), 4.30 (s, 1H, 5-H), 4.90 (d, J = 7.6 Hz, 1H, 4a-H), 5.43 (d, J = 7.6 Hz, 1H, 10b-H), 6.75 (s, 2H, 2′-H, 6′-H), 6.95 (d, J = 8.0 Hz, 1H, 7-H), 6.99 (t, J = 7.6, 7.2 Hz, 1H, 9-H), 7.23 (t, J = 8.0, 7.2 Hz, 1H, 8-H), 7.40 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, CDCl3) δ: 50.5 (C-3), 56.3 (2 × C-OCH3), 60.9 (C-OCH3), 61.7 (C-5), 68.2 (C-2), 79.0 (C-10b), 90.2 (C-4a), 104.2 (C-2′, C-6′), 110.6 (C-7), 121.6 (C-9), 124.8 (C-10a), 126.5 (C-10), 131.1 (C-8), 137.2 (C-4′), 139.3 (C-1′), 153.4 (C-3′, C-5′), 160.2 (C-6a); HRMS (ESI) calcd. for C20H23NO5 [M + H]+ 358.1649; found 358.1649
(±)-(4aR*,5R*,10bR*)-5-(3,4,5-trimethoxyphenyl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5R*,10aR*)-2e]: White crystals, yield 7%, mp 164–166 °C; 1H-NMR (400 MHz, acetone-d6) δ: 2.66 (d, J = 12.0 Hz, 1H, 3-Ha), 2.77 (m, 1H, 3-Hb), 3.37 (m, 4 H, 2-Ha, 2-Hb, 4a-H, NH), 3.74 (s, 3H, OCH3), 3.85 (s, 6H, 2 × OCH3), 5.10 (d, J = 4.0 Hz, 1H, 10b-H), 5.22 (s, 1H, 5-H), 6.87 (s, 3H, 2′-H, 6′-H, 7-H), 6.96 (m, 1H, 9-H), 7.18 (m, 1H, 8-H), 7.42 (d, J = 7.6 Hz, 1H, 10-H); 13C-NMR (100 MHz, acetone-d6) δ: 46.4 (C-3), 55.0 (C-4a), 56.4 (2 × C-OCH3), 60.5 (C-OCH3), 61.2 (C-2), 70.6 (C-10b), 79.2 (C-5), 104.5 (C-2′, C-6′), 117.0 (C-7), 121.7 (C-9), 122.5 (C-10a), 128.2 (C-10), 129.2 (C-8), 134.9 (C-1′), 154.3 (C-3′, C-5′), 156.2 (C-6a); HRMS (ESI) calcd. for C20H23NO5 [M + H]+ 358.1649; found 358.1648.
(±)-(4aR*,5S*,10bS*)-5-(naphthalen-1-yl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine hydrochloride [rac-(4aR*,5S*,10aS*)-2f]: White crystals, yield 53%, mp 241–244 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.22 (s, 2H, 3-Ha, 3-Hb), 4.19 (m, 3H, 2-Ha, 2-Hb, 4a-H), 5.52 (s, 1H, 10b-H), 6.43 (bs, 1H, 5-H), 6.87 (s, 1H, 7-H), 7.05 (t, J =7.6 Hz, 1H, 9-H), 7.27 (s, 1H, 8-H), 7.45 (d, J = 7.6 Hz, 1H, 10-H), 7.60 (s, 3H, 2′-H, 3′-H, 7′-H), 7.94 (s, 1H, 6′-H), 8.04 (m, 2H, 4′-H, 5′-H), 8.53 (s, 2H, 8′-H, NH2-Ha), 11.06 (bs, 1H, NH2-Hb); 13C-NMR (100 MHz, DMSO-d6) δ: 44.0 (C-3), 55.6 (C-4a), 63.4 (C-2), 70.8 (C-5), 71.9 (C-10b), 116.0 (C-7), 120.6 (C-10a), 121.3 (C-9), 124.2 (C-8′), 125.5 (C-10), 126.0 (C-2′, C-3′, C-7′), 126.7 (C-6′), 128.8 (C-5′), 129.6 (C-8), 130.7 (C-4′); HRMS (ESI) calcd. for C21H19NO2 [M + H]+ 318.1489; found 318.1486.
(±)-(4aR*,5S*,10bS*)-5-naphthalen-2-yl-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aS*)-2g]: White crystals, yield 51%, mp 102–104 °C; 1H-NMR (360 MHz, CDCl3) δ: 1.55 (bs, 1H, NH), 2.82 (m, 2H, 3-Ha, 3-Hb), 3.02 (t, J = 9.7, 9.4 Hz,1H, 4a-H), 3.86 (m, 1H, 2-Ha), 4.03 (dd, J = 11.5, 2.2 Hz, 1H, 2-Hb), 4.62 (d, J = 9.4 Hz, 1H, 10b-H), 5.11 (d, J = 9.7 Hz, 1H, 5-H), 6.90 (d, J = 8.3 Hz, 1H, 7-H), 6.98 (m, 1H, 9-H), 7.15 (m, 1H, 8-H), 7.46 (m, 3H, 10-H, 6′-H, 7′-H), 7.59 (dd, J = 8.3, 1.4 Hz, 1H, 3′-H), 7.85 (m, 4 H, 1′-H, 4′-H, 5′-H, 8′-H); 13C-NMR (90 MHz, CDCl3) δ: 46.3 (C-3), 59.0 (C-4a), 67.5 (C-2), 76.1 (C-10b), 80.6 (C-5), 116.2 (C-7), 121.1 (C-8), 122.3 (C-10a), 124.3 (C-1′), 125.5 (C-8′), 126.6 (C-3′), 126.7 (C-7′), 127.4 (C-9), 127.9 (C-6′), 128.2 (C-5′), 129.1 (C-10, C-4′), 133.2 (C-4a’), 133.8 (C-8a’), 134.3 (C-2′), 153.6 (C-6a); HRMS (ESI) calcd. for C21H19NO2 [M + H]+ 318.1489; found 318.1486.
(±)-(4aR,5S,10bR)-5-(naphthalen-2-yl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5S*,10aR*)-2g]: White crystals, yield 44%, mp 154–156 °C; 1H-NMR (360 MHz, CDCl3) δ: 2.26 (bs, 1H, NH), 3.05 (m, 3H, 2-Ha, 3-Ha, 3-Hb), 3.70 (m, 1H, 2-Hb), 4.64 (s, 1H, 5-H), 5.11 (d, J = 7.9 Hz, 1H, 4a-H), 5.56 (d, J = 7.6 Hz, 1H, 10b-H), 7.04 (m, 2H, 7-H, 9-H), 7.39 (m, 1H, 8-H), 7.51 (m, 3H, 6′-H, 7′-H, 10-H), 7.70 (dd, J = 8.6, 1.8 Hz, 1H, 3′-H), 7.90 (m, 3H, 4′-H, 5′-H, 8′-H), 8.06 (s, 1H, 1′-H); 13C-NMR (90 MHz, CDCl3) δ: 50.7 (C-3), 61.4 (C-5), 68.5 (C-2), 79.1 (C-10b), 90.2 (C-4a), 110.6 (C-7), 121.5 (C-8), 124.9 (C-10a), 125.5 (C-1′), 125.6 (C-8′), 125.8 (C-3′), 126.2 (C-7′), 126.6 (C-9), 127.7 (C-6′), 128.2 (C-5′), 128.4 (C-4′), 131.1 (C-10), 132.7 (C-4a’), 133.5 (C-8a’), 140.8 (C-2′), 160.3 (C-6a); HRMS (ESI) calcd. for C21H19NO2 [M + H]+ 318.1489; found 318.1488.
(±)-(4aR*,5R*,10bR*)-5-(naphthalen-2-yl)-2,3,4,4a,5,10b-hexahydrochromeno[4,3-b][1,4]oxazine [rac-(4aR*,5R*,10aR*)-2g]: White crystals, yield 11%, mp 117–119 °C; 1H-NMR (360 MHz, acetone-d6) δ: 2.63 (d, J = 11.9 Hz, 1H, 3-Ha), 2.75 (m, 2H, 3-Hb, NH), 3.38 (m, 2H, 2-Ha, 2-Hb), 3.52 (d, J = 4.7 Hz, 1H, 4a-H), 5.19 (d, J = 4.0 Hz, 1H, 10b-H), 5.48 (s, 1H, 5-H), 6.95 (d, J = 8.3 Hz, 1H, 7-H), 7.00 (t, J = 7.6 Hz, 1H, 9-H), 7.22 (t, J = 7.2 Hz, 1H, 8-H), 7.46 (d, J = 7.6 Hz, 1H, 10-H), 7.52 (m, 2H, 6′-H, 7′-H), 7.66 (d, J = 8.6 Hz, 1H, 3′-H), 7.94 (m, 3H, 4′-H, 5′-H, 8′-H), 8.09 (s, 1H, 1′-H); 13C-NMR (90 MHz, acetone-d6) δ: 45.4 (C-3), 53.9 (C-4a), 60.3 (C-2), 69.6 (C-10b), 78.3 (C-5), 116.1 (C-7), 120.9 (C-8), 121.6 (C-10a), 124.2 (C-1′), 125.0 (C-8′), 125.9 (C-3′), 126.1 (C-7′), 127.4 (C-9), 127.6 (C-6′), 127.8 (C-5′), 128.0 (C-4′), 128.4 (C-10), 133.1 (C-4a’), 133.2 (C-8a’), 136.1 (C-2′), 155.3 (C-6a); HRMS (ESI) calcd. for C21H19NO2 [M + H]+ 318.1489; found 318.1488.
2.8. General Procedure for the Synthesis of Acetamide Derivatives rac-cis-24a-e,g and rac-trans-24a-g
3-Aminoflavanone hydrochloride salts rac-cis-1a-e, g or rac-trans-1a-g (0.655 mmol) were suspended in anhydrous THF (5 mL) under inert atmosphere. Under stirring, Et3N (230 µL, 1.64 mmol) was added to the suspension at room temperature or at 0 °C. After 10 min, acetyl chloride (56 µL, 0.786 mmol) was added dropwise to the reaction mixture and stirred for additional 10 min. Extraction with ethyl acetate and water, drying over MgSO4, and concentration under reduced pressure provided the crude product, which was purified by column chromatography using hexane/ethyl acetate 1:1 as eluent.
(±)-N-[(2S*,3R*)-4-oxo-2-phenyl-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-cis-24a): White crystals, yield 69%, mp 169–171 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.02 (s, 3H, CH3), 5.42 (t, J = 6.0 Hz, 1H, 3-H), 6.10 (d, J = 6.8 Hz, 1H, 2-H), 6.30 (d, J = 5.2 Hz, 1H, NH), 7.03 (m, 2H, 3′-H, 5′-H), 7.13 (m, 2H, 6-H, 8-H), 7.25 (m, 3H, 2′-H, 4′-H, 6′-H), 7.54 (m, 1H, 7-H), 7.82 (dd, J = 8.0, 1.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 23.2 (CH3), 56.5 (C-3), 79.7 (C-2), 118.1 (C-8), 120.1 (C-4a), 121.6 (C-6), 126.9 (C-5), 127.2 (C-2′, C-6′), 128.7 (C-3′, C-5′), 128.9 (C-4′), 135.1 (C-1′), 137.2 (C-7), 160.5 (C-8a), 170.5 (amide carbonyl), 189.4 (C-4); HRMS (ESI) calcd. for C17H15NaNO3 [M+Na]+ 304.095; found 304.096.
(±)-N-[(2S*,3R*)-4-oxo-2-(4-methoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-cis-24b): White crystals, yield 72%, mp 147–149 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.02 (s, 3H, CH3), 3.74 (s, 3H, OCH3), 5.40 (t, J = 6.4 Hz, 1H, 3-H), 6.06 (d, J = 6.4 Hz, 1H, 2-H), 6.28 (d, J = 5.2 Hz, 1H, NH), 6.75 (d, 2H, 3′-H, 5′-H), 6.99 (m, 4 H, 6-H, 8-H, 2′-H, 6′-H), 7.50 (m, 1H, 7-H), 7.82 (dd, J = 7.6, 1.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 23.5 (C-CH3), 55.6 (C-OCH3), 56.9 (C-3), 79.9 (C-2), 114.4 (C-3′, C-5′), 118.5 (C-8), 120.4 (C-4a), 121.8 (C-6), 127.2 (C-5), 127.4 (C-1′), 129.0 (C-2′, C-6′), 137.4 (C-4), 160.3 (C-4′), 160.7 (C-8a), 170.7 (amide carbonyl), 190.0 (C-4); HRMS (ESI) calcd. for C18H17NaNO4 [M+Na]+ 334.105; found 334.107.
(±)-N-[(2S*,3R*)-4-oxo-2-(3,4-dimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-cis-24c): White crystals, yield 75%, mp 183–185 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.03 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 5.41 (t, J = 6.4 Hz, 1H, 3-H), 6.06 (d, J = 6.4 Hz, 1H, 2-H), 6.30 (bs, 1H, NH), 6.68 (m, 3H, 2′-H, 5′-H, 6′-H), 7.01 (m, 2H, 6-H, 8-H), 7.51 (m, 1H, 7-H), 7.83 (dd, J = 7.6, 1.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 23.1 (C-CH3), 55.7 (C-OCH3), 55.8 (C-OCH3), 56.5 (C-3), 79.7 (C-2), 110.8 (C-5′), 111.0 (C-2′), 118.1 (C-8), 119,4 (C-6′), 120.0 (C-4a), 121.5 (C-6), 126.7 (C-5), 127.3 (C-1′), 137.1 (C-7), 148.9 (C-4′), 149.4 (C-3′), 160.2 (C-8a), 170.4 (amide carbonyl), 189.5 (C-4); HRMS (ESI) calcd. for C19H19NaNO5 [M+Na]+ 364.116; found 364.118.
(±)-N-[(2S*,3R*)-4-oxo-2-(3,5-dimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-cis-24d): White crystals, yield 67%, mp 144–147 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.03 (s, 3H, CH3), 3.65 (s, 6H, 2 × OCH3), 5.39 (t, J = 6.4 Hz, 1H, 3-H), 6.00 (d, J = 6.4 Hz, 1H, 2-H), 6.30 (d, 2H, 2′-H, 6′-H), 6.34 (d, J = 2.0 Hz, 1H, 4′-H), 6.43 (d, J = 5.6 Hz, 1H, NH), 7.01 (m, 2H, 6-H, 8-H), 7.50 (m, 1H, 7-H), 7.80 (dd, J = 8.0, 1.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 23.1 (C-CH3), 55.3 (2 × C-OCH3), 56.3 (C-3), 79.6 (C-2), 100.4 (C-4′), 105.4 (C-2′, C-6′), 118.0 (C-8), 120.1 (C-4a), 121.6 (C-6), 126.8 (C-5), 137.0 (C-1′), 137.2 (C-7), 160.3 (C-8a), 160.8 (C-3′, C-5′), 170.4 (amide carbonyl), 189.2 (C-4); HRMS (ESI) calcd. for C19H19NO5 [M + H]+ 342.134; found 342.134.
(±)-N-[(2S*,3R*)-4-oxo-(3,4,5-trimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-cis-24e): White crystals, yield 64%, mp 170–172 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.04 (s, 3H, CH3), 3.67 (s, 6H, 2 × OCH3), 3.79 (s, 3H, OCH3), 5.41 (t, J = 6.0 Hz, 1H, 3-H), 6.03 (d, J = 6.4 Hz, 1H, 2-H), 6.38 (s, 2H, 2′-H, 6′-H), 6.42 (d, J = 5.6 Hz, 1H, NH), 7.04 (m, 2H, 6-H, 8-H), 7.53 (m, 1H, 7-H), 7.83 (dd, J = 8.0, 1.2 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 23.1 (C-CH3), 56.0 (2 × C-OCH3), 56.4 (C-3), 60.8 (C-OCH3), 79.9 (C-2), 104.4 (C-2′, C-6′), 118.0 (C-8), 120.0 (C-4a), 121.6 (C-6), 126.7 (C-5), 130.4 (C-1′, C-4′), 137.1 (C-7), 153.2 (C-3′, C-5′), 160.3 (C-8a), 170.4 (amide carbonyl), 189.3 (C-4); HRMS (ESI) calcd. for C20H21NaNO6 [M+Na]+ 394.126; found 394.128.
(±)-N-[(2S*,3S*)-4-oxo-2-phenyl-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-trans-24a): White crystals, yield 75%, mp 192–194 °C; 1H-NMR (400 MHz, CDCl3) δ: 1.86 (s, 3H, CH3), 5.02 (dd, J = 12.0, 8.4 Hz, 1H, 3-H), 5.38 (d, J = 12.4 Hz, 1H, 2-H), 6.03 (d, J = 8.4 Hz, 1H, NH), 7.02 (m, 2H, 6-H, 8-H), 7.39 (m, 3H, 3′-H, 5′-H, 7-H), 7.49 (m, 3H, 2′-H, 4′-H, 6′-H), 7.88 (dd, J = 7.6, 0.8 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 22.9 (C-CH3), 58.3 (C-3), 83.2 (C-2), 118.1 (C-8), 120.2 (C-4a), 122.1 (C-6), 127.7 (C-5), 127.8 (C-2′, C-6′), 128.6 (C-3′, C-5′), 129.4 (C-4′), 136.2 (C-1′), 136.6 (C-7), 161.4 (C-8a), 170.3 (amide carbonyl), 191.0 (C-4); HRMS (ESI) calcd. for C17H15NaNO3 [M+Na]+ 304.095; found 304.096.
(±)-N-[(2S*,3S*)-4-oxo-2-(4-methoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-trans-24b): White crystals, yield 71%, mp 189–191 °C; 1H-NMR (400 MHz, CDCl3) δ: 1.88 (s, 3H, CH3), 3.82 (s, 3H, OCH3), 5.08 (dd, J = 12.4, 8.4 Hz, 1H, 3-H), 5.31 (d, J = 12.4 Hz, 1H, 2-H), 5.90 (d, J = 8.4 Hz, 1H, NH), 6.92 (d, 2H, 3′-H, 5′-H), 7.00 (d, J = 8.4 Hz, 1H, 8-H), 7.05 (t, J = 7.2 Hz, 1H, 6-H), 7.42 (d, 2H, 2′-H, 6′-H), 7.49 (m, 1H, 7-H), 7.88 (dd, J = 7.2, 1.2 Hz, 1H, 5-H); 13C-NMR (100 MHz, CDCl3) δ: 23.0 (C-CH3), 55.4 (C-OCH3), 58.0 (C-3), 83.1 (C-2), 114.0 (C-3′, C-5′), 118.1 (C-8), 120.2 (C-4a), 122.0 (C-6), 127.6 (C-5), 128.2 (C-1′), 129.3 (C-2′, C-6′), 136.6 (C-7), 160.4 (C-4′), 161.4 (C-8a), 170.2 (amide carbonyl), 191.3 (C-4); HRMS (ESI) calcd. for C18H17NaNO4 [M+Na]+ 334.105; found 334.107.
(±)-N-[(2S*,3S*)-4-oxo-2-(3,4-dimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-trans-24c): White crystals, yield 82%, mp 180–181 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 1.72 (s, 3H, CH3), 3.76 (d, 6 H, 2 × OCH3), 4.92 (dd, J = 12.4, 8.4 Hz, 1H, 3-H), 5.49 (d, J = 12.4 Hz, 1H, 2-H), 6.94 (m, 2H, 2′-H, 5′-H), 7.06 (d, J = 8.4 Hz, 1H, 8-H), 7.11 (m, 2H, 6-H, 6′-H), 7.58 (m, 1H, 7-H), 7.79 (dd, J = 7.6, 1.2 Hz, 1H, 5-H), 8.14 (d, J = 8.4 Hz, 1H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 22.3 (C-CH3), 55.4 (C-OCH3), 55.5 (C-OCH3), 57.4 (C-3), 81.5 (C-2), 111.1 (C-5′), 111.2 (C-2′), 118.0 (C-8), 120.0 (C-4a), 120.7 (C-6′), 121.8 (C-6), 126.9 (C-5), 129.3 (C-1′), 136.3 (C-7), 148.4 (C-4′), 149.2 (C-3′), 160.8 (C-8a), 169.1 (amide carbonyl), 190.5 (C-4); HRMS (ESI) calcd. for C19H19NaNO5 [M+Na]+ 364.116; found 364.118.
(±)-N-[(2S*,3S*)-4-oxo-2-(3,5-dimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-trans-24d): White crystals, yield 72%, mp 192–193 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 1.74 (s, 3H, CH3), 3.75 (s, 6H, 2 × OCH3), 4.85 (dd, J = 12.0, 8.4 Hz, 1H, 3-H), 5.51 (d, J = 12.0 Hz, 1H, 2-H), 6.51 (t, J = 2.0 Hz, 1H, 4′-H), 6.66 (d, 2H, 2′-H, 6′-H), 7.07 (d, J = 8.4 Hz, 1H, 8-H), 7.12 (t, J = 7.6 Hz, 1H, 6-H), 7.59 (m, 1H, 7-H), 7.79 (dd, J = 8.0, 1.6 Hz, 1H, 5-H), 8.18 (d, J = 8.4 Hz, 1H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 22.3 (C-CH3), 55.3 (2 × C-OCH3), 57.5 (C-3), 81.3 (C-2), 100.4 (C-4′), 105.8 (C-2′, C-6′), 118.0 (C-8), 120.0 (C-4a), 121.9 (C-6), 126.9 (C-5), 136.3 (C-7), 139.2 (C-1′), 160.2 (C-3′, C-5′), 160.7 (C-8a), 169.2 (amide carbonyl), 190.2 (C-4); HRMS (ESI) calcd. for C19H19NO5 [M + H]+ 342.134; found 342.134.
(±)-N-[(2S*,3S*)-4-oxo-2-(3,4,5-trimethoxyphenyl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-trans-24e): White crystals, yield 74%, mp 147–149 °C; 1H-NMR (400 MHz, CD3OD) δ: 1.89 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 3.87 (s, 6H, 2 × OCH3), 5.04 (d, J = 12.4 Hz, 1H, 3-H), 5.42 (d, J = 12.4 Hz, 1H, 2-H), 6.84 (s, 2H, 2′-H, 6′-H), 7.07 (d, J = 8.4 Hz, 1H, 8-H), 7.13 (m, 1H, 6-H), 7.58 (m, 1H, 7-H), 7.89 (dd, J = 8.0, 1.6 Hz, 1H, 5-H); 13C-NMR (100 MHz, CD3OD) δ: 22.3 (C-CH3), 56.7 (2 × C-OCH3), 59.3 (C-3), 61.1 (C-OCH3), 83.9 (C-2), 106.3 (C-2′, C-6′), 119.1 (C-8), 121.3 (C-4a), 123.1 (C-6), 128.3 (C-5), 133.8 (C-1′), 137.7 (C-7), 139.6 (C-4′) 154.4 (C-3′, C-5′), 162.7 (C-8a), 173.2 (amide carbonyl), 192.1 (C-4); HRMS (ESI) calcd. for C20H21NaNO6 [M+Na]+ 394.126; found 394.128.
(±)-N-[(2S*,3S*)-4-oxo-2-(naphthalen-1-yl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-trans-24f): White crystals, yield 81%, mp 248–250 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 1.58 (s, 3H, CH3), 5.12 (t, J = 11.6 Hz, 1H, 3-H), 6.42 (d, J = 12.0 Hz, 1H, 2-H), 7.07 (d, J = 8.0 Hz, 1H, 8-H), 7.17 (t, J = 8.0 Hz, 1H, 6-H), 7.21 (m, 4 H, 7-H, 2′-H, 3′-H, 7′-H), 7.81 (d, J = 5.6 Hz, 1H, 6′-H), 7.87 (dd, J = 8.0, 1.6 Hz, 1H, 5-H), 7.98 (m, 2H, 4′-H, 5′-H), 8.20 (m, 2H, 8′-H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 22.1 (C-CH3), 57.5 (C-3), 78.3 (C-2), 118.0 (C-6), 120.3 (C-4a), 122.1 (C-8), 123.5 (C-8′), 125.2 (C-5), 125.8 (C-2′, C-3′), 126.4 (C-7′), 127.0 (C-6′), 128.7 (C-5′), 129.4 (C-4′), 131.3 (C-1′), 132.8 (C-4a’), 133.3 (C-8a’), 136.4 (C-7), 160.9 (C-8a), 169.4 (amide carbonyl), 190.2 (C-4); HRMS (ESI) calcd. for C21H17NaNO3 [M+Na]+ 354.110; found 354.113.
(±)-N-[(2S*,3S*)-4-oxo-2-(naphthalen-2-yl)-3,4-dihydro-2H-chromen-3-yl]acetamide (rac-trans-24g): White crystals, yield 73%, mp 200–202 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 1.68 (s, 3H, CH3), 5.00 (dd, J = 12.4, 8.4 Hz, 1H, 3-H), 5.76 (d, J = 12.4 Hz, 1H, 2-H), 7.11 (d, J = 8.4 Hz, 1H, 8-H), 7.15 (m, 1H, 6-H), 7.55 (m, 2H, 3′-H, 7′-H), 7.61 (m, 1H, 7-H), 7.68 (dd, J = 8.4, 1.6 Hz, 1H, 5-H), 7.84 (dd, J = 8.0, 1.6 Hz, 1H, 6′-H), 7.95 (m, 3H, 4′-H, 5′-H, 8′), 8.02 (s, 1H, 1′-H), 8.21 (d, J = 8.4 Hz, 1H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 22.2 (C-CH3), 57.7 (C-3), 81.7 (C-2), 118.0 (C-8), 120.1 (C-4a), 122.0 (C-6), 125.1 (C-5), 126.4 (C-1′), 126.5 (C-8′), 126.9 (C-3′), 127.2 (C-7′), 127.6 (C-6′), 127.8 (C-5′), 128.0 (C-4′), 132.4 (C-1′), 133.1 (C-4a’), 134.5 (C-8a’), 136.4 (C-7), 160.8 (C-8a), 169.2 (amide carbonyl), 190.2 (C-4); HRMS (ESI) calcd. for C21H17NO3 [M + H]+ 332.128; found 332.128.
2.9. General Procedure for the Synthesis of Condensed Thiazole Derivatives 3a-g
Under inert atmosphere, acetamide derivatives rac-cis-24a-e, g or rac-trans-24a-g (0.355 mmol) and Lawesson’s reagent (0.355 mmol) were dissolved in anhydrous toluene (5 mL). The mixture was stirred for 4 h at 70 °C. After cooling, toluene was evaporated and the crude product was purified by column chromatography using hexane/ethyl acetate 8:1 or toluene/ethyl acetate 8:1 as eluent, which provided the pure product.
(±)-2-methyl-4-phenyl-4H-chromeno[3,4-d][1,3]thiazole (3a): White crystals, yield 82%, mp 101–103 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.67 (s, 3H, CH3), 6.54 (s, 1H, 4-H), 6.89 (m, 2H, 6-H, 8-H), 7.12 (m, 2H, 7-H, 9-H), 7.28 (m, 3H, 2′-H, 4′-H, 6′-H), 7.37 (m, 2H, 3′-H, 5′-H); 13C-NMR (100 MHz, CDCl3) δ: 19.6 (CH3), 78.7 (C-4), 117.0 (C-6), 118.2 (C-5a), 121.9 (C-8), 124.3 (C-9), 126.2 (C-9b), 127.3 (C-3′, C-5′), 128.6 (C-4′), 128.7 (C-2′, C-6′), 129.3 (C-7), 139.8 (C-1′), 147.4 (C-3a), 151.2 (C-9a), 165.2 (C-2); HRMS (ESI) calcd. for C17H13NaNOS [M+Na]+ 362.061; found 362.067.
(4R)-3a: tR = 3.20 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (−3.68), 293 (−9.35), 280sh (−8.29), 243 (40.45), 218 (−169.42).
(4S)-3a: tR = 3.78 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (4.65), 293 (10.30), 280 (9.23), 243 (−43.81), 218 (173.68).
(±)-2-methyl-4-(4-methoxyphenyl)-4H-chromeno[3,4-d][1,3]thiazole (3b): White crystals, yield 52%. mp 108–109 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.69 (s, 3H, CH3), 3.74 (s, 3H, OCH3), 6.49 (s, 1H, 4-H), 6.82 (d, 2H, 3′-H, 5′-H), 6.89 (m, 2H, 6-H, 8-H), 7.09 (m, 2H, 7-H, 9-H), 7.26 (d, 2H, 2′-H, 6′-H); 13C-NMR (100 MHz, CDCl3) δ: 19.6 (C-CH3), 55.3 (C-OCH3), 78.5 (C-4), 114.1 (C-3′, C-5′), 117.1 (C-6), 118.3 (C-5a), 121.8 (C-8), 124.3 (C-9), 126.3 (C-9b), 128.9 (C-2′, C-6′), 129.2 (C-7), 131.9 (C-1′), 147.7 (C-4′), 151.2 (C-3a), 159.9 (C-9a), 165.2 (C-2); HRMS (ESI) calcd. for C18H15NaNO2S [M+Na]+ 332.072; found 332.070.
(4R)-3b: tR = 4.36 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (−1.96), 292 (−11.31), 280sh (−9.34), 257 (32.56), 223 (−86.35).
(4S)-3b: tR = 5.02 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (1.87), 292 (7.45), 280sh (5.86), 257 (−17.30), 223 (50.79).
(±)-2-methyl-4-(3,4-dimethoxyphenyl)-4H-chromeno[3,4-d][1,3]thiazole (3c): White crystals, yield 66%, mp 119–121 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.71 (s, 3H, CH3), 3.81 (d, 6H, 2xOCH3), 6.48 (s, 1H, 4-H), 6.77 (d, J = 8.0 Hz, 1H, 6-H), 6.84 (d, J = 8.4 Hz, 1H, 8-H), 6.92 (m, 3H, 2′-H, 5′-H, 6′-H), 7.11 (m, 2H, 7-H, 9-H); 13C-NMR (100 MHz, CDCl3) δ: 19.6 (C-CH3), 55.9 (2xC-OCH3), 78.8 (C-4), 110.7 (C-5′), 111.1 (C-2′), 117.1 (C-6), 118.3 (C-5a), 120.0 (C-6′), 121.9 (C-8), 124.3 (C-9), 126.3 (C-9b), 129.3 (C-7), 132.2 (C-1′), 147.7 (C-3a), 149.1 (C-4′), 149.4 (C-3′), 151.2 (C-9a), 165.2 (C-2); HRMS (ESI) calcd. for C19H17NaNO3S [M+Na]+ 362.082; found 362.080.
(4S)-3c: tR = 7.25 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (5.55), 294 (15.61), 263sh (−8.66), 241 (−22.57), 217 (103.93).
(4R)-3c: tR = 7.58 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (−4.29), 294 (−14.08), 263sh (10.85), 241 (25.19), 217 (−98.22).
(±)-2-methyl-4-(3,5-dimethoxyphenyl)-4H-chromeno[3,4-d][1,3]thiazole (3d): White crystals, yield 70%, mp 75–76 °C; 1H-NMR (360 MHz, CDCl3) δ: 2.67 (s, 3H, CH3), 3.69 (s, 6H, 2xOCH3), 6.36 (s, 1H, 4′-H), 6.47 (s, 1H, 4-H), 6.54 (d, 2H, 2′-H, 6′-H), 6.87 (m, 2H, 6-H, 8-H), 7.09 (m, 2H, 7-H, 9-H); 13C-NMR (90 MHz, CDCl3) δ: 19.5 (C-CH3), 55.3 (2 × C-OCH3), 78.4 (C-4), 100.2 (C-4′), 105.3 (C-2′, C-6′), 116.9 (C-6), 118.1 (C-5a), 121.9 (C-8), 124.3 (C-9), 126.2 (C-9b), 129.2 (C-7), 141.9 (C-1′), 147.1 (C-3a), 151.1 (C-9a), 160.8 (C-3′, C-5′), 165.2 (C-2); HRMS (ESI) calcd. for C19H17NaNO3S [M+Na]+ 362.082; found 362.081.
(4R)-3d: tR = 4.80 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (−6.17), 294 (−10.80), 283sh (−3.78), 244 (29.05), 218 (−143.69).
(4S)-3d: tR = 5.78 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (5.29), 294 (8.26), 283sh (2.36), 244 (−24.94), 218 (114.38).
(±)-2-methyl-4-(3,4,5-trimethoxyphenyl)-4H-chromeno[3,4-d][1,3]thiazole (3e): White crystals, yield 84%, mp 113–115 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.72 (s, 3H, CH3), 3.77 (m, 9H, 3 × OCH3), 6.46 (s, 1H, 4-H), 6.62 (s, 2H, 2′-H, 6′-H), 6.93 (m, 2H, 6-H, 8-H), 7.16 (m, 2H, 7-H, 9-H); 13C-NMR (101 MHz, CDCl3) δ: 20.0 (C-CH3), 56.5 (2 × C-OCH3), 61.2 (C-OCH3), 79.4 (C-4), 104.9 (C-2′, C-6′), 117.3 (C-6), 118.5 (C-5a), 122.4 (C-8), 124.7 (C-9), 126.7 (C-9b), 129.7 (C-7), 135.5 (C-1′, C-4′), 147.8 (C-3a), 151.6 (C-9a), 153.7 (C-3′, C-5′), 165.6 (C-2); HRMS (ESI) calcd. for C20H19NaNO4S [M+Na]+ 392.093; found 392.095.
(4R)-3e: tR = 6.51 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (−4.54), 293 (−9.61), 285sh (−4.80), 263sh (7.01), 240 (13.85), 218 (−75.26).
(4S)-3e: tR = 6.67 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 326sh (4.66), 293 (8.98), 285sh (4.36), 263sh (−6.88), 240 (−14.89), 218 (67.60).
(±)-2-methyl-4-(naphthalen-1-yl)-4H-chromeno[3,4-d][1,3]thiazole (3f): White crystals, yield 60%, mp 173–175 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.69 (s, 3H, CH3), 6.77 (d, J = 8.0 Hz, 1H, 6-H), 6.90 (t, J = 7.2 Hz, 1H, 8-H), 7.04 (s, 2H, 4-H, 7-H), 7.20 (m, 3H, 9-H, 2′-H, 3′-H), 7.50 (t, J = 7.6 Hz, 1H, 6′-H), 7.55 (t, J = 7.6 Hz, 1H, 7′-H), 7.77 (d, J = 8.0 Hz, 1H, 4′-H), 7.84 (d, J = 7.6 Hz, 1H, 5′-H), 8.45 (d, J = 8.4 Hz, 1H, 8′-H); 13C-NMR (100 MHz, CDCl3) δ: 19.7 (C-CH3), 76.2 (C-4), 117.3 (C-6), 118.5 (C-5a), 122.0 (C-8), 124.4 (C-9), 124.5 (C-8′), 125.1 (C-3′), 125.9 (C-6′), 126.6 (C-2′), 126.8 (C-7′), 127.4 (C-9b), 128.7 (C-5′), 129.2 (C-4′), 129.8 (C-7), 131.7 (C-8a’), 134.1 (C-1′), 134.3 (C-4a’), 147.1 (C-3a), 151.1 (C-9a), 165.2 (C-2); HRMS (ESI) calcd. for C21H15NaNOS [M+Na]+ 352.077; found 352.076.
(4R)-3f: tR = 4.10 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 316 (−1.29), 274sh (−5.17), 239sh (−6.11), 223 (−8.99), 216 (9.89).
(4S)-3f: tR = 5.62 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 316 (0.40), 274sh (3.19), 239sh (2.89), 223 (4.81), 216 (−6.90).
(±)-2-methyl-4-(naphthalen-2-yl)-4H-chromeno[3,4-d][1,3]thiazole (3g): White crystals, yield 55%, mp 129–130 °C; 1H-NMR (400 MHz, CDCl3) δ: 2.67 (s, 3H, CH3), 6.70 (s, 1H, 4-H), 6.89 (m, 2H, 7-H, 9-H), 7.10 (m, 2H, 6-H, 8-H), 7.40 (m, 2H, 6′-H, 7′-H), 7.52 (d, J = 8.8 Hz, 1H, 3′-H), 7.74 (m, 4 H, 1′-H, 4′-H, 5′-H, 8′-H); 13C-NMR (100 MHz, CDCl3) δ: 19.6 (C-CH3), 79.0 (C-4), 117.0 (C-6), 118.2 (C-5a), 122.0 (C-8), 124.4 (C-9), 125.0 (C-8′), 126.2 (C-3′), 126.4 (C-6′), 126.4 (C-9b), 126.5 (C-1′), 127.7 (C-7′), 128.4 (C-5′), 128.6 (C-4′), 129.3 (C-7), 133.2 (C-4a’), 133.5 (C-8a’), 137.1 (C-2′), 147.4 (C-3a), 151.3 (C-9a), 165.3 (C-2); HRMS (ESI) calcd. for C21H15NaNOS [M+Na]+ 352.077; found 352.077.
(4R)-3g: tR = 3.79 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 239 (6.99), 225 (−28.42), 214 (7.25).
(4S)-3g: tR = 4.52 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕϕ)}:239 (−5.03), 225 (12.80), 214 (−1.62).
2.10. General Procedure for the Knorr Reaction Affording the Pyrrole-Condensed Derivatives 4a-g
3-Aminoflavanone derivatives rac-cis-1a-e, g or rac-trans-1a-g (0.363 mmol) were dissolved in a mixture of 96% ethanol (4 mL) and water (2 mL). Under stirring, ethyl acetoacetate (55 µL, 0.436 mmol) and NaOAc x 3 H2O (300 mg, 2.178 mmol) were added to the reaction. The mixture was refluxed for 3 h and then water was added. Extraction with CH2Cl2, drying of the combined organic phases over MgSO4, and concentration under reduced pressure provided the crude product as orange oil. Column chromatography using toluene/ethyl acetate 4:1 with 0.1% Et3N as eluent and subsequent trituration in cold Et2O afforded the pure product.
(±)-ethyl 2-methyl-4-phenyl-3,4-dihydrochromeno[3,4-b]pyrrole-1-carboxylate (4a): Off-white crystals, yield 53%, mp 133–136 °C; 1H-NMR (400 MHz, CDCl3) δ: 1.37 (t, 3H, CH3), 2.42 (s, 3H, 2-CH3), 4.31 (m, 2H, CH2), 6.10 (s, 1H, 4-H), 6.91 (dd, J = 8.0, 1.6 Hz, 1H, 6-H), 6.97 (m, 1H, 8-H), 7.04 (m, 1H, 7-H), 7.40 (m, 5H, 2′-H, 3′-H, 4′-H, 5′-H, 6′-H), 7.73 (bs, 1H, NH), 8.27 (dd, J = 8.0, 2.0 Hz, 1H, 9-H); 13C-NMR (100 MHz, CDCl3) δ: 14.5 (2xCH3), 60.0 (C-CH2), 75.8 (C-4), 109.0 (C-5), 114.9 (C-9b), 116.8 (C-6), 121.6 (C-9a), 122.0 (C-8), 125.7 (C-3a), 126.1 (C-9), 126.9 (C-4), 128.0 (C-2′, C-6′), 129.1 (C-3′, C-5′), 129.4 (C-7), 136.7 (C-2), 138.2 (C-1′), 152.0 (C-5a), 165.9 (ester carbonyl); HRMS (ESI) calcd. for C21H19NO3 [M + H]+ 334.1438; found 334.1438.
(4R)-4a: tR = 3.20 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 300 (−3.47), 250 (−1.88), 235 (8.76), 214 (−28.73).
(4S)-4a: tR = 3.43 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕϕ)}: 300 (3.06), 250 (2.22), 235 (−10.08), 214 (30.02).
(±)-ethyl 2-methyl-4-(4-methoxyphenyl)-3,4-dihydrochromeno[3,4-b]pyrrole-1-carboxylate (4b): White crystals, yield 32%, mp 169–171 °C; 1H-NMR (400 MHz, CDCl3) δ: 1.36 (t, 3H, CH3), 2.40 (s, 3H, 4-CH3), 3.76 (s, 3H, OCH3), 4.26 (m, 2H, CH2), 6.01 (s, 1H, 4-H), 6.86 (m, 3H, 6-H, 3′-H, 5′-H), 6.94 (t, J = 7.2 Hz, 1H, 7-H), 7.01 (t, J = 6.8 Hz, 1H, 8-H), 7.30 (d, 2H, 2′-H, 6′-H), 7.86 (bs 1H, NH), 8.25 (d, J = 6.8 Hz, 1H, 9-H); 13C-NMR (100 MHz, CDCl3) δ: 14.4 (C-CH3), 14.5 (C-CH3), 55.4 (C-OCH3), 60.0 (C-CH2), 75.4 (C-4), 108.9 (C-1), 114.4 (C-3′, C-5′), 114.9 (C-9b), 116.8 (C-6), 121.6 (C-9a), 121.9 (C-8), 125.9 (C-3a), 126.0 (C-9), 126.8 (C-7), 129.5 (C-2′, C-6′), 130.2 (C-1′), 136.7 (C-2), 152.0 (C-5a), 160.4 (C-4′), 165.9 (ester carbonyl); HRMS (ESI) calcd. for C22H21NaNO4 [M+Na]+ 364.1543; found 364.1546.
(4R)-4b: tR = 7.13 min on Chiralpak IA column (hexane/2-propanol 90:10), HPLC-ECD {λ [nm] (ϕ)}: 310sh (−4.28), 277 (−5.65), 237 (33.61), 219 (−31.69), 203 (25.66).
(4S)-4b: tR = 7.68 min on Chiralpak IA column (hexane/2-propanol 90:10), HPLC-ECD {λ [nm] (ϕ)}: 310 (3.55), 277 (4.50), 237 (−27.21), 219 (23.67), 203 (−26.07).
(±)-ethyl 2-methyl-4-(3,4-dimethoxyphenyl)-3,4-dihydrochromeno[3,4-b]pyrrole-1-carboxylate (4c): White crystals, yield 48%, mp 214–216 °C; 1H-NMR (400 MHz, CDCl3) δ: 1.40 (t, 3H, CH3), 2.45 (s, 3H, 4-CH3), 3.78 (m, 6H, 2 × OCH3), 4.35 (m, 2H, CH2), 6.02 (s, 1H, 4-H), 6.80 (d, J = 6.8 Hz, 1H, 5′-H), 6.93 (m, 5H, 6-H, 7-H, 8-H 2′-H, 6′-H), 8.04 (bs, 1H, NH), 8.28 (d, J = 5.6 Hz, 1H, 9-H); 13C-NMR (100 MHz, CDCl3) δ: 14.4 (C-CH3), 14.5 (C-CH3), 55.9 (2 × C-OCH3), 60.2 (C-CH2), 75.9 (C-4), 109.0 (C-1), 110.8 (C-2′), 111.0 (C-5′), 115.0 (C-9b), 116.8 (C-6′), 120.8 (C-6), 121.7 (C-9a), 122.0 (C-8), 126.0 (C-3a), 126.1 (C-9), 126.8 (C-7), 130.5 (C-1′), 136.8 (C-2), 149.4 (C-4′,), 149.8 (C-3′), 152.2 (C-5a), 165.9 (ester carbonyl); HRMS (ESI) calcd. for C23H23NaNO5 [M+Na]+ 416.1468; found 416.1466.
(4R)-4c: tR = 9.60 min on Chiralpak IA column (hexane/2-propanol 90:10), HPLC-ECD {λ [nm] (ϕ)}: 308 (−2.89), 238 (10.00), 215 (−15.99), 206 (−9.40).
(4S)-4c: tR = 9.96 min on Chiralpak IA column (hexane/2-propanol 90:10), HPLC-ECD {λ [nm] (ϕ)}: 308 (2.20), 238 (−8.41), 215 (10.48), 206 (−12.66).
(±)-ethyl 2-methyl-4-(3,5-dimethoxyphenyl)-3,4-dihydrochromeno[3,4-b]pyrrole-1-carboxylate (4d): White crystals, yield 35%, mp 156–158 °C; 1H-NMR (400 MHz, CDCl3) δ: 1.38 (t, 3H, CH3), 2.45 (s, 3H, 4-CH3), 3.79 (s, 6H, 2 × OCH3), 4.30 (m, 2H, CH2), 6.04 (s, 1H, 4-H), 6.49 (s, 1H, 4′-H), 6.63 (d, 2H, 2′-H, 6′-H), 6.96 (m, 3H, 6-H, 7-H, 8-H), 7.60 (bs, 1H, NH), 8.27 (dd, J = 7.6, 1.2 Hz, 1H, 9-H); 13C-NMR (100 MHz, CDCl3): δ: 14.6 (C-CH3), 55.6 (2 × C-OCH3), 60.1 (C-CH2), 75.8 (C-4), 101.3 (C-4′), 105.6 (C-2′, C-6′), 109.2 (C-1), 114.9 (C-9a), 116.9 (C-6), 121.6 (C-9a), 122.2 (C-8), 125.6 (C-3a), 126.2 (C-9), 126.9 (C-7), 136.6 (C-2), 140.4 (C-1′), 152.2 (C-5a), 161.5 (C-3′, C-5′), 165.8 (ester carbonyl); HRMS (ESI) calcd. for C23H23NaNO5 [M+Na]+ 416.1468; found 416.1464.
(4R)-4d: tR = 4.53 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 310 (−4.70), 254 (−2.22), 237 (10.34), 216 (−20.58), 205 (27.90).
(4S)-4d: tR = 4.80 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 310 (4.06), 254 (2.07), 237 (−9.85), 216 (19.44), 20 5(−21.99).
(±)-ethyl 2-methyl-4-(3,4,5-trimethoxyphenyl)-3,4-dihydrochromeno[3,4-b]pyrrole-1-carboxylate (4e): Pale yellow crystals, yield 55%, mp 199–201 °C; 1H-NMR (400 MHz, CDCl3): δ: 1.40 (t, 3H, CH3), 2.51 (s, 3H, 4-CH3), 3.77 (m, 9H, 3 × OCH3), 4.31 (m, 2H, CH2), 6.02 (s, 1H, 4-H), 6.66 (s, 2H, 2′-H, 6′-H), 6.93 (d, J = 7.6 Hz, 1H, 6-H), 6.99 (t, J = 7.6 Hz, 1H, 7-H), 7.05 (t, J = 7.2 Hz, 1H, 8-H), 8.28 (d, J = 7.2 Hz, 1H, 9-H), 8.44 (bs, 1H, NH); 13C-NMR (100 MHz, CDCl3) δ: 14.4 (C-CH3), 14.6 (C-CH3), 56.1 (2 × C-OCH3), 60.0 (C-CH2), 60.7 (C-OCH3), 76.6 (C-4), 105.1 (C-2′, C-6′), 108.9 (C-1), 115.0 (C-9b), 116.7 (C-6), 121.7 (C-9a), 122.1 (C-8), 125.8 (C-3a), 126.1 (C-9), 126.8 (C-7), 134.0 (C-1′), 136.8 (C-2), 138.1 (C-4′),152.2 (C-5a), 153.4 (C-3′, C-5′), 166.0 (ester carbonyl); HRMS (ESI) calcd. for C24H25NaNO6 [M+Na]+ 446.1574; found 446.1571.
(4R)-4e: tR = 11.57 min on Chiralpak IC column (hexane/2-propanol 70:30), HPLC-ECD {λ [nm] (ϕ)}: 311 (−6.94), 291sh (−6.42), 240 (21.81), 217 (−36.71).
(4S)-4e: tR = 16.28 min on Chiralpak IC column (hexane/2-propanol 70:30), HPLC-ECD {λ [nm] (ϕ)}: 311 (9.81), 291sh (9.30), 240 (−30.91), 217 (51.74).
(±)-ethyl 2-methyl-4-(naphthalen-1-yl)-3,4-dihydrochromeno[3,4-b]pyrrole-1-carboxylate (4f): White crystals, yield 51%, mp 204–207 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 1.37 (t, 3H, CH3), 2.50 (s, 3H, 4-CH3), 4.32 (q, 2H, CH2), 6.68 (m, 1H, 6-H), 6.94 (m, 2H, 7-H, 8-H), 7.07 (d, J = 6.8 Hz, 1H, 2′-H), 7.15 (s, 1H, 4-H), 7.41 (t, J = 7.6 Hz, 1H, 3′-H), 7.59 (t, J = 7.2 Hz, 1H, 6′-H), 7.66 (t, J = 7.2 Hz, 1H, 7′-H), 7.95 (d, J = 8.0 Hz, 1H, 4′-H), 8.01 (d, J = 8.0 Hz, 1H, 5′-H), 8.38 (m, 1H, 9-H), 8.55 (d, J = 8.0 Hz, 1H, 8′-H), 11.50 (bs, 1H, NH); 13C-NMR (100 MHz, DMSO-d6) δ: 14.07 (C-CH3), 14.4 (C-CH3), 59.3 (C-CH2), 72.0 (C-4), 107.5 (C-1), 114.0 (C-9b), 116.6 (C-6), 121.4 (C-8), 121.7 (C-9a), 124.4 (C-8′), 125.2 (C-3′), 125.2 (C-3a), 125.6 (C-9), 126.0 (C-6′), 126.2 (C-7), 126.6 (C-2′), 126.8 (C-7′), 128.6 (C-5′), 129.6 (C-2), 131.2 (C-4a’), 133.7 (C-8a’), 133.8 (C-1′), 137.2 (C-4), 150.6 (C-5a), 165.2 (ester carbonyl); HRMS (ESI) calcd. for C25H21NaNO3 [M+Na]+ 406.1414; found 406.1411.
(4R)-4f: tR = 3.66 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 323 (0.91), 271 (−6.26), 238sh (−10.92), 225 (−73.35), 212 (47.44).
(4S)-4f: tR = 4.38 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 323 (−1.22), 271 (7.61), 238sh (12.80), 225 (78.13), 212 (−57.70).
(±)-ethyl 2-methyl-4-(naphthalen-2-yl)-3,4-dihydrochromeno[3,4-b]pyrrole-1-carboxylate (4g): White crystals, yield 60%, mp 149–151 °C; 1H-NMR (400 MHz, CDCl3) δ: 1.35 (s, 3H, CH3), 2.34 (s, 3H, 4-CH3), 4.29 (s, 2H, CH2), 6.21 (s, 1H, 4-H), 6.93 (m, 3H, 6-H, 8-H, 3′-H), 7.48 (m, 3H, 7-H, 6′-H, 7′-H), 7.80 (m, 5H, 9-H, 4′-H, 5′-H, 8′-H, NH), 8.28 (d, J = 6.0 Hz, 1H, 1′-H); 13C-NMR (100 MHz, CDCl3) δ: 14.4 (C-CH3), 14.5 (C-CH3), 60.0 (C-CH2), 75.9 (C-4), 109.0 (C-1), 114.9 (C-9b), 116.8 (C-6), 121.6 (C-9a), 122.2 (C-8), 125.2 (C-8′), 125.6 (C-3a), 126.1 (C-3′), 126.6 (C-9), 126.8 (C-6′), 126.9 (C-7), 127.4 (C-1′), 127.9 (C-7′), 128.3 (C-5′), 129.2 (C-4′), 133.2 (C-4a’), 133.8 (C-8a’), 135.5 (C-2′), 136.9 (C-2), 152.0 (C-5a), 165.9 (ester carbonyl); HRMS (ESI) calcd. for C25H21NaNO3 [M+Na]+ 406.1414; found 406.1414.
(4R)-4g: tR = 3.86 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 317 (−6.27), 273 (−6.98), 236 (17.06), 222 (−37.60), 207 (55.29).
(4S)-4g: tR = 4.20 min on Chiralpak IA column (hexane/2-propanol 80:20), HPLC-ECD {λ [nm] (ϕ)}: 317 (5.90), 273 (6.95), 236 (−18.81), 222 (35.02), 207 (−37.51).
2.11. X-Ray Diffraction Analysis
Single crystals of
17e have been obtained with the slow evaporation of its solution in chloroform. Data collection was carried out at 298 K using Mo-Kα radiation (λ = 0.71073 Å) with a Burker-Nonius MACH3 diffractometer equipped with point detector. The structure could be solved by SIR-92 program [
20] and refined by full-matrix least-squares method on F
2 using the SHELX program [
21]. Non-hydrogen atoms were refined anisotropically, hydrogen atoms were placed into geometric positions and methyl protons were fixed using the riding model. Publication material was prepared with the WINGX-suite [
22] and publCIF software [
23]. ORTEP view of the structure with selected geometric parameters are shown in
Figure S342, other data are in the expected range. Further crystallographic information is compiled in
Table S2. The structure is deposited in the Cambridge Crystallographic Data Centre under CCDC 2016874.
2.12. MTT Assay
The number of viable cells was indirectly determined by measuring the conversion of the tetrazolium salt MTT (3-{4,5-dimethilthiasol-2-il}-2,5-diphenyltetrasolium bromide, Sigma-Aldrich) to formazan by mitochondrial dehydrogenases. Cells were plated in 96-well multi-titer plates (10,000 cells per well density) in quadruplicates and were cultured for 3 days and treated by the compounds daily. Negative control group was treated with equal amount of vehicle solvent (DMSO) and positive control group was treated with 1 µg/mL doxorubicin. Cells were then incubated with 5 mg/mL MTT for 3 h, precipitated formazan crystals were dissolved in acidic isopropanol (10% 1 M HCl in isopropanol supplemented with 10% Triton X 100), and concentration of formazan was assessed colorimetrical way measuring absorbance at 565 nm.
Determination of IC50
Logistic dose-response curves were fitted using the equation y = A2 + (A1-A2)/(1 + (x/x0)^p) where the parameters are: A1: initial value (ymin), A2: final value (ymax), x0: center (EC/IC50), and p is the calculated power. Fittings were carried out and parameters were calculated using Origin 8.6 (OriginLab Corporation, Northampton, MA, USA).
2.13. MitoProbeTM DilC1(5) Assay and SYTOX Green Labeling
Decrease in mitochondrial membrane potential is an early hallmark of apoptosis and the disruption of the plasma membrane integrity is characteristic for cellular necrosis. These events were investigated simultaneously using MitoProbeTM DilC1(5) assay kit and SYTOX green (both from Molecular Probes/ThermoiFisher) staining, respectively. DilC1(5) (1,1′,3,3,3′,3′-hexamethylindodicarbo - cyanine iodide) is a fluorescent cyanine dye which penetrates cytoplasm with intact membrane and accumulates primarily in mitochondria depending on the mitochondrial membrane potential. Since decrease in mitochondrial membrane potential is an early marker of apoptosis, the DilC1(5) staining intensity is typically decreased in apoptotic cells. SYTOX Green is a nucleic acid stain impermeant to live cells with intact plasma membrane, but it penetrates the compromised membrane of necrotic, dead cells resulting in a bright green fluorescent nuclear staining.
Cells were plated in 96-well multi-titer plates (10,000 cells per well density) in quadruplicates and were incubated for 24 h with various concentrations of rac-19g. Negative control group was treated with equal amount of vehicle solvent (DMSO) and positive control groups were shortly treated with 50 µM carbonyl cyanide 3- chlorophenylhydrazone (CCCP) or lysis buffer (20 mM Tris HCl, 5 mM EDTA in H2O) to disrupt mitochondrial membrane potential or to lyse cells and disrupt membrane integrity, respectively. Then supernatant were removed and cells were incubated with DilC1(5) and SYTOX Green following the manufacturer’s protocol. Finally, the excess of the dyes was removed, cells were gently washed in PBS, and fluorescence of DilC1(5) and SYTOX Green was measured at 630/680 nm and 490/520 nm (excitation/emission), respectively, using a FlexStation 3 (Molecular Devices) multimodal microplate reader.
2.14. CyQUANT® Cell Proliferation Assay
CyQUANT assay assesses the cellular proliferation indirectly by directly determining the DNA content in a cell population using CyQUANT fluorescent dye. The dye exhibits strong fluorescence enhancement when bound to cellular DNA and the fluorescent intensity is proportional to the amount of bound DNA in a high dynamic range. Therefore, it is suitable to asses DNA synthesis associated with cellular proliferation. Cells were plated in 96-well multi-titer plates (10,000 cells per well density) in quadruplicates and were incubated for 24 h with various concentrations of rac-19g, then assayed for cellular proliferation following the manufacturer’s protocol. Briefly, supernatants were gently removed and the plate was snap frozen and stored at −70 °C. Then plate was thawed, cells were lysed and incubated with CyQUANT dye. The excess of the dye was removed and fluorescence was measured at 490/520 nm (excitation/emission) using a FlexStation 3 (Molecular Devices) multimodal microplate reader.
2.15. Computational Methods
Mixed torsional/low-frequency mode conformational searches were carried out by means of the Macromodel 10.8.011 software using the Merck Molecular Force Field (MMFF) with an implicit solvent model for CHCl
3 [
24]. Geometry reoptimizations were carried out at the B3LYP/6-31+G(d,p) level in vacuo, the CAM-B3LYP/TZVP [
25] and the ωB97X/TZVP [
26] levels with the PCM solvent model for CHCl
3. TDDFT ECD calculations were run with various functionals (B3LYP, BH&HLYP, CAM-B3LYP, PBE0) and the TZVP basis set as implemented in the Gaussian 09 package with the same or no solvent model as in the preceding DFT optimization step [
27]. ECD spectra were generated as sums of Gaussians with 1500–3000 cm
−1 widths at half-height, using dipole-velocity-computed rotational strength values [
28]. Ball-and-stick representations of the conformers were generated by using the Molekel software [
29].

 ,
, 
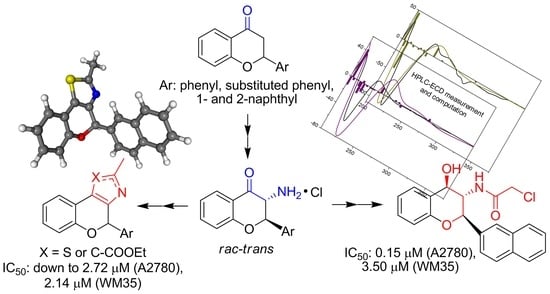
















{kind=link}
{kind=link}
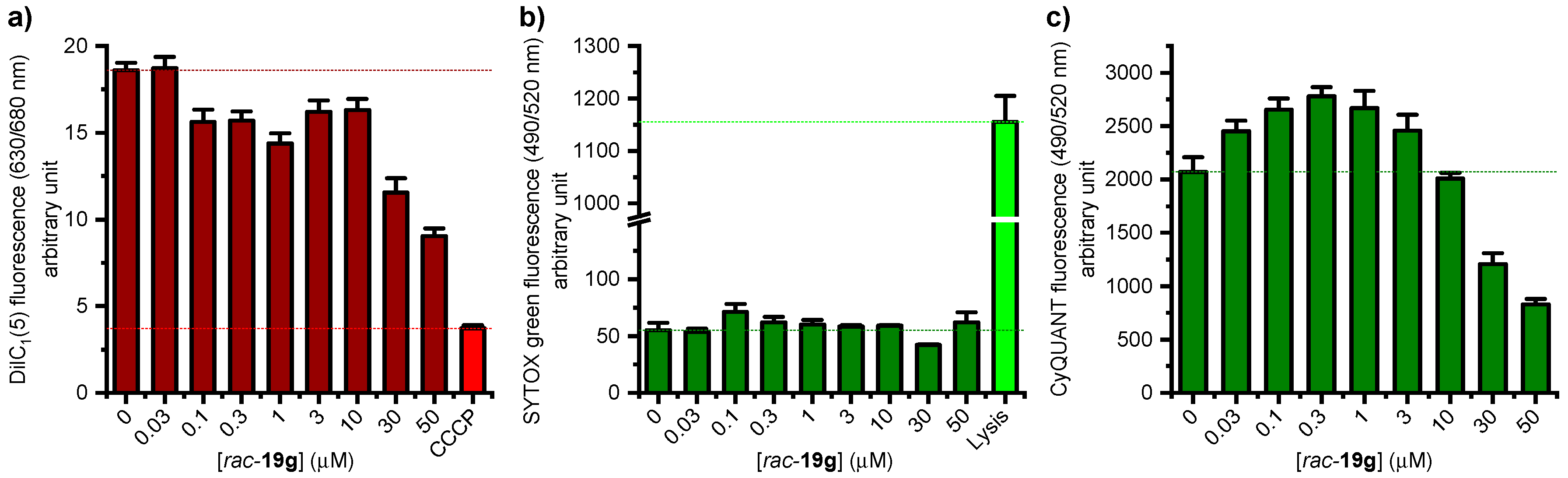{kind=link}
{kind=link}
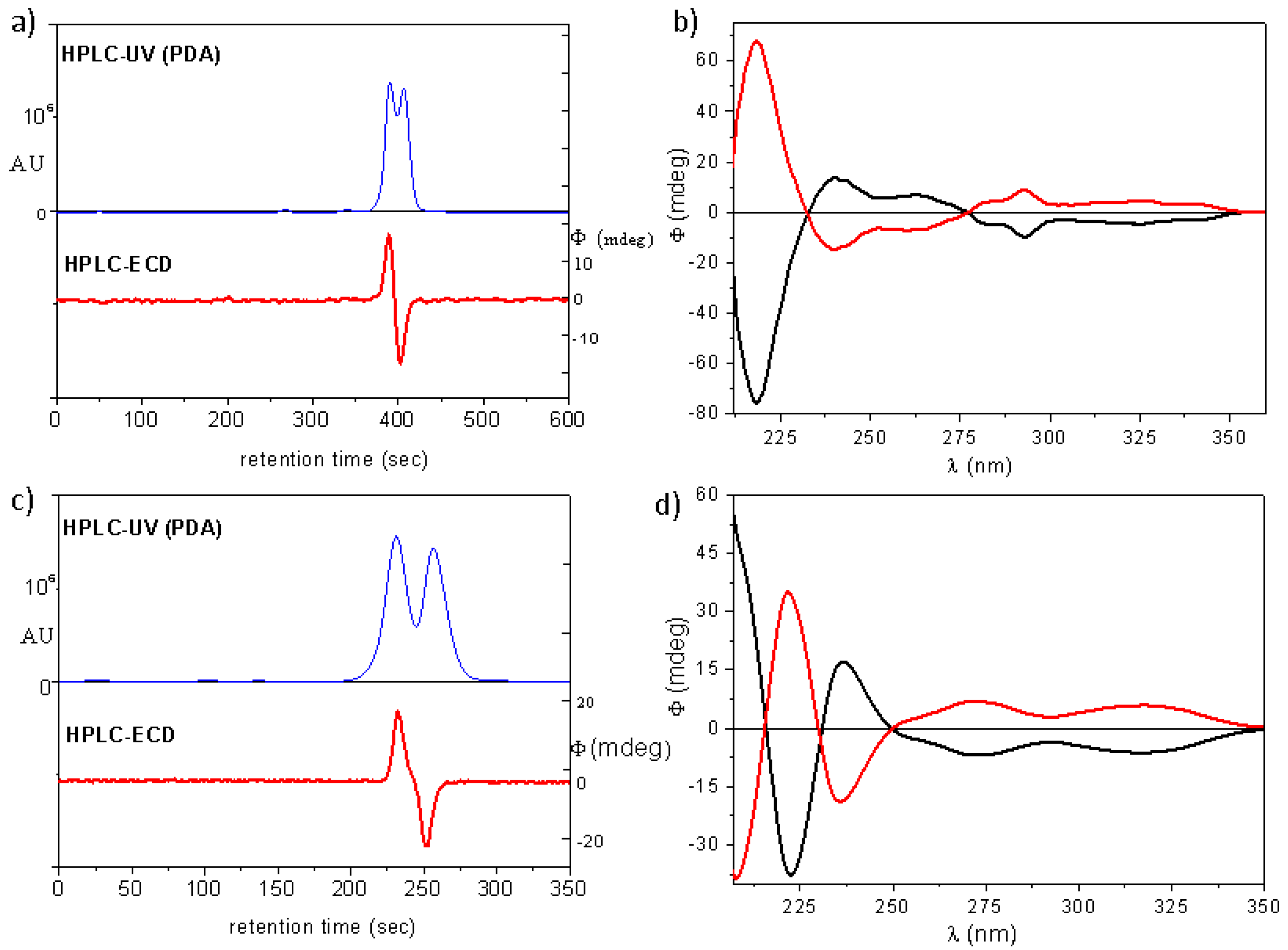{kind=link}
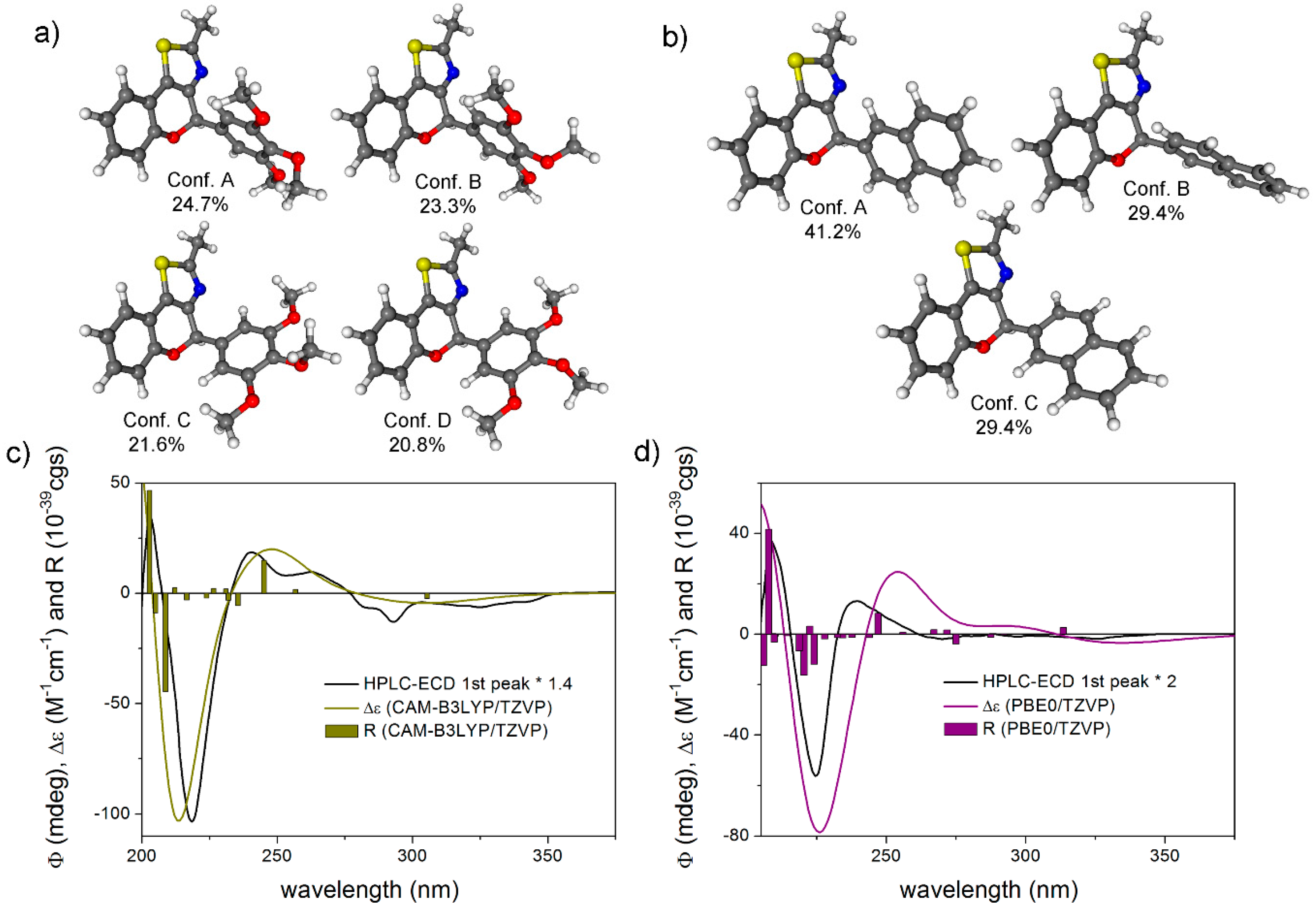{kind=link}
{kind=link}
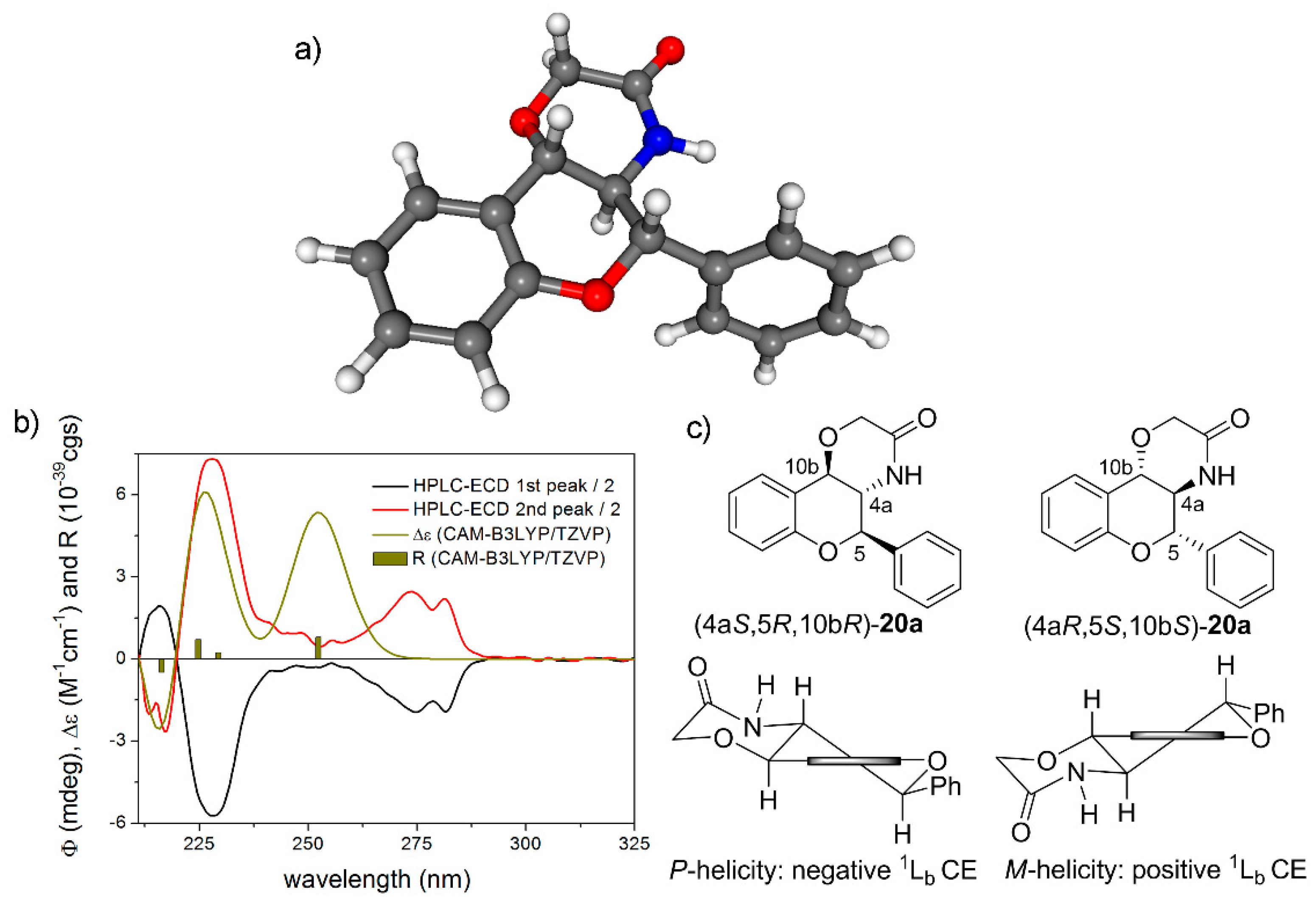{kind=link}
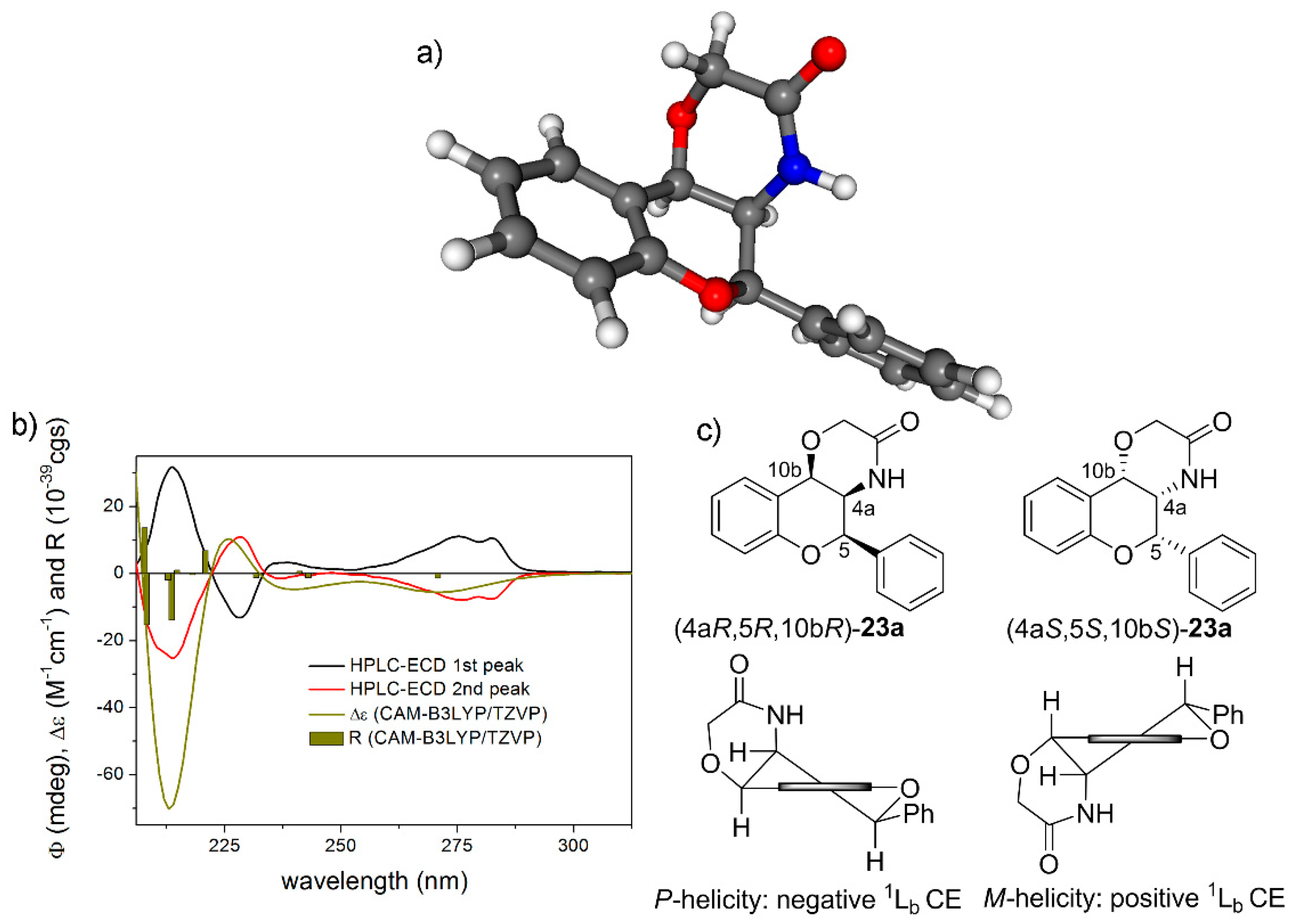{kind=link}
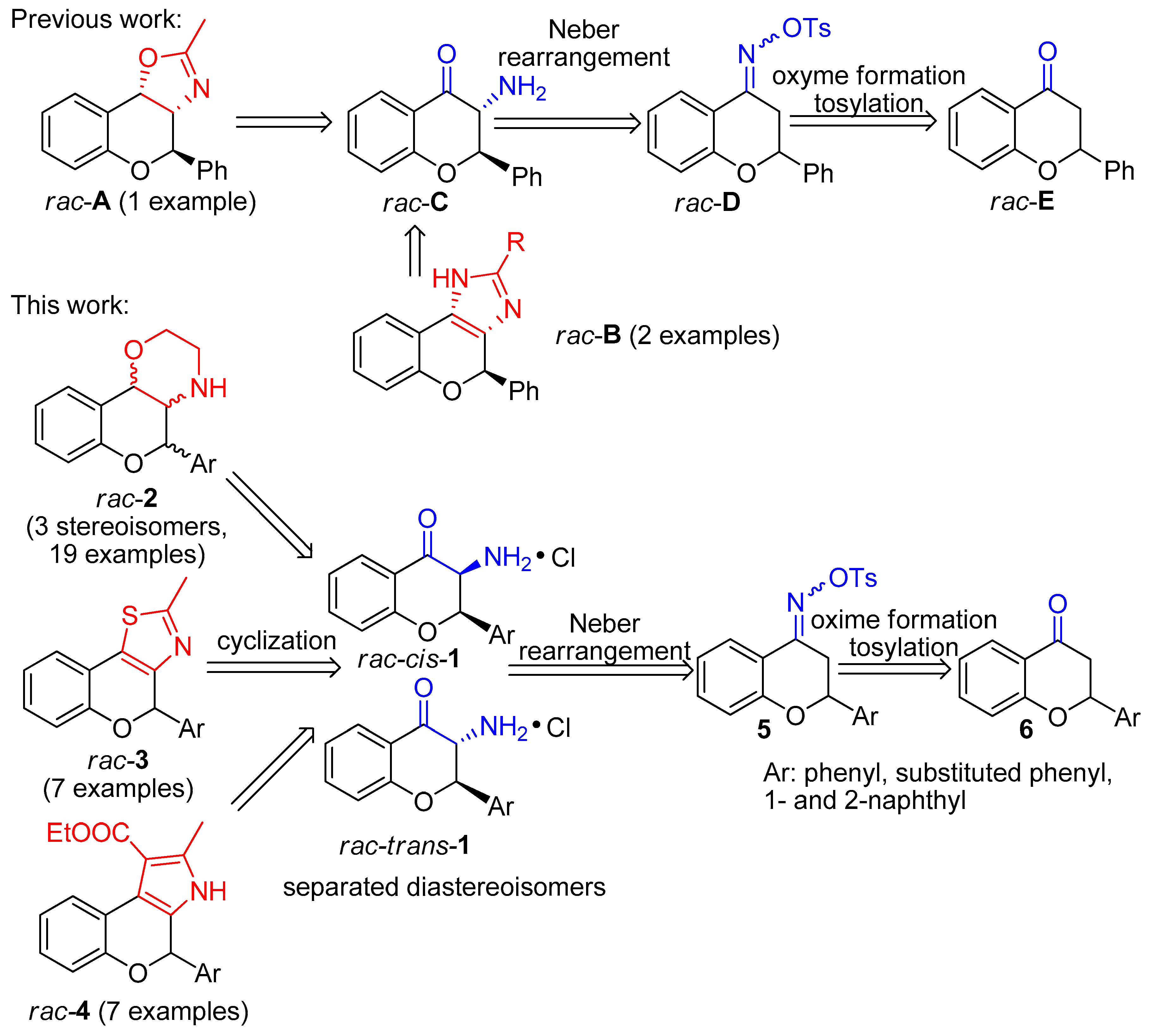{kind=link}
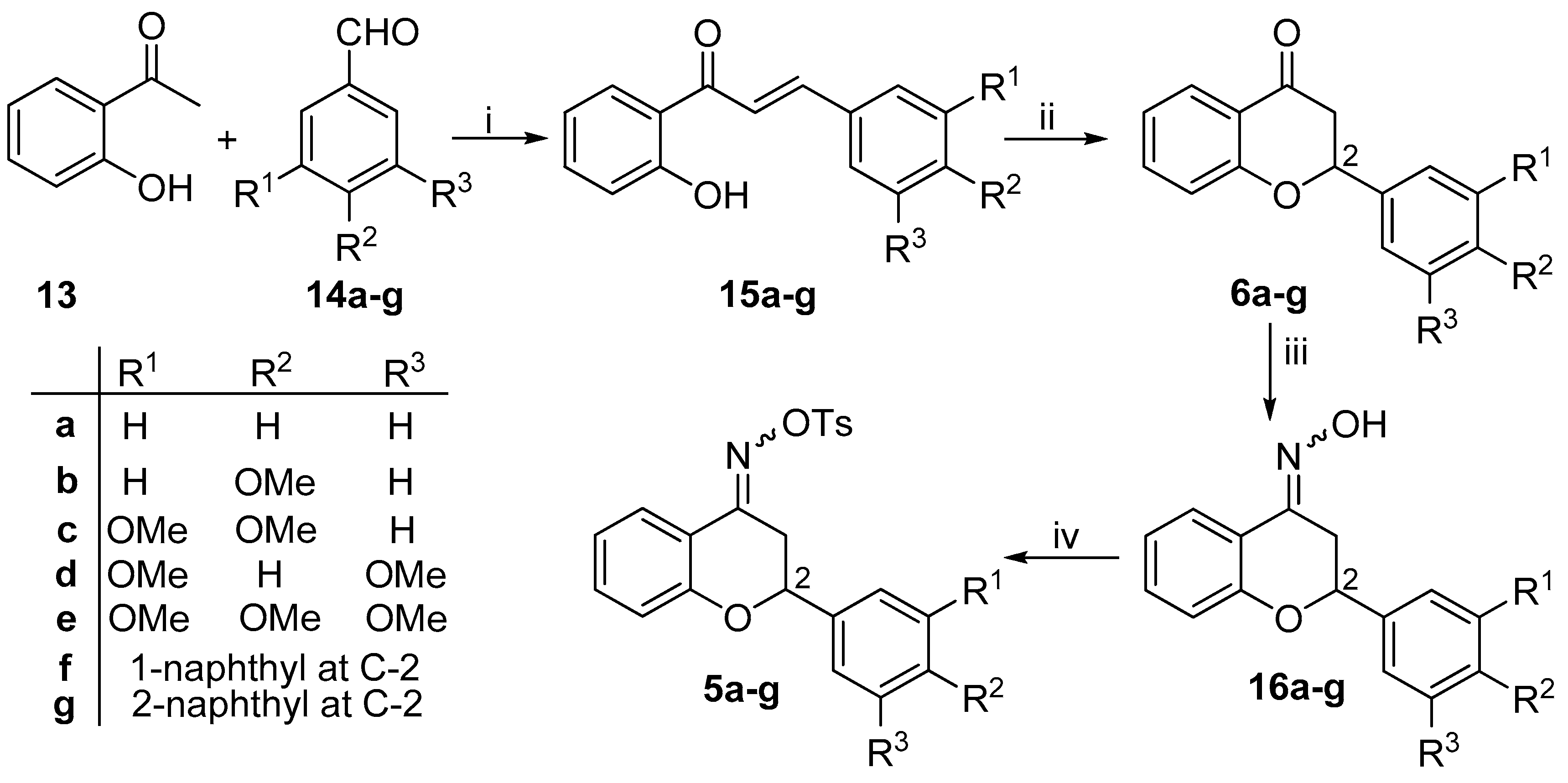{kind=link}
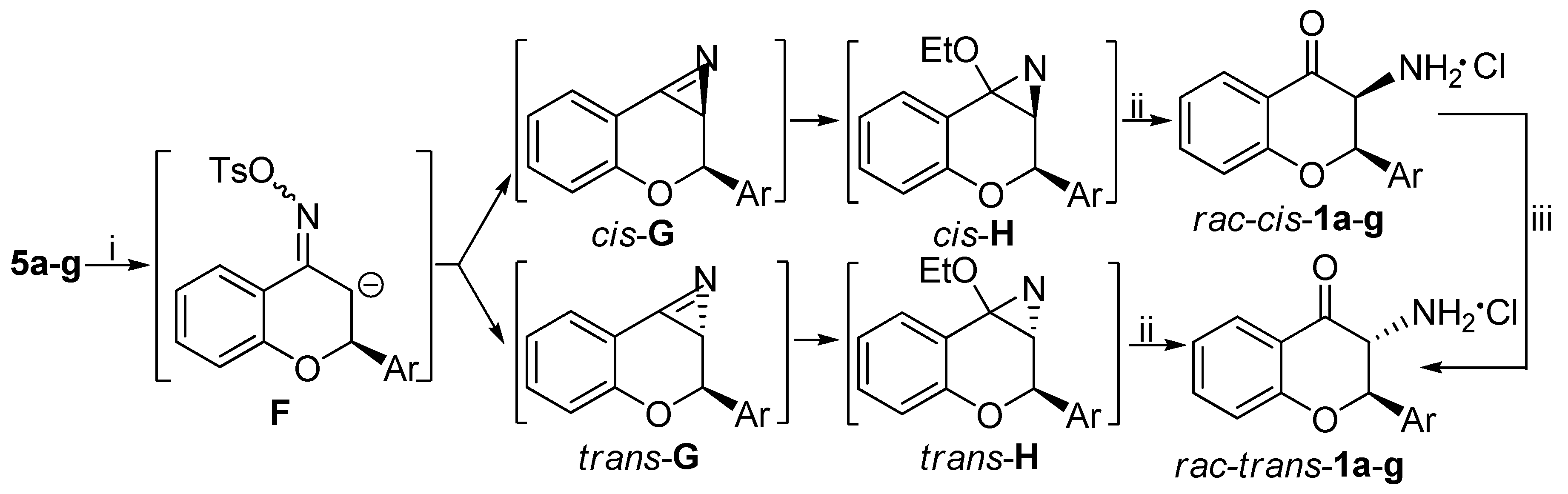{kind=link}
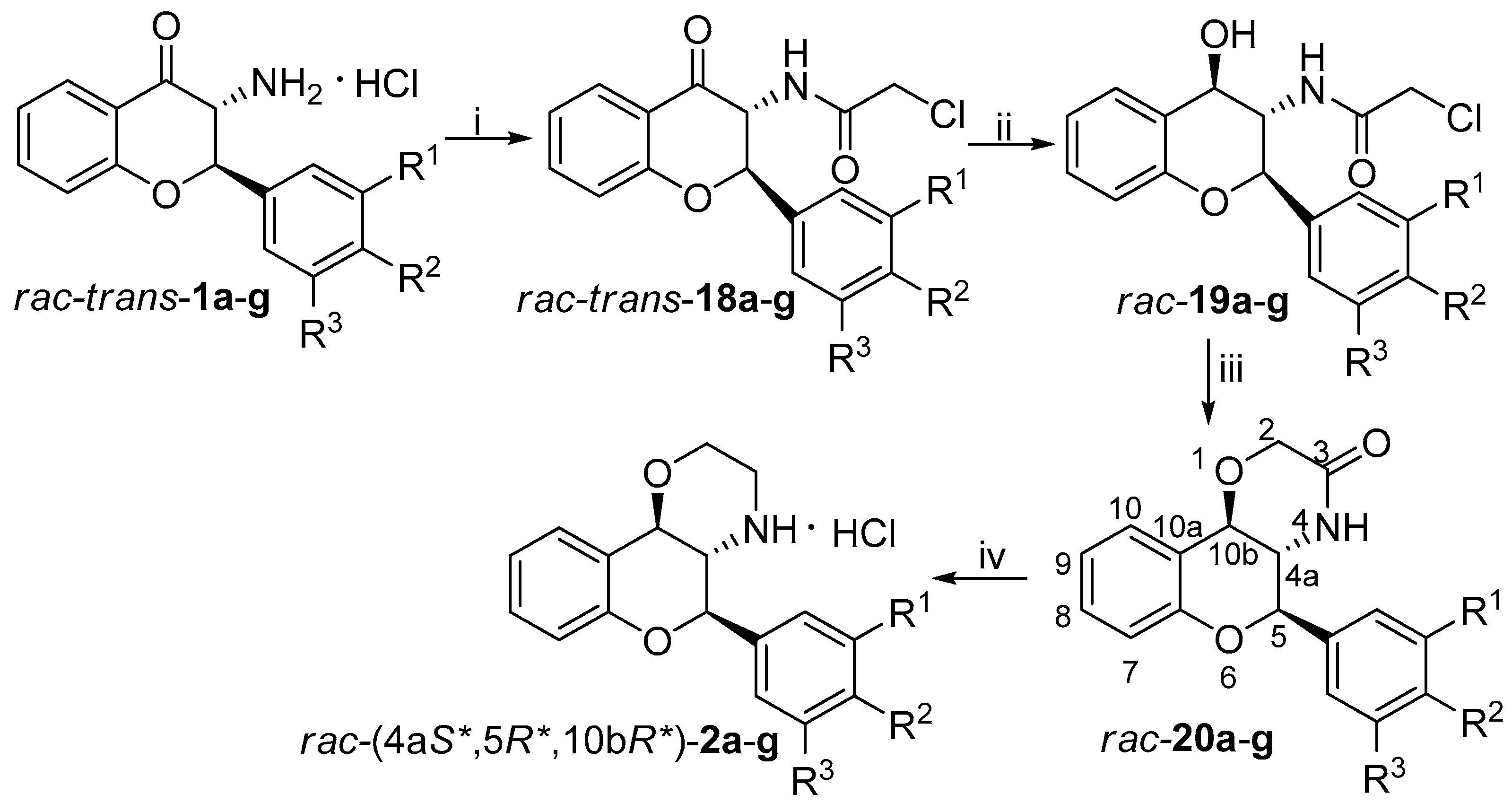{kind=link}
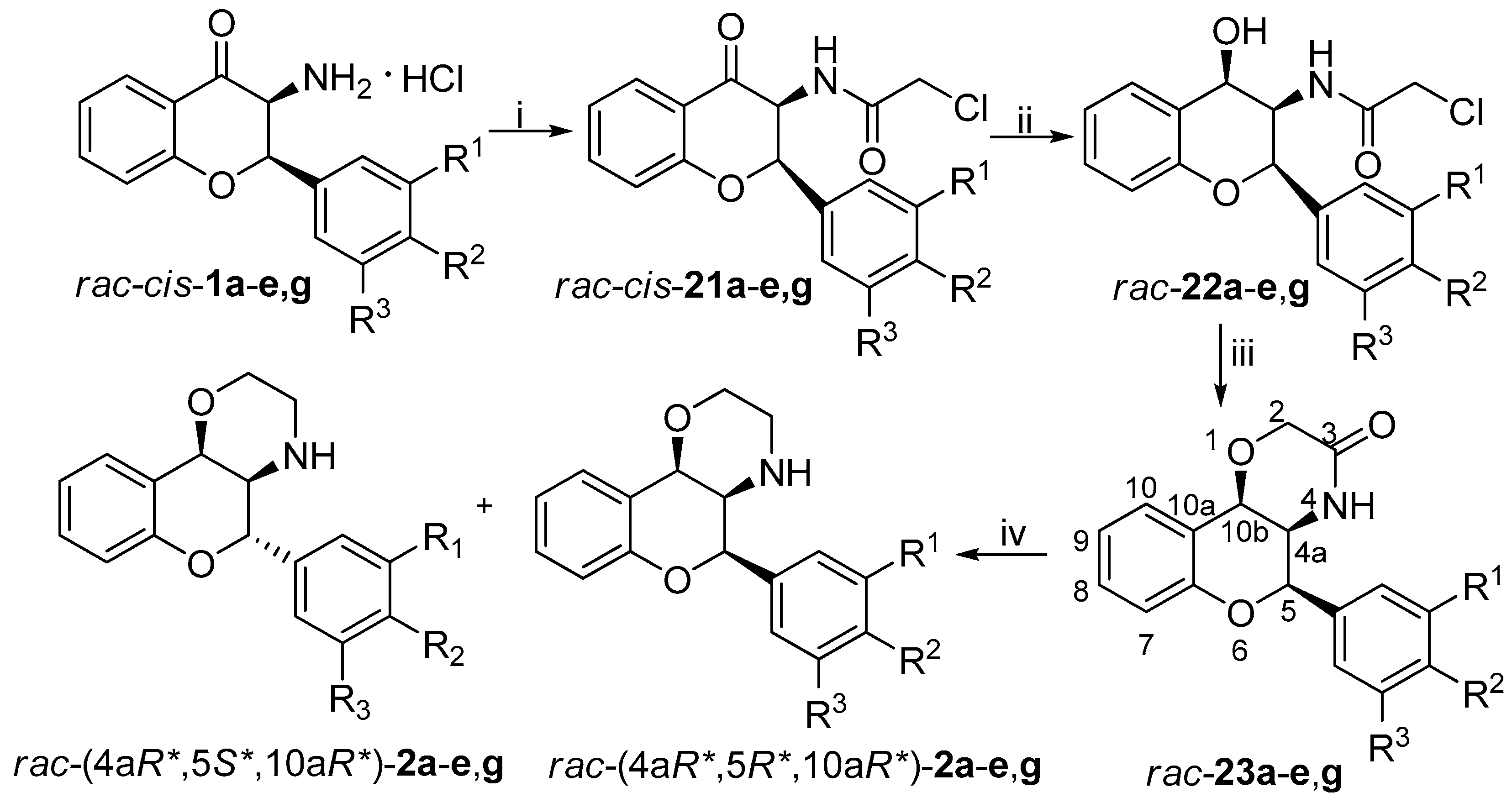{kind=link}
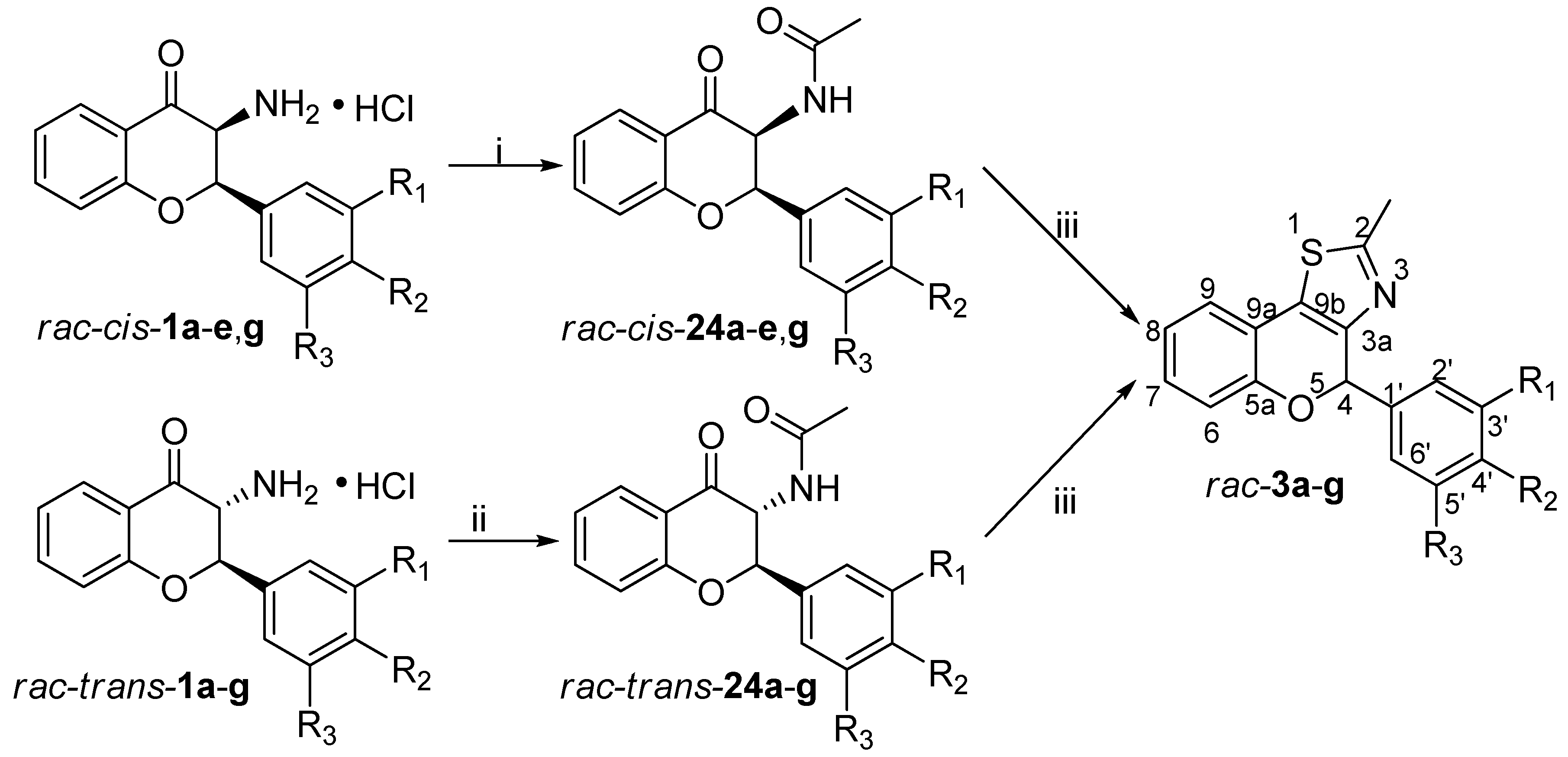{kind=link}
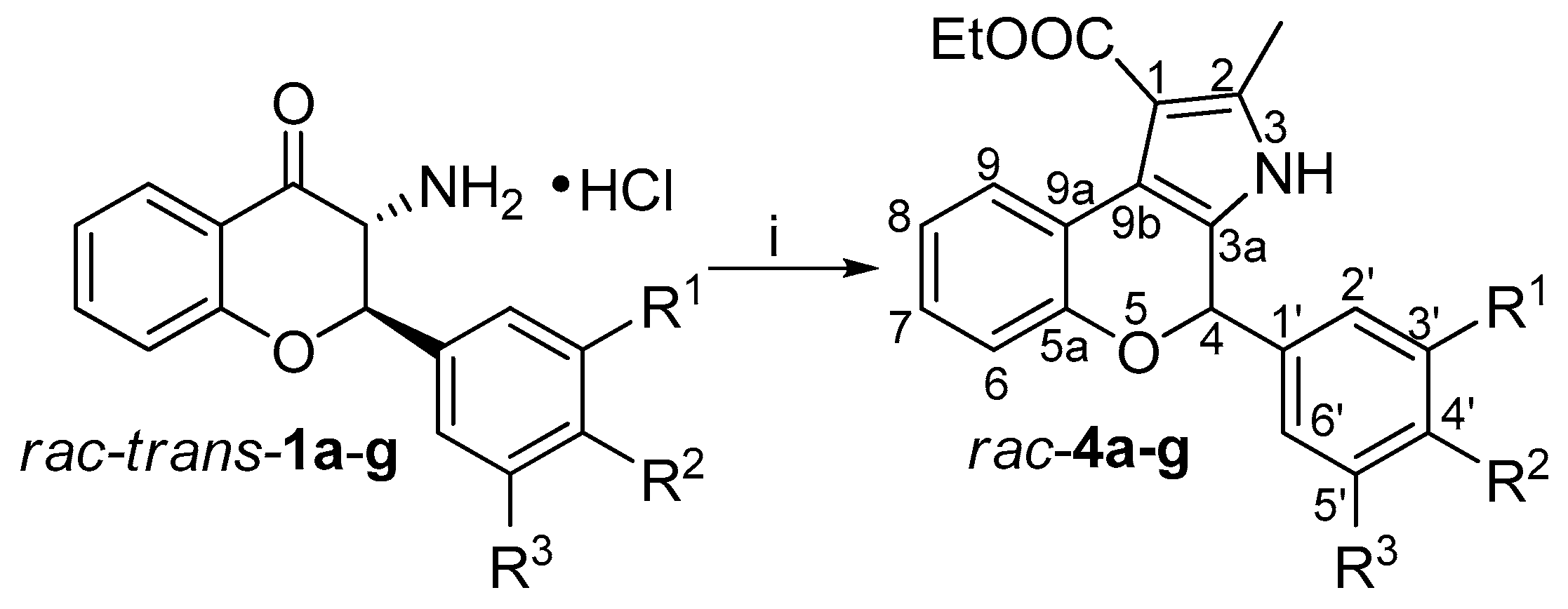{kind=link}
