4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects

 ,
,  , ,
, ,  , ,
, ,  ,
,  , , , ,
, , , ,
Abstract
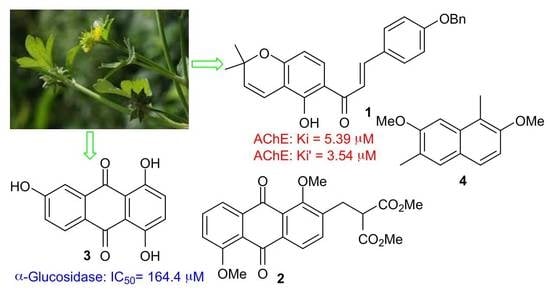
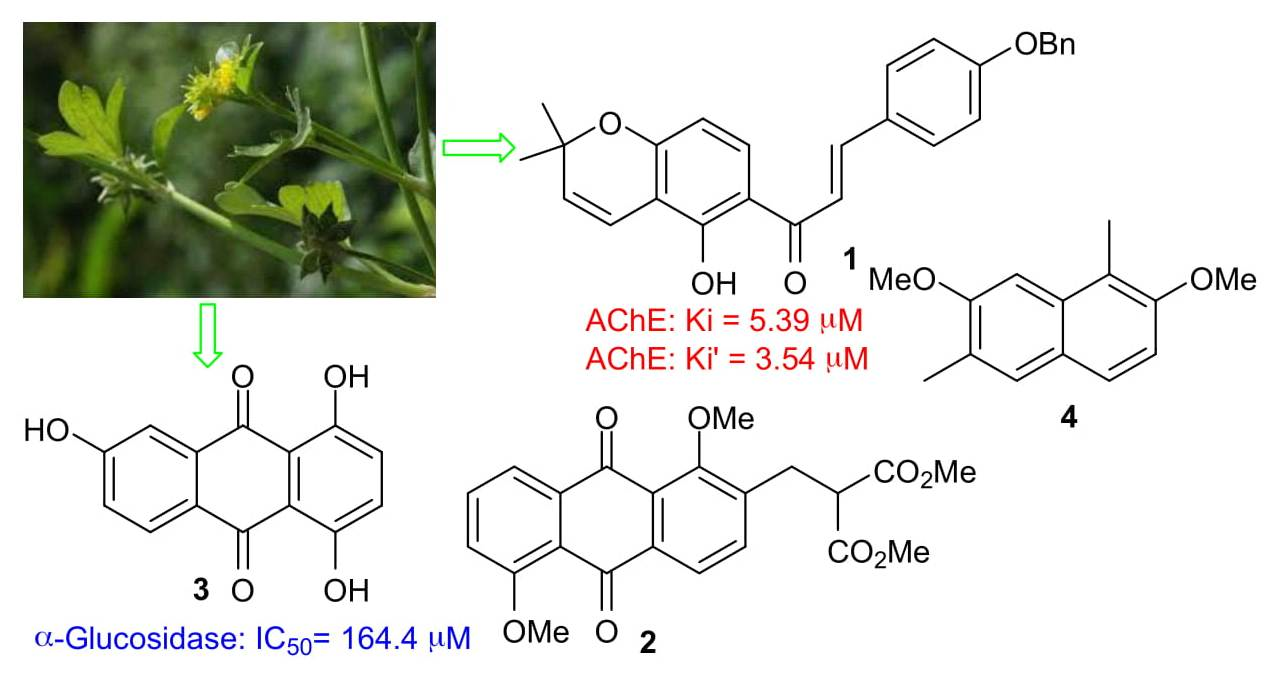
:
1. Introduction
2. Material and Methods
2.1. General Experimental Procedures and Chemicals
2.2. Plant Material
2.3. Extraction and Isolation
2.4. Solutions Preparation for AChE and BChE
2.5. Cholinesterase Assay
2.6. α-Glucosidase Assay
2.7. Sulforhodamine B Assay
2.8. Molecular Docking
3. Results
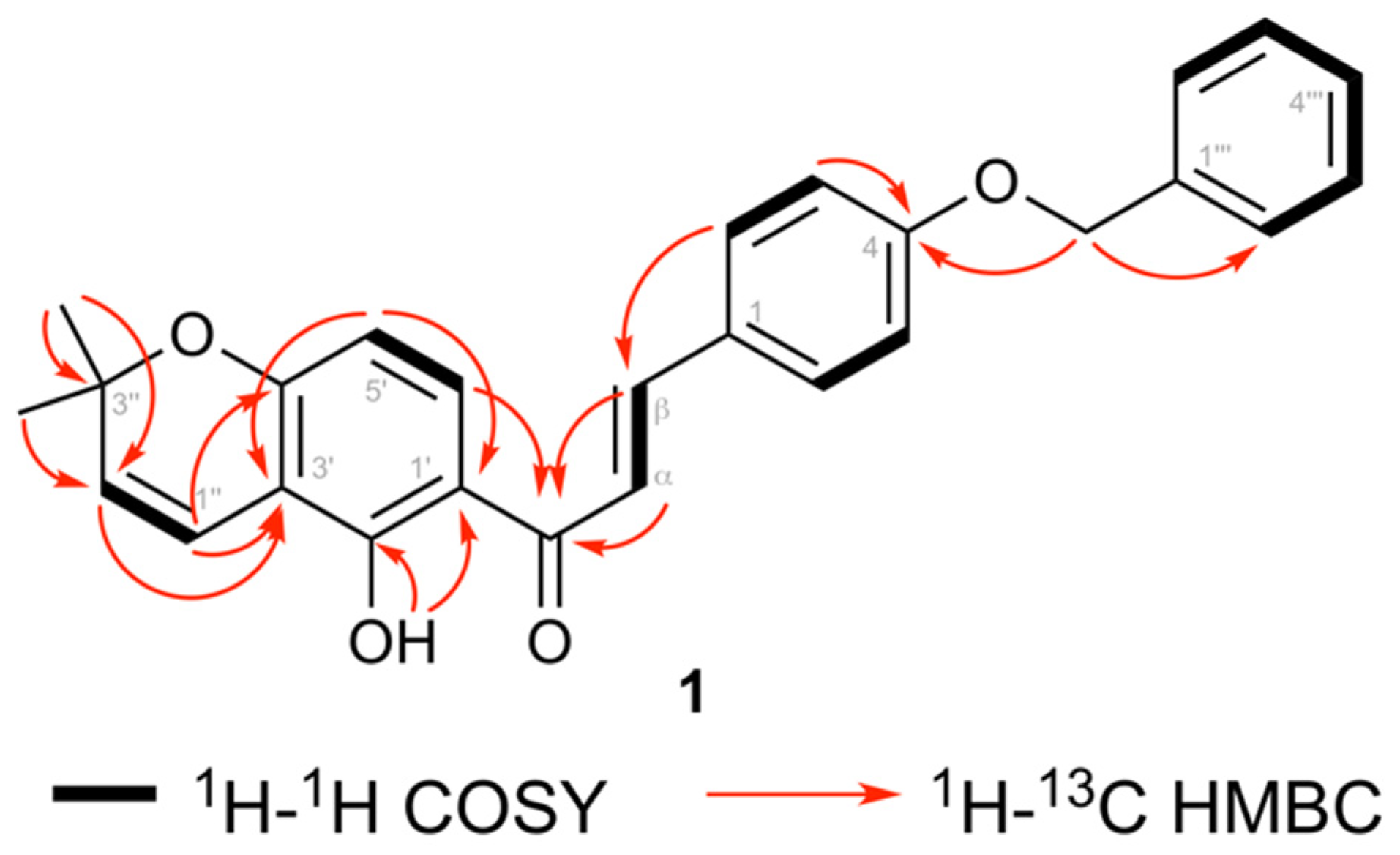
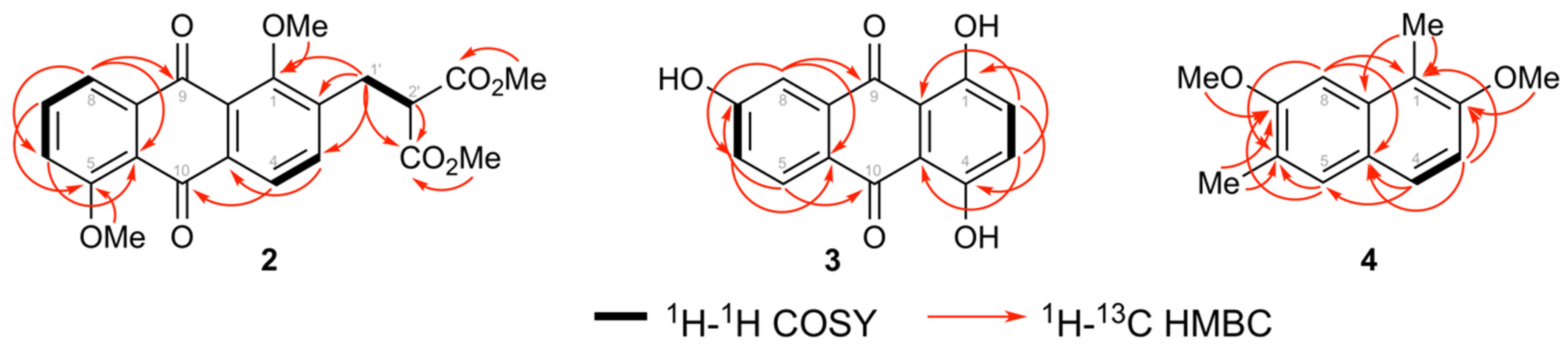
3.1. Structure Elucidation
3.2. Biological Evaluation
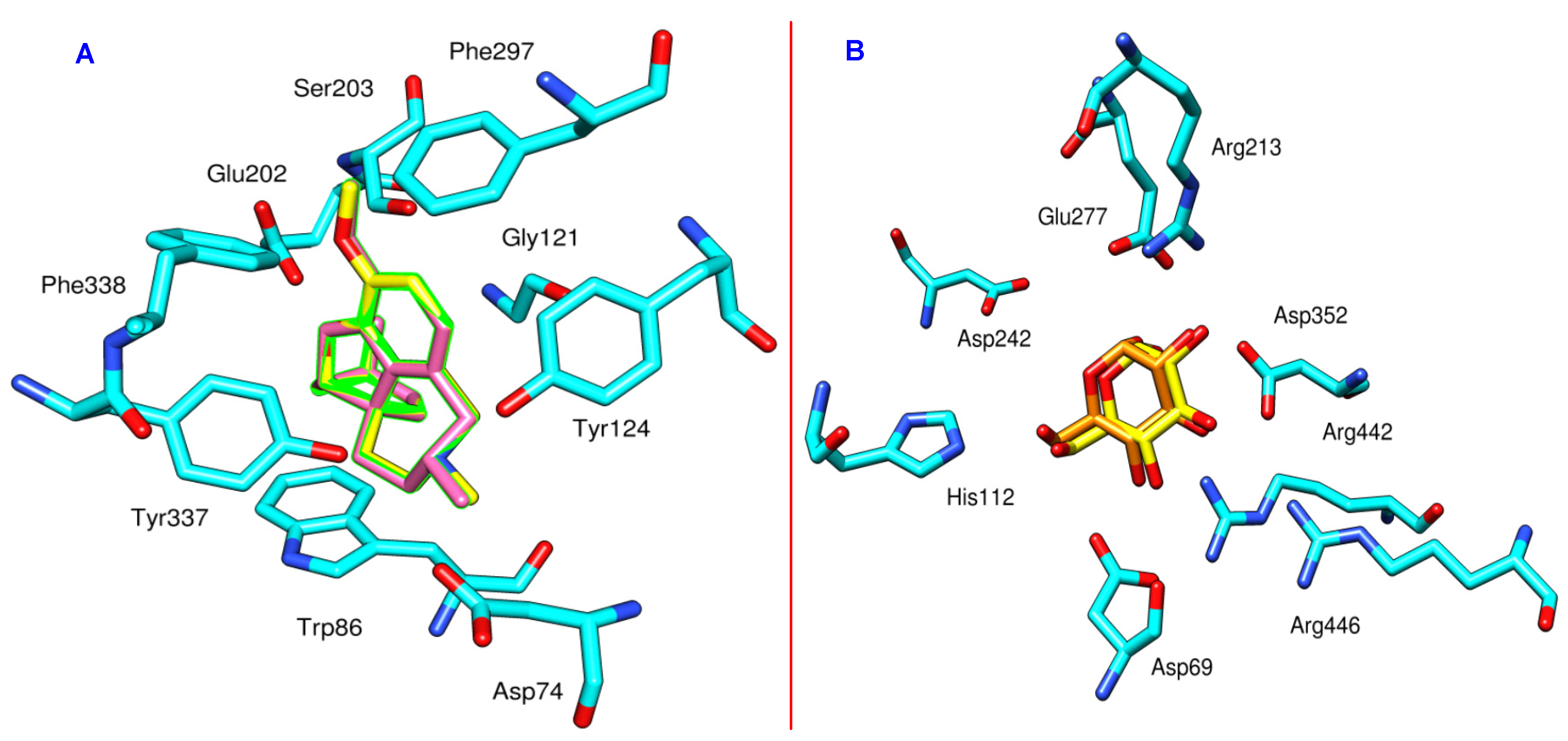
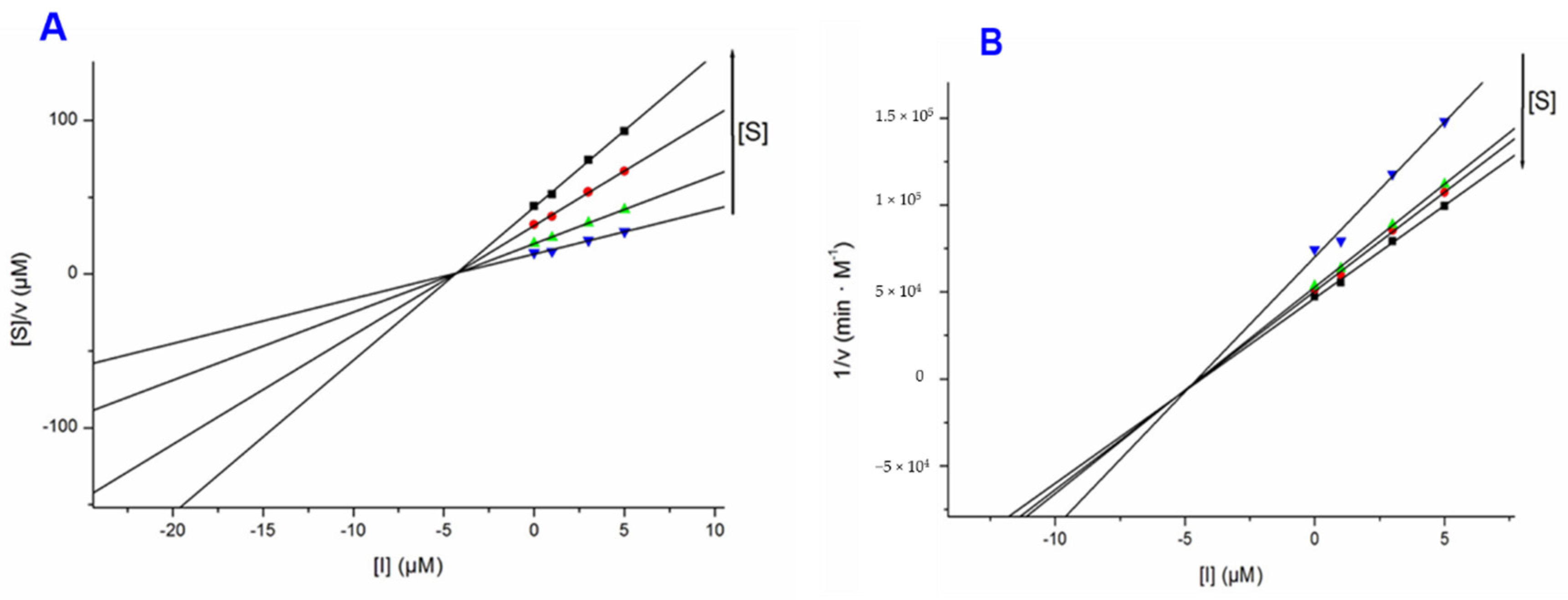
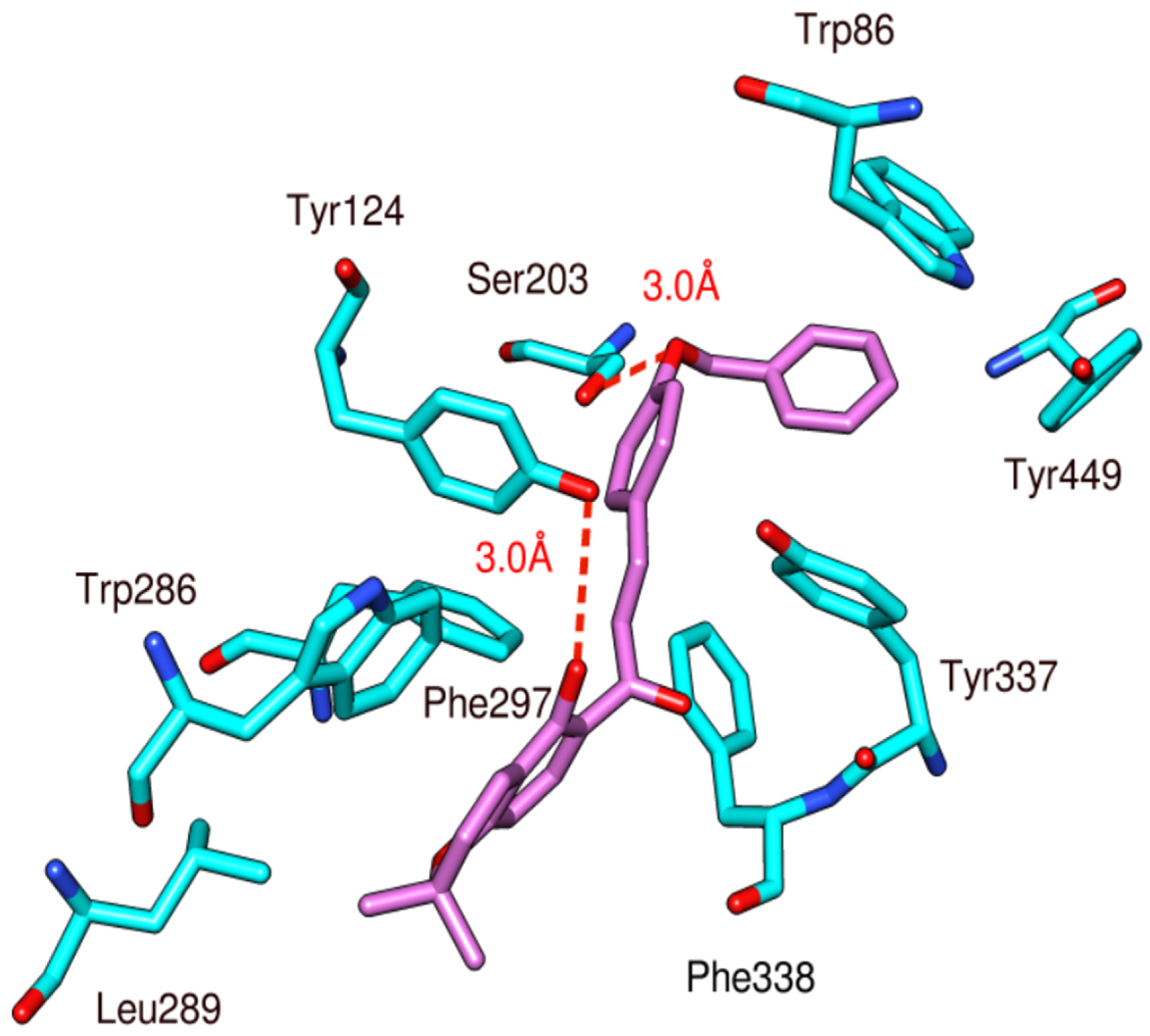
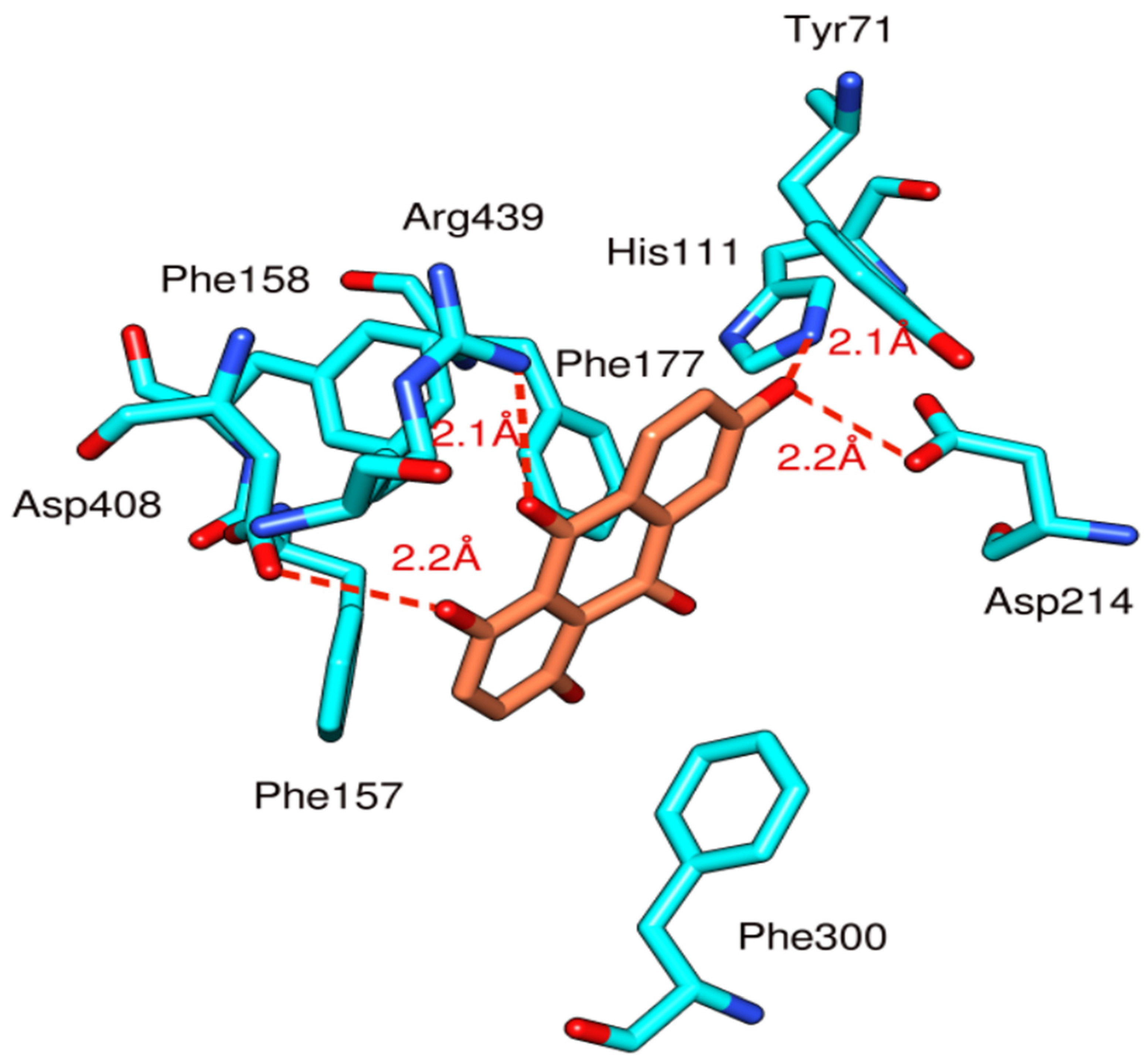
3.3. Docking Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef]
- Clardy, J.; Fischbach, M.A.; Currie, C.R. The natural history of antibiotics. Curr. Biol. 2009, 19, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Giddings, L.-A. Natural products as leads to antitumor drugs. Phytochem. Rev. 2013, 13, 123–137. [Google Scholar] [CrossRef]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Qin, F.; Zhou, G. Studies on chemical constituents of Ranunculus muricatus Linn. Nat. Prod. Res. Dev. 2013, 25, 736–741. [Google Scholar]
- Raziq, N.; Saeed, M.; Ali, M.S.; Lateef, M.; Shahid, M.; Akbar, S.; Zafar, S. Muriolide, a novel antioxidant lactone from Ranunculus muricatus. Nat. Prod. Res. 2020, 30. in print. [Google Scholar] [CrossRef]
- Khan, J.; Khan, R.; Qureshi, R. Ethnobotanical study of commonly used weeds of District Bannu, Khyber Pakhtunkhwa (Pakistan). J. Med. Plant Stud. 2013, 1, 1–6. [Google Scholar]
- Iqbal, H.; Sher, Z.; Khan, Z.U. Medicinal plants from salt range Pind Dadan Khan, district Jhelum, Punjab, Pakistan. J. Med. Plant Res. 2011, 5, 2157–2168. [Google Scholar]
- Ullah, M.; Khan, M.U.; Mahmood, A.; Malik, R.N.; Hussain, M.; Wazir, S.M.; Daud, M.; Shinwari, Z.K. An ethnobotanical survey of indigenous medicinal plants in Wana district South Waziristan agency. Pak. J. Ethnopharmacol. 2013, 150, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Chaudhry, B.A.; Ijaz, H.; Qadir, M.I. Caffeoyl-β-d-glucopyranoside and 1,3-dihydroxy-2-tetracosanoylamino-4-(E)-nonadecene isolated from Ranunculus muricatus exhibit antioxidant activity. Sci. Rep. 2019, 9, 15613. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.U.; Ijaz, F.; Iqbal, Z.; Afzal, A.; Ali, N.; Afzal, M.; Khan, M.A.; Muhammad, S.; Qadir, G.; Asif, M. A novel survey of the ethno medicinal knowledge of dental problems in Manoor valley (Northern Himalaya), Pakistan. J. Ethnopharmacol. 2016, 194, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Ahmad, T.; Mushtaq, M.N.; Malik, M.N.H.; Naz, H.; Ahsan, H.; Asif, H.; Noor, N.; Rahman, M.S.U.; Dar, U.; et al. Phytochemical analysis and cardiotonic activity of methanolic extract of Ranunculus muricatus Linn. In isolated rabbit heart. Acta Pol. Pharm. 2016, 73, 949–954. [Google Scholar]
- Khan, F.A.; Zahoor, M.; Khan, E. Chemical and biological evaluation of Ranunculus muricatus. Pak. J. Pharm. Sci. 2016, 29, 503–510. [Google Scholar]
- Nazir, S.; Tahir, K.; Naz, R.; Khan, Z.; Khan, A.; Islam, R.; Rehman, A.U. In vitro screening of Ranunculus muricatus for potential cytotoxic and antimicrobial activities. Glob. J. Pharmacol. 2014, 8, 427–431. [Google Scholar]
- Lal, S.D.; Yadav, B.K. Folk medicines of Kurukshetra district (Haryana). India. Econ. Bot. 1983, 37, 299–305. [Google Scholar] [CrossRef]
- Wang, A.W.; Wang, M.; Yuan, J.-R.; Tian, J.-K.; Wu, L.-M.; Geng, H. The study on antitumour effects in vitro of different extracts in Radix Ranunculus Ternati. J. Nat. Prod. Res. Dev. 2004, 16, 529–531. [Google Scholar]
- Xie, J.P.; He, Y.; Yue, J.; Hu, C.H.; Wang, H.-H. Identification of differential expression proteins of Mycobacterium tuberculosis strain isolated from clinical species treated with Radix Ranuncoli Ternati Extracts by comparative proteomics. Chin. Biochem. Mol. Biol. 2006, 22, 63–69. [Google Scholar]
- Qasem, J.R. Fungicidal activity of Ranunculus asiaticus and other weeds against Fusarium oxysporum f. sp. lycopersici. Ann. Appl. Biol. 1996, 128, 533–540. [Google Scholar] [CrossRef]
- Ibrar, M.; Samreen, U. Phytochemical screening and evaluation of cytotoxic and phytotoxic effects of Ranunculus muricatus L. Pak. J. Plant Sci. 2012, 18, 35–45. [Google Scholar]
- Aslam, M.S.; Choudhary, B.A.; Uzair, M.; Ijaz, A.S. Phytochemical study of Ariel parts of Ranunculus muricatus for the pharmacological active compounds. J. Appl. Pharm. 2013, 5, 827–832. [Google Scholar]
- Cavdar, H.; Senturk, M.; Guney, M.; Durdagi, S.; Kayik, G.; Supuran, C.T.; Ekinci, D. Inhibition of acetylcholinesterase and butyrylcholinesterase with uracil derivatives: Kinetic and computational studies. J. Enzyme Inhib. Med. Chem. 2019, 34, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, G.; Wu, D.; Yu, Y.; Hu, N.; Wang, H.; Li, X.; Wu, Y. Emerging strategies for the activity assay and inhibitor screening of alpha-glucosidase. Food Funct. 2020, 11, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.S.; Lauridsen, M.B.; Dragsted, L.O.; Nielsen, J.; Staerk, D. Development of a bioassay-coupled HPLC-SPE-ttNMR platform for identification of α-glucosidase inhibitors in apple peel (Malus domestica Borkh). Food Chem. 2012, 135, 1692–1699. [Google Scholar] [CrossRef]
- Shah, S.; Javaid, K.; Zafar, H.; Khan, K.M.; Khalil, R.; Ul-Haq, Z.; Choudhary, M.I. Synthesis, and In vitro and in silico α-glucosidase inhibitory studies of 5-chloro-2-aryl benzo [d] thiazoles. Bioorg. Chem. 2018, 78, 269–279. [Google Scholar] [CrossRef]
- Chemical Computing Group (CCG) Inc. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2019. [Google Scholar]
- Kuete, V.; Noumedem, J.A.K.; Nana, F. Chemistry and Pharmacology of 4-Hydroxylonchocarpin: A Review. Chin. J. Integr. Med. 2013, 19, 475–480. [Google Scholar] [CrossRef]
- Krohn, K.; Farooq, U.; Hussain, H.; Ahmed, I.; Rheinheimer, J.; Draeger, S.; Schulz, B.; van Ree, T. Phomosines H–J, Novel Highly Substituted Biaryl Ethers, Isolated from the Endophytic Fungus Phomopsis sp. from Ligustrum vulgare. Nat. Prod. Commun. 2011, 6, 1907–1912. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Wang, S.; Lin, S.; Zhu, C.; Yue, Z.; Yu, Y.; Liu, B.; Wu, X.; Yang, Y.; Li, Y.; et al. Natural and unnatural anthraquinones isolated from the ethanol extract of the roots of Knoxia valerianoides. Acta Pharm. Sin. B 2012, 2, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Farina, F.; Vega, J.C.; Prados, P. 1,4,6-Trihydroxyantbraquinone. J. Am Chem. Soc. 1918, 40, 404–406. [Google Scholar]
- Farina, F.; Vega, J.C.; Prados, P. Polycyclic hydroxyquinones. X. Synthesis of substituted tetrahydroquinizarins by Diels-Alder reaction with naphthazarin and its diacetate. Quim. Org. Y Bioquim. 1982, 78, 344–453. [Google Scholar]
- Echavarren, A.; Prados, P.; Farina, F. Polycyclic hydroxyquinones. Part 17. Regiospecific Diels-Alder cycloadditions with chloronaphthoquinones as model reactions for regiospecific construction of the A-ring of anthracyclinones. J. Chem. Res. Synop. 1986, 364–365. [Google Scholar] [CrossRef]
- Ng’ang’a, M.M.; Hussain, H.; Chhabra, S.; Langat-Thoruwa, C.; Krohn, K.; Hussain, J.; Al-Harrasi, A.; Green, I. Eucleanal: A newnaphthalene derivative from Euclea divinorum. Nat. Prod. Commun. 2012, 7, 193–194. [Google Scholar]
- Ng’ang’a, M.M.; Hussain, H.; Chhabra, S.; Langat-Thoruwa, C.; Krohn, K.; Hussain, J.; Al-Harrasi, A.; Green, I. Eucleanal A and B: Two newnapthalene derivatives from Euclea divinorum. Chin. Chem. Lett. 2012, 23, 576–578. [Google Scholar] [CrossRef]
- Mahabusarakam, W.; Hemtasin, C.; Chakthong, S.; Voravuthikunchai, S.P.; Olawumi, I.B. Naphthoquinones, Anthraquinones and Naphthalene Derivatives from the Bulbs of Eleutherine americana. Planta Med. 2010, 76, 345–349. [Google Scholar] [CrossRef]
- Lin, C.N.; Lu, C.M.; Lin, H.C.; Ko, F.N.; Teng, C.M. Novel antiplatelet naphthalene from Rhamnus nakaharai. J. Nat. Prod. 1995, 58, 1934–1940. [Google Scholar] [CrossRef]
- Ganapaty, S.; Thomas, P.S.; Karagianis, G.; Waterman, P.G.; Brun, R. Antiprotozoal and cytotoxic naphthalene derivatives from Diospyros assimilis. Phytochemistry 2006, 67, 1950–1956. [Google Scholar] [CrossRef]
- Ngadjui, B.T.; Kapche, G.W.; Tamboue, H.; Abegaz, B.M.; Connolly, J.D. Prenylated flavonoids and a dihydro-4-phenylcoumarinfrom Dorstenia poinsettifolia. Phytochemistry 1999, 51, 119–123. [Google Scholar] [CrossRef]
- Habib, M.R.; Nikkon, F.; Rahman, M.; Haque, M.E.; Karim, M.R. Isolation of stigmasterol and beta-sitosterol from methanolic extract of root bark of Calotropis gigantea (Linn). Pak. J. Biol. Sci. 2007, 10, 4174–4176. [Google Scholar]
- Seo, S.; Tomita, Y.; Tori, K.; Yoshimura, Y. Determination of the absolute configuration of a secondary hydroxy group in a chiral secondary alcohol using glycosidation shifts in carbon-13 nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1978, 100, 3331–3339. [Google Scholar] [CrossRef]
- Burmaoglu, S.; Yilmaz, A.O.; Polat, M.F.; Kaya, R.; Gulcin, İ.; Algul, O. Synthesis and biological evaluation of novel tris-chalcones as potent carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and α-glycosidase inhibitors. Bioorg. Chem. 2019, 85, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Aslan, H.E.; Demir, Y.; Özaslan, M.S.; Türkan, F.; Beydemir, Ş.; Küfrevioğlu, Ö.I. The behavior of some chalconeson acetylcholinesterase and carbonic anhydrase activity. Drug Chem. Toxicol. 2019, 42, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Liua, H.R.; Liua, X.J.; Fana, H.Q.; Tanga, J.J.; Gaob, X.H.; Liu, W.K. Design, synthesis and pharmacological evaluation of chalconederivatives as acetylcholinesterase inhibitors. Bioorg. Med. Chem. 2014, 22, 6124–6133. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Rangarajan, T.M.; Chaudhary, R.; Singh 2, R.P.; Singh, M.; Singh 3, R.P.; Tondo 6, A.R.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Novel Class of Chalcone Oxime Ethers as Potent Monoamine Oxidase-B and Acetylcholinesterase Inhibitors. Molecules 2020, 25, 2356. [Google Scholar] [CrossRef] [PubMed]
- Sribuhoma, T.; Saraphona, C.; Decharchoocharta, P.; Boonyaratb, C.; Yenjai, C. Acetylcholinesterase inhibition and cytotoxicity of flavonoids and chalcones from Derris indica. ScienceAsia 2016, 42, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Fosso, M.Y.; LeVine, H., 3rd; Green, K.D.; Tsodikov, O.V.; Garneau-Tsodikov, S. Effects of structural modifications on the metal binding, anti-amyloid activity, and cholinesterase inhibitory activity of chalcones. Org. Biomol. Chem. 2015, 13, 9418–9426. [Google Scholar] [CrossRef]
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef]
- Mohd, S.; Rizvi, D.; Shaikh, S.; Naaz, D.; Shakil, S.; Ahmad, A.; Haneef, M.; Abuzenada, A.M. Kinetics and Molecular Docking Study of an Anti-diabetic Drug Glimepiride as Acetylcholinesterase Inhibitor: Implication for Alzheimer’s Disease-Diabetes Dual Therapy. Neurochem. Res. 2016, 41, 1475–1482. [Google Scholar]
- Forstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef]
- Hitzeman, N. Cholinesterase inhibitors for Alzheimer’s disease. Am. Fam. Physician. 2006, 74, 747–759. [Google Scholar] [PubMed]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N. Drug development for Alzheimer’s disease: Where are we now and where are we headed. Am. J. Geriatr. Pharmacother. 2009, 7, 167–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowarah, J.; Singh, V.P. Anti-diabetic drugs recent approaches and advancements. Bioorg. Med. Chem. 2020, 28, 115263. [Google Scholar] [CrossRef]
- Jörgens, V.; Grüsser, M. Happy Birthday, Claude Bernard. Diabetes 2013, 62, 2181–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, H.; Abbas, G.; Green, I.R.; Ali, I. Dipeptidyl peptidase IV inhibitors as a potential target for diabetes: Patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 535–553. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; Wiley: Hobokin, NJ, USA, 2013. [Google Scholar]
- Chiasson, J.L. Acarbose for the prevention of diabetes, hypertension, and cardiovascular disease in subjects with impaired glucose tolerance: The Study to Prevent Non-Insulin-Dependent Diabetes Mellitus (STOP-NIDDM) Trial. Endocr. Pract. 2006, 12, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, F.A.; Lucassen, P.L.; Akkermans, R.P.; van de Lisdonk, E.H.; Rutten, G.E.; van Weel, C. α-Glucosidase Inhibitors for Patients With Type 2 Diabetes. Diabetes Care 2005, 28, 154–163. [Google Scholar]
- Abbas, G.; Hassan, Z.; Al-Harrasi, A.; Muhammad, S.A.; Al-Quraini, A.J.; Al-Maani, Z.K.; Al-Adawai, A.M. Synthesis, molecular docking, and pharmacological evaluation of halobenzodithiophene derivatives against alpha-glucosidase, urease, and free radical production. Turk. J. Chem. 2018, 42, 1113–1123. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | ||||
|---|---|---|---|---|---|
| No | δH (nH, Multiplicity, J in Hz) | δC | No | δH (nH, Multiplicity, J in Hz) | δC |
| 1′ | - | 109.4 | 1 | - | 158.7 |
| 2′ | - | 160.9 | 2 | - | 138.4 |
| 3′ | - | 114.1 | 3 | 7.60 (1H, d, J = 8.0 Hz) | 136.4 |
| 4′ | - | 159.6 | 4 | 8.00 (1H, d, J = 8.0 Hz) | 123.2 |
| 5′ | 6.37 (1H, d, J = 9.0 Hz) | 108.1 | 4a | - | 137.0 |
| 6′ | 7.72 (1H, d, J = 9.0 Hz) | 130.5 | 5 | - | 159.8 |
| α | 7.45 (1H, d, J = 16 Hz, 1H) | 127.8 | 6 | 7.30 (1H, dd, J = 8.0, 2.0 Hz) | 117.1 |
| β | 7.86 (1H, d, J = 16.0 Hz, 1H) | 144.0 | 7 | 7.70 (1H, t, J = 8.0 Hz) | 135.0 |
| keto | - | 191.9 | 8 | 7.90 (1H, dd, J = 8.0, 2.0 Hz) | 119.6 |
| OH-2′ | 13.77 | 8a | - | 137.0 | |
| 1 | - | 128.0 | 9 | - | 182.7 |
| 2,6 | 7.60 (2H, BB′) | 130.3 | 9a | - | 124.8 |
| 3,5 | 7.00 (2H, AA′) | 115.3 | 10 | - | 182.1 |
| 4 | - | 160.9 | 10a | - | 120.9 |
| 1′′ | 6.76 (1H, d, J = 10 Hz) | 115.9 | 1′ | 3.34 (1H, d, J = 8.0 Hz) | 30.1 |
| 2′′ | 5.59 (1H, d, J = 10 Hz) | 127.4 | 2′ | 3.90 (1H, t, J = 8.0 Hz) | 51.3 |
| 3′′ | - | 77.7 | CO2Me | 3.70 (6H, s) | 52.6 |
| 4′′,5′′ | 1.47 (6H, s) | 28.3 | CO2Me | - | 169.0 |
| 1′′′ | - | 136.3 | 1-OMe | 3.97 (3H, s) | 62.0 |
| 2′′′–6′′′ | 7.46 (2H, m) | 127.4 | 5-OMe | 4.04 (3H, s) | 56.3 |
| 3′′′,5′′′ | 7.40 (2H, m) | 128.6 | |||
| 4′′′ | 7.32 (1H, m) | 127.8 | |||
| CH2Bn | 5.12 (2H, s) | 70.1 |
| 3 | 4 | ||||
|---|---|---|---|---|---|
| No | δH (nH, Multiplicity, J in Hz) | δC | No | δH (nH, Multiplicity, J in Hz) | δC |
| 1 | - | 156.4 | 1 | - | 117.9 |
| 2 | 7.28 (1H, m) | 128.3 | 2 | - | 154.2 |
| 3 | 7.28 (1H, m) | 129.2 | 3 | 7.09 (1H, d, J = 8.0 Hz) | 111.0 |
| 4 | 156.5 | 4 | 7.56 (1H, d, J = 8.0 Hz) | 125.9 | |
| 4a | 112.0 | 4a | 124.3 | ||
| 5 | 7.98 (1H, d, J = 8.0 Hz) | 129.5 | 5 | 7.49 (1H, s) | 129.1 |
| 6 | 7.20 (1H, dd, J = 8.0, 2.0 Hz) | 121.8 | 6 | - | 125.6 |
| 7 | 163.6 | 7 | - | 157.2 | |
| 8 | 7.40 (1H, d, J = 2.0 Hz) | 112.1 | 8 | 7.08 (1H, s) | 100.3 |
| 8a | - | 134.8 | 8a | - | 133.5 |
| 9 | - | 186.3 | 1-Me | 2.50 (3H, s) | 10.7 |
| 9a | - | 112.5 | 6-Me | 2.33 (3H, s) | 16.5 |
| 10 | - | 185.3 | 7-OMe | 3.95 (3H, s) | 55.1 |
| 10a | - | 124.3 | 2-OMe | 3.91 (3H, s) | 56.6 |
| OH-1 | 12.53 (1H, s) | - | |||
| OH-4 | 12.84 (1H, s) | - | |||
| OH-7 | 11.18 (1H, s) | - |
| AChE | BChE | α-Glucosidase | |||
|---|---|---|---|---|---|
| Compound | % Inhibition a | Ki (µM) (Ki’ (µM)) | % Inhibition a | % Inhibition a | IC50 (µM) |
| 1 | 97.4 | 5.39 ± 0.51 (3.54 ± 0.24) | 27.3 | n.d. | n.d. |
| 2 | n.d. | n.d. | n.d. | 13.4 | n.d. |
| 3 | n.d. | n.d. | n.d. | 80.4 | 164.46 ± 83.04 |
| 4 | n.d. | n.d. | n.d. | n.d. | n.d. |
| 5 | 49.9 | n.d. | 8.7 | 36.1 | n.d. |
| GH | 90.5 | 0.54 | 54.8 | Acarbose | 1072.5 ± 453.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, H.; Ali, I.; Wang, D.; Mamadalieva, N.Z.; Hussain, W.; Csuk, R.; Loesche, A.; Fischer, L.; Staerk, D.; Anam, S.; et al. 4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects. Biomolecules 2020, 10, 1562. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10111562
Hussain H, Ali I, Wang D, Mamadalieva NZ, Hussain W, Csuk R, Loesche A, Fischer L, Staerk D, Anam S, et al. 4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects. Biomolecules. 2020; 10(11):1562. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10111562
Chicago/Turabian StyleHussain, Hidayat, Iftikhar Ali, Daijie Wang, Nilufar Z. Mamadalieva, Wahid Hussain, René Csuk, Anne Loesche, Lucie Fischer, Dan Staerk, Syariful Anam, and et al. 2020. "4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects" Biomolecules 10, no. 11: 1562. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10111562