Connexin 43 Deficiency Is Associated with Reduced Myocardial Scar Size and Attenuated TGFβ1 Signaling after Transient Coronary Occlusion in Conditional Knock-Out Mice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Protocol
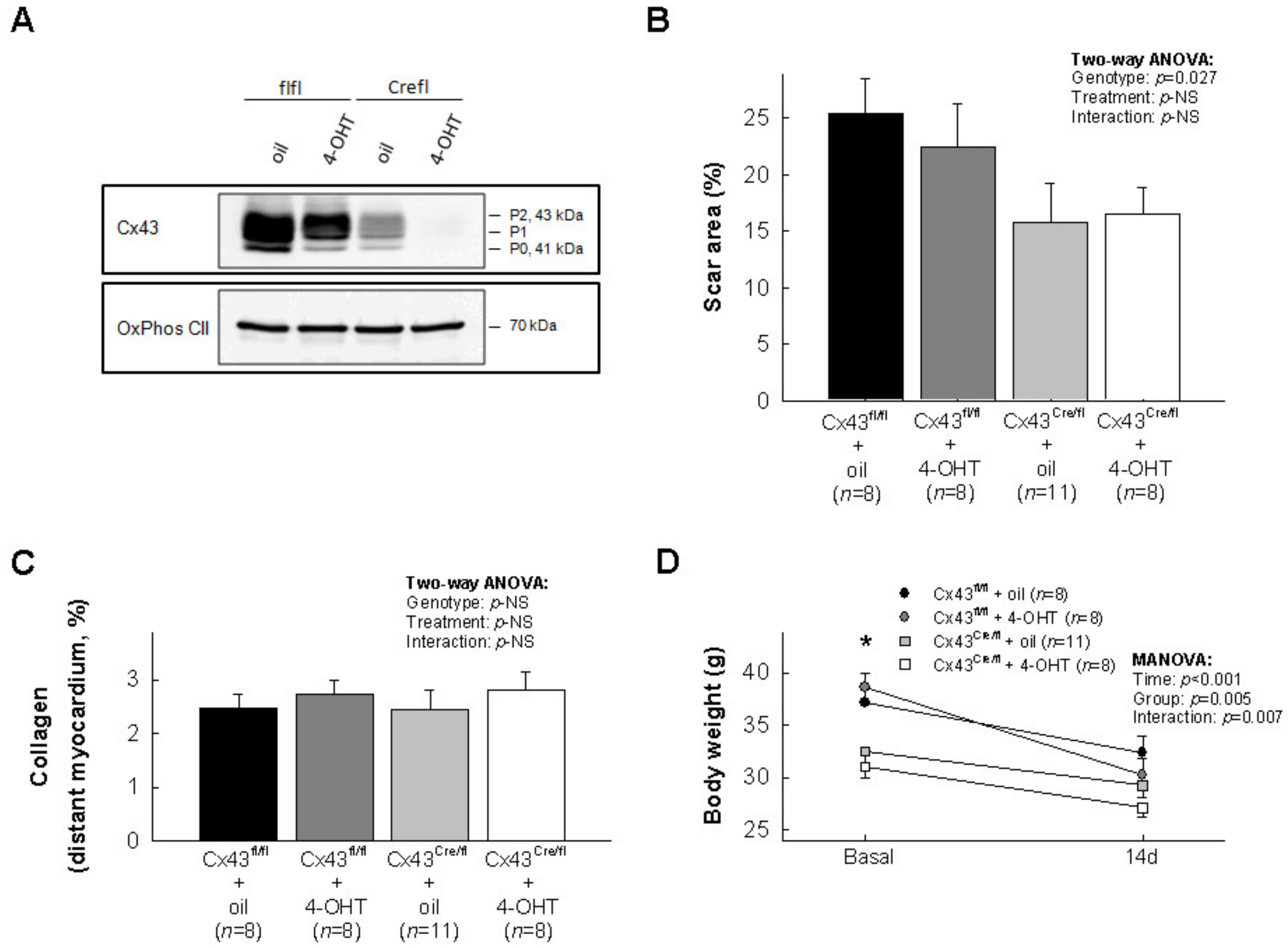
2.3. Myocardial Scar Size
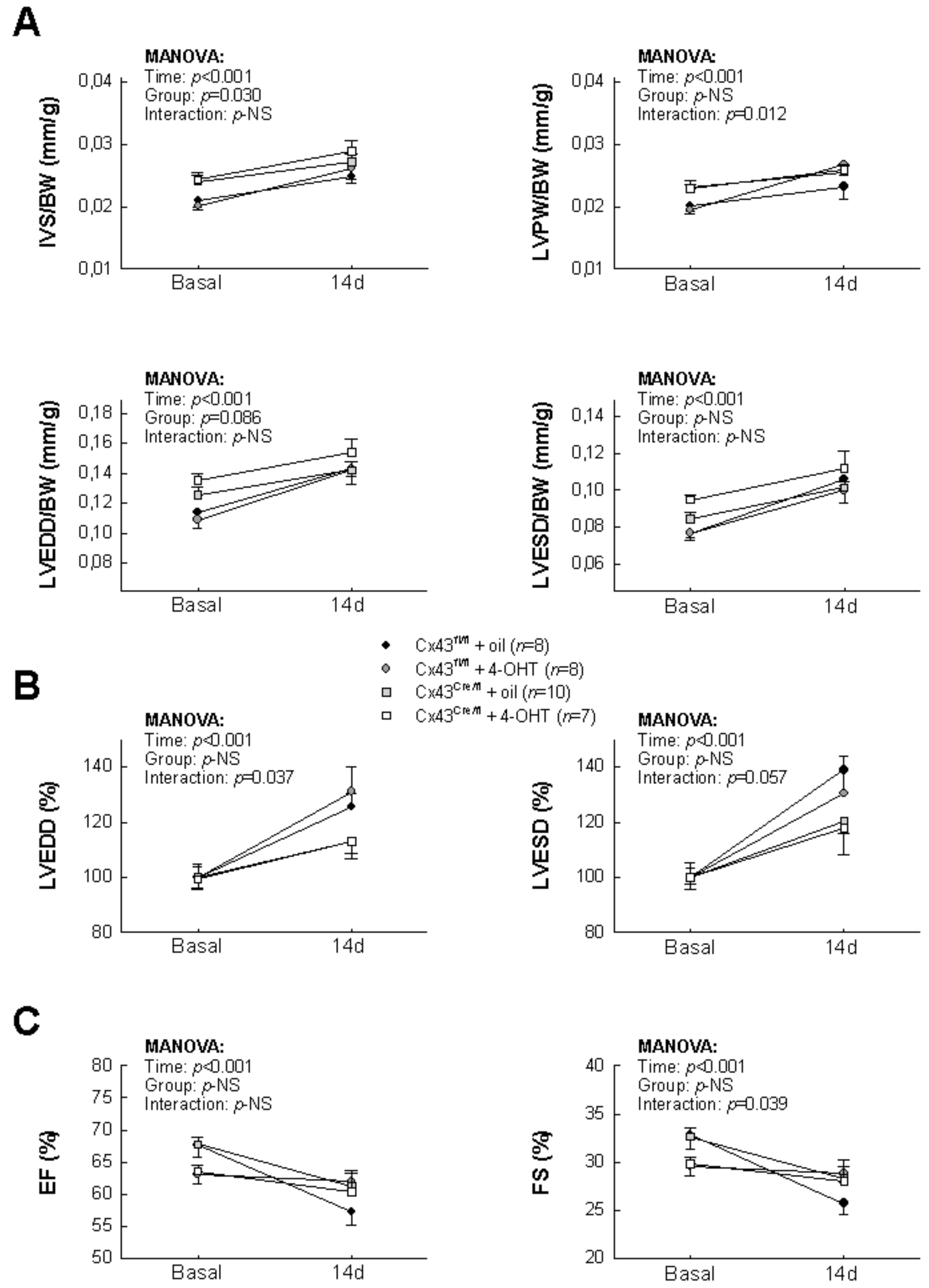
2.4. Transthoracic Echocardiography
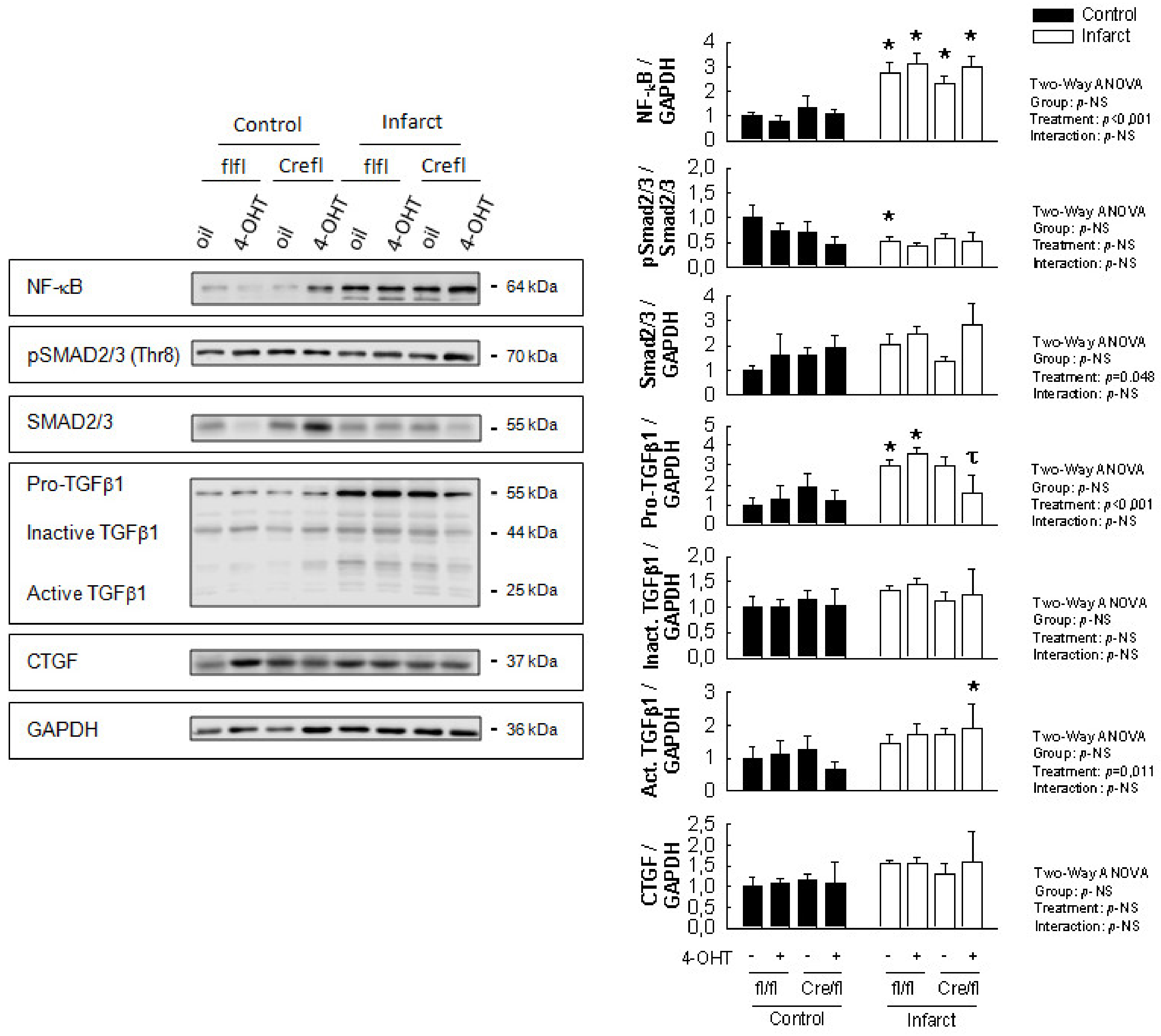
2.5. Western Blot
2.6. Statistics
3. Results
3.1. Effects of Cx43 Deficiency on Myocardial Scar Size after Transient Coronary Occlusion
3.2. Effects of Cx43 Deficiency on Post-Infartion Left Ventricular Remodeling
3.3. Changes in Expression of Selected Fibrotic and Remodeling Markers 14 Days after Transient Coronary Occlusion
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lambiase, P.D.; Tinker, A. Connexins in the heart. Cell Tissue Res. 2014, 360, 675–684. [Google Scholar] [CrossRef]
- Sosinsky, G.E.; Nicholson, B. Structural organization of gap junction channels. Biochimica et Biophysica Acta (BBA) Biomembranes 2005, 1711, 99–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kléber, A.G.; Saffitz, J.E. Role of the intercalated disc in cardiac propagation and arrhythmogenesis. Front. Physiol. 2014, 5, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Dorado, D.; Rodriguez-Sinovas, A.; Ruiz-Meana, M. Gap junction-mediated spread of cell injury and death during myocardial ischemia–reperfusion. Cardiovasc. Res. 2004, 61, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorado, D.; Inserte, J.; Ruiz-Meana, M.; Gonzaález, M.A.; Solares, J.; Juliá, M.; Barrabés, J.A.; Soler-Soler, J. Gap Junction Uncoupler Heptanol Prevents Cell-to-Cell Progression of Hypercontracture and Limits Necrosis During Myocardial Reperfusion. Circulation 1997, 96, 3579–3586. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Garcia-Dorado, D.; Hofstaetter, B.; Piper, H.M.; Soler-Soler, J. Propagation of cardiomyocyte hypercontracture by passage of Na(+) through gap junctions. Circ. Res. 1999, 85, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Sinovas, A.; García-Dorado, D.; Ruiz-Meana, M.; Soler-Soler, J. Enhanced effect of gap junction uncouplers on macroscopic electrical properties of reperfused myocardium. J. Physiol. 2004, 559, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Sinovas, A.; García-Dorado, D.; Ruiz-Meana, M.; Soler-Soler, J. Protective effect of gap junction uncouplers given during hypoxia against reoxygenation injury in isolated rat hearts. Am. J. Physiol. Circ. Physiol. 2006, 290, H648–H656. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Sinovas, A.; Sánchez, J.A.; González-Loyola, A.; Barba, I.; Morente, M.; Aguilar, R.; Agulló, E.; Miró-Casas, E.; Esquerda, N.; Ruiz-Meana, M.; et al. Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischaemia and preconditioning protection. J. Physiol. 2010, 588, 1139–1151. [Google Scholar] [CrossRef]
- Sánchez, J.A.; Rodriguez-Sinovas, A.; Barba, I.; Miró-Casas, E.; Fernández-Sanz, C.; Ruiz-Meana, M.; Alburquerque-Béjar, J.J.; García-Dorado, D. Activation of RISK and SAFE pathways is not involved in the effects of Cx43 deficiency on tolerance to ischemia–reperfusion injury and preconditioning protection. Basic Res. Cardiol. 2013, 108, 351. [Google Scholar] [CrossRef]
- Bhatt, A.S.; Ambrosy, A.P.; Velazquez, E.J. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Curr. Cardiol. Rep. 2017, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Kovacs, A.; Kanter, E.M.; Yamada, K.A. Reduced expression of Cx43 attenuates ventricular remodeling after myocardial infarction via impaired TGF-beta signaling. Am. J. Physiol. Circ. Physiol. 2009, 298, H477–H487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, S.; Kovacs, A.; Yamada, K.A.; Saffitz, J.E. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J. Am. Coll. Cardiol. 2003, 41, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Nofi, C.; Bogatyryov, Y.; Dedkov, E. Preservation of Functional Microvascular Bed Is Vital for Long-Term Survival of Cardiac Myocytes Within Large Transmural Post-Myocardial Infarction Scar. J. Histochem. Cytochem. 2017, 66, 99–120. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, Y.; Hu, C.; Du, Y.; Liu, Y.; Liu, J.; Zhang, J.; Cheng, G.; Han, H.; Zhao, Q.; et al. Significant association between serum resistin and hypersensitive troponin I levels in patients with a first ST-segment elevation myocardial infarction. Nat. Rev. Dis. Prim. 2019, 5, 39. [Google Scholar] [CrossRef]
- Kristensen, S.D.; Laut, K.G.; Fajadet, J.; Kaifoszova, Z.; Kala, P.; Di Mario, C.; Wijns, W.; Clemmensen, P.; Agladze, V.; Antoniades, L.; et al. Reperfusion therapy for ST elevation acute myocardial infarction 2010/2011: Current status in 37 ESC countries. Eur. Heart J. 2014, 35, 1957–1970. [Google Scholar] [CrossRef] [Green Version]
- Eckardt, D. Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J. Mol. Cell. Cardiol. 2004, 36, 101–110. [Google Scholar] [CrossRef]
- Van Rijen, H.V.; Eckardt, M.; Degen, J.; Theis, M.; Ott, T.; Willecke, K.; Jongsma, H.J.; Opthof, T.; De Bakker, J.M. Slow Conduction and Enhanced Anisotropy Increase the Propensity for Ventricular Tachyarrhythmias in Adult Mice with Induced Deletion of Connexin43. Circulation 2004, 109, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, J.A.; Rodriguez-Sinovas, A.; Fernández-Sanz, C.; Ruiz-Meana, M.; Garcia-Dorado, D. Effects of a reduction in the number of gap junction channels or in their conductance on ischemia-reperfusion arrhythmias in isolated mouse hearts. Am. J. Physiol. Circ. Physiol. 2011, 301, H2442–H2453. [Google Scholar] [CrossRef] [Green Version]
- Shekarforoush, S.; Fatahi, Z.; Safari, F. The effects of pentobarbital, ketamine–pentobarbital and ketamine–xylazine anesthesia in a rat myocardial ischemic reperfusion injury model. Lab. Anim. 2015, 50, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poncelas, M.; Inserte, J.; Aluja, D.; Hernando, V.; Vilardosa, Ú.; García-Dorado, D. Delayed, oral pharmacological inhibition of calpains attenuates adverse post-infarction remodelling. Cardiovasc. Res. 2017, 113, 950–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; García-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, F.L.; Wu, C.O.; Tian, X.; O’Connor, C.; Rich, M.W.; Burg, M.M.; Sheps, D.; Raczynski, J.; Somers, V.K.; Jaffe, A.S. Weight Change after Myocardial Infarction—the Enhancing Recovery in Coronary Heart Disease patients (ENRICHD) Experience. Am. Hear. J. 2008, 155, 478–484. [Google Scholar] [CrossRef] [Green Version]
- Valls-Lacalle, L.; Negre-Pujol, C.; Rodríguez, C.; Varona, S.; Valera-Cañellas, A.; Consegal, M.; Martínez-González, J.; Rodríguez-Sinovas, A. Opposite Effects of Moderate and Extreme Cx43 Deficiency in Conditional Cx43-Deficient Mice on Angiotensin II-Induced Cardiac Fibrosis. Cells 2019, 8, 1299. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Coutinho, P.; Frank, S.; Franke, S.; Law, L.-Y.; Martin, P.; Green, C.; Becker, D. Targeting connexin43 expression accelerates the rate of wound repair. Curr. Boil. 2003, 13, 1697–1703. [Google Scholar] [CrossRef] [Green Version]
- Ongstad, E.L.; O’Quinn, M.P.; Ghatnekar, G.S.; Yost, M.J.; Gourdie, R.G. A Connexin43 Mimetic Peptide Promotes Regenerative Healing and Improves Mechanical Properties in Skin and Heart. Adv. Wound Care 2013, 2, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef] [Green Version]
- Gilmartin, D.J.; Soon, A.; Thrasivoulou, C.; Phillips, A.R.J.; Jayasinghe, S.; Becker, D.L. Sustained Release of Cx43 Antisense Oligodeoxynucleotides from Coated Collagen Scaffolds Promotes Wound Healing. Adv. Heal. Mater. 2016, 5, 1786–1799. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, J.; Ghatnekar, G.; Grek, C.L.; Moyer, K.; Gourdie, R.G. Connexin 43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [Green Version]
- Elbadawy, H.; Mirabelli, P.; Xeroudaki, M.; Parekh, M.; Bertolin, M.; Breda, C.; Cagini, C.; Ponzin, D.; Lagali, N.; Ferrari, S. Effect of connexin 43 inhibition by the mimetic peptide Gap27 on corneal wound healing, inflammation and neovascularization. Br. J. Pharmacol. 2016, 173, 2880–2893. [Google Scholar] [CrossRef] [PubMed]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A Peptide Mimetic of the Connexin43 Carboxyl-Terminus Reduces Gap Junction Remodeling and Induced Arrhythmia Following Ventricular Injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, J.A.; Van Veen, T.A.; De Jong, S.; Van Der Nagel, R.; Van Stuijvenberg, L.; Driessen, H.; Labzowski, R.; Oefner, C.M.; Bosch, A.A.; Nguyen, T.Q.; et al. Reduced Cx43 Expression Triggers Increased Fibrosis Due to Enhanced Fibroblast Activity. Circ. Arrhythmia Electrophysiol. 2012, 5, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Leask, A. Getting to the Heart of the Matter. Circ. Res. 2015, 116, 1269–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Rodriguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-β signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horbelt, D.; Denkis, A.; Knaus, P. A portrait of Transforming Growth Factor beta superfamily signalling: Background matters. Int. J. Biochem. Cell Biol. 2012, 44, 469–474. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| IVS | LVPW | LVEDD | LVESD | |||||
|---|---|---|---|---|---|---|---|---|
| Baseline | 14d | Baseline | 14d | Baseline | 14d | Baseline | 14d | |
| Cx43fl/fl + oil | 0.77 ± 0.02 | 0.80 ± 0.03 | 0.74 ± 0.02 | 0.73 ± 0.04 | 4.21 ± 0.16 | 4.59 ± 0.20 * | 2.82 ± 0.09 | 3.42 ± 0.19 * |
| (n = 8) | ||||||||
| Cx43fl/fl + 4-OHT | 0.77 ± 0.01 | 0.77 ± 0.02 | 0.75 ± 0.03 | 0.79 ± 0.01 | 4.17 ± 0.11 | 4.23 ± 0.17 | 2.93 ± 0.08 | 2.97 ± 0.15 |
| (n = 8) | ||||||||
| Cx43Cre/fl + oil | 0.77 ± 0.02 | 0.78 ± 0.02 | 0.74 ± 0.02 | 0.73 ± 0.02 | 4.02 ± 0.11 | 4.05 ± 0.10 | 2.70 ± 0.08 | 2.90 ± 0.09 * |
| (n = 10) | ||||||||
| Cx43Cre/fl + 4-OHT | 0.76 ± 0.02 | 0.79 ± 0.04 | 0.72 ± 0.02 | 0.71 ± 0.02 | 4.26 ± 0.26 | 4.26 ± 0.26 | 2.98 ± 0.15 | 3.09 ± 0.25 |
| (n = 7) | ||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valls-Lacalle, L.; Consegal, M.; Ruiz-Meana, M.; Benito, B.; Inserte, J.; Barba, I.; Ferreira-González, I.; Rodríguez-Sinovas, A. Connexin 43 Deficiency Is Associated with Reduced Myocardial Scar Size and Attenuated TGFβ1 Signaling after Transient Coronary Occlusion in Conditional Knock-Out Mice. Biomolecules 2020, 10, 651. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040651
Valls-Lacalle L, Consegal M, Ruiz-Meana M, Benito B, Inserte J, Barba I, Ferreira-González I, Rodríguez-Sinovas A. Connexin 43 Deficiency Is Associated with Reduced Myocardial Scar Size and Attenuated TGFβ1 Signaling after Transient Coronary Occlusion in Conditional Knock-Out Mice. Biomolecules. 2020; 10(4):651. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040651
Chicago/Turabian StyleValls-Lacalle, Laura, Marta Consegal, Marisol Ruiz-Meana, Begoña Benito, Javier Inserte, Ignasi Barba, Ignacio Ferreira-González, and Antonio Rodríguez-Sinovas. 2020. "Connexin 43 Deficiency Is Associated with Reduced Myocardial Scar Size and Attenuated TGFβ1 Signaling after Transient Coronary Occlusion in Conditional Knock-Out Mice" Biomolecules 10, no. 4: 651. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040651