Insights into the Conserved Regulatory Mechanisms of Human and Yeast Aging


 ,
,  , and
, and 
Abstract
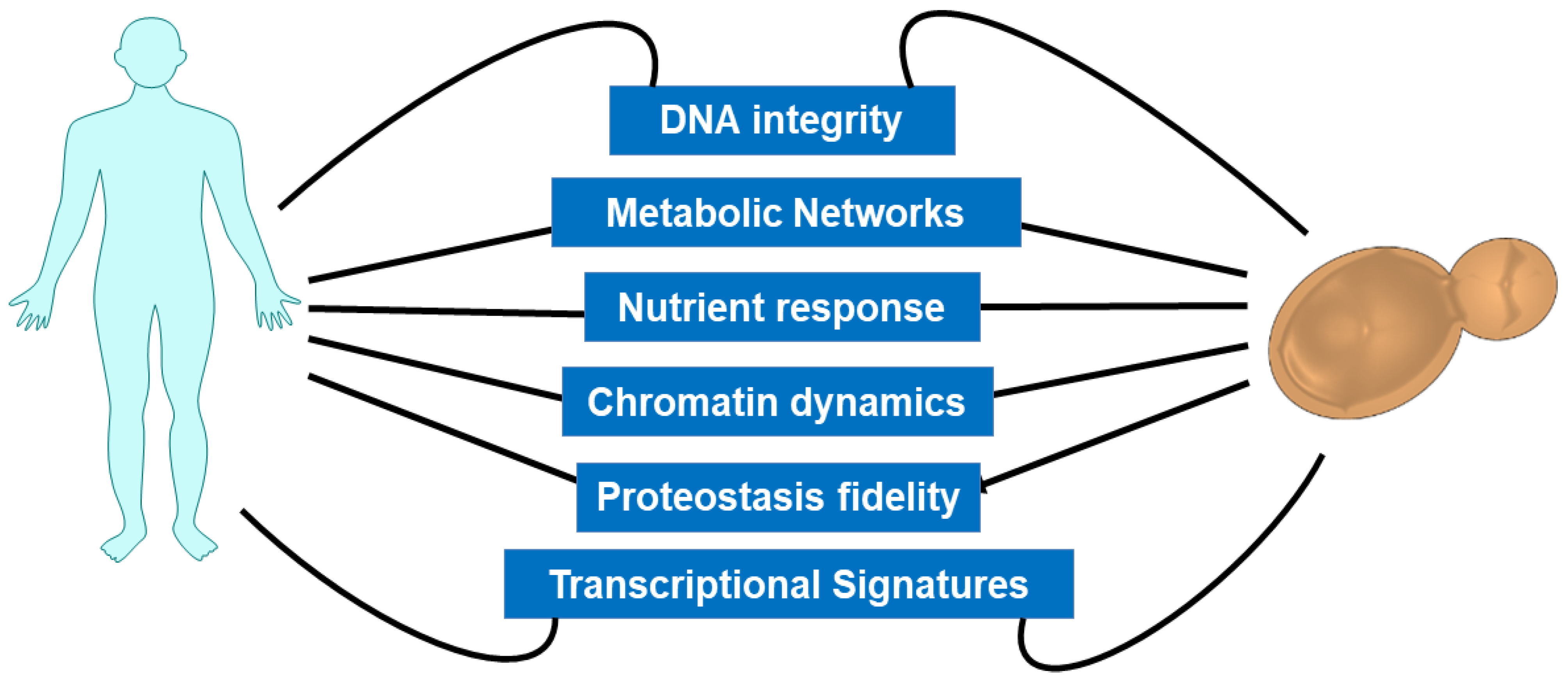
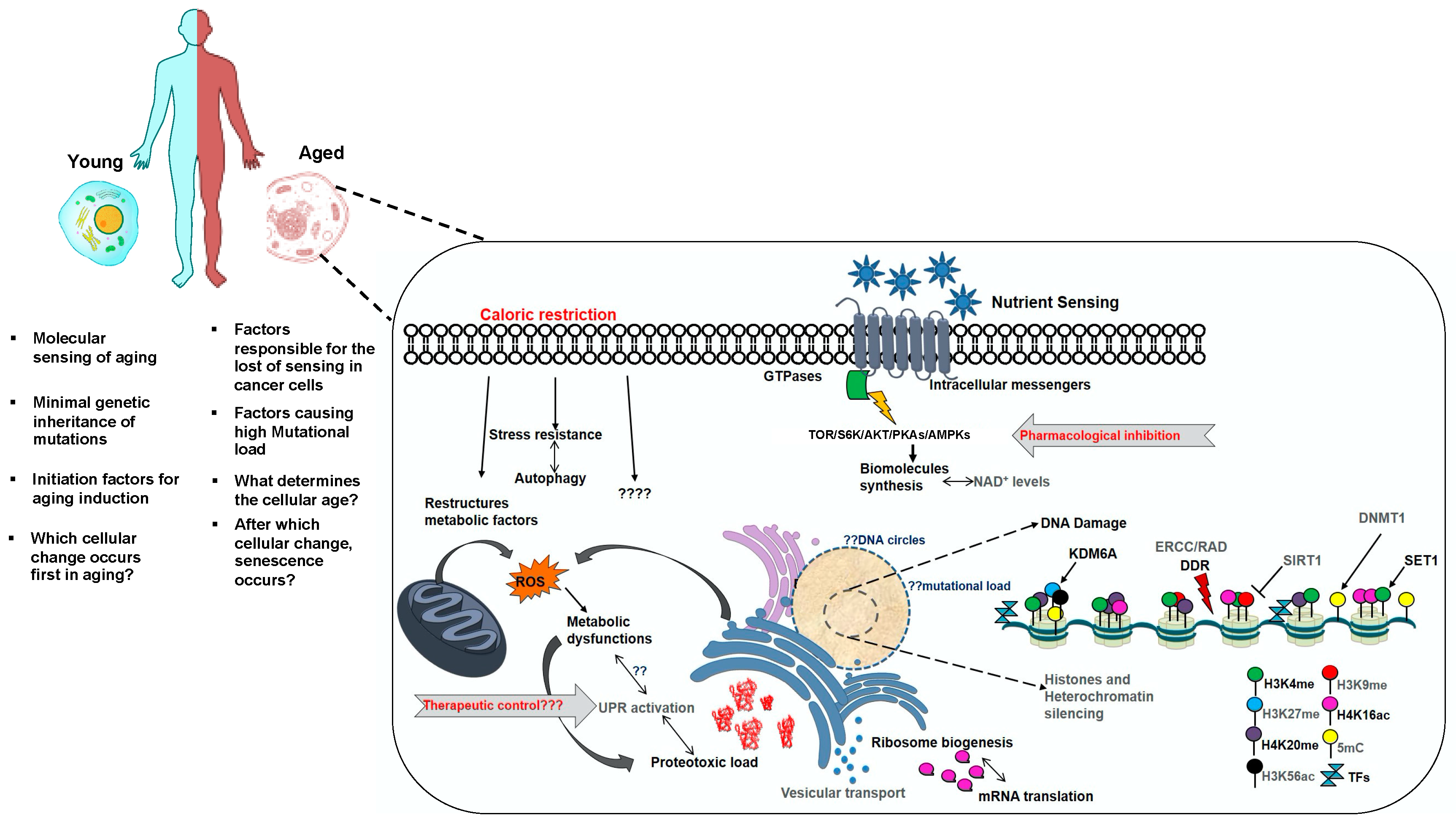
:1. Introduction
2. Integrative Biology of Yeast Aging: Pro-Longevity Interventions
3. Epigenomic Landscape and Dynamics Underlying Aging
4. Histone Acetylation and Sir2 Roles in Aging
5. Associating Aging with Genomic Instability
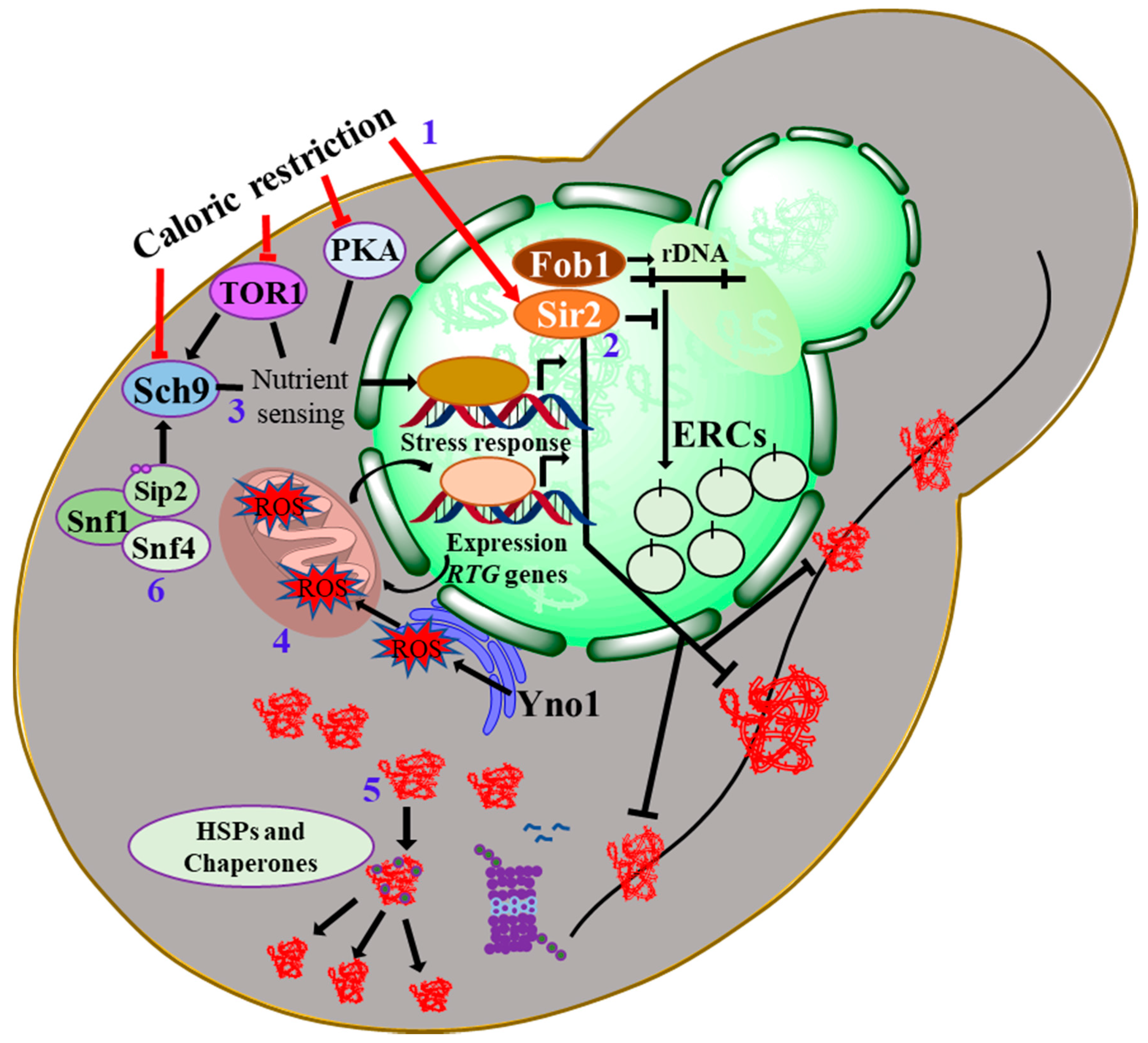
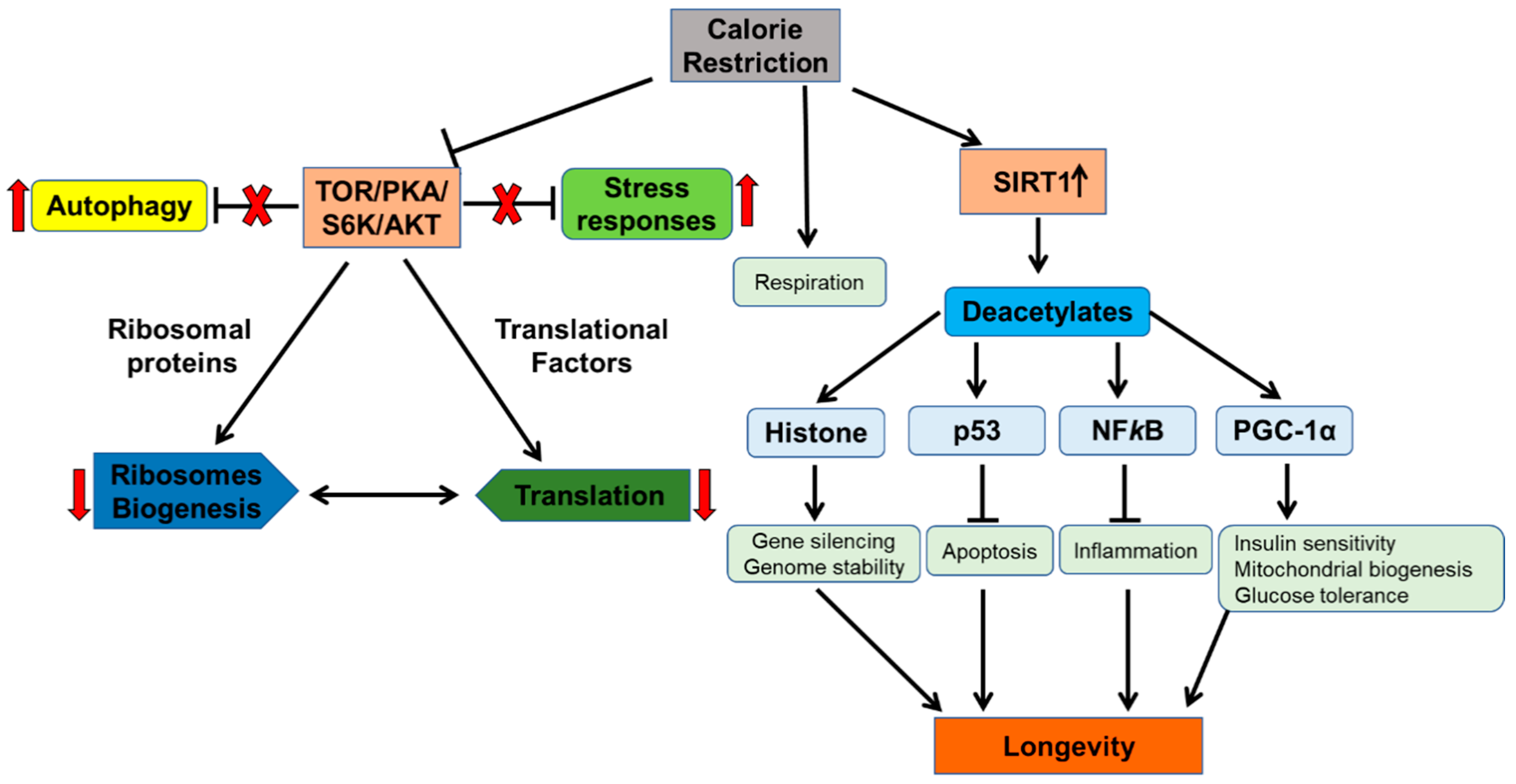
6. Calorie Restriction as an Anti-Aging Intervention: Molecular Mechanisms and Multitude Effects
6.1. Signalling Nexus of Protein kinase-A in Cellular Aging
6.2. The Target of Rapamycin and Sch9
7. Loss of Proteostasis Networks in Aging
8. Aging and Telomeres Dysfunction
9. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Guarente, L.; Kenyon, C. Genetic pathways that regulate ageing in model organisms. Nature 2000, 408, 255–262. [Google Scholar] [CrossRef]
- Ram, J.L.; Costa, A.J., II. Invertebrates as Model Organisms for Research on Aging Biology. In Conn’s Handbook of Models for Human Aging; Academic Press: London, UK, 2018; pp. 445–452. [Google Scholar]
- Vanhooren, V.; Libert, C. The mouse as a model organism in aging research: Usefulness, pitfalls and possibilities. Ageing Res. Rev. 2013, 12, 8–21. [Google Scholar] [CrossRef]
- Pun, P.B.L.; Gruber, J.; Tang, S.Y.; Schaffer, S.; Ong, R.L.S.; Fong, S.; Ng, L.F.; Cheah, I.; Halliwell, B. Ageing in nematodes: Do antioxidants extend lifespan in Caenorhabditiselegans? Biogerontology 2010, 11, 17–30. [Google Scholar] [CrossRef]
- Mortimer, R.K.; Johnston, J.R. Life span of individual yeast cells. Nature 1959, 183, 1751–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, B.; Steffen, K.; Kaeberlein, M. Ruminations on dietary restriction and aging. Cell. Mol. Life Sci. 2007, 64, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Weindruch, R. The caloric restriction paradigm: Implications for healthy human aging. Am. J. Hum. Biol. 2012, 24, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeberlein, M.; Powers, R.W.; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabrizio, P.; Pozza, F.; Pletcher, S.D.; Gendron, C.M.; Longo, V.D. Regulation of longevity and stress resistance by Sch9 in yeast. Science 2001, 292, 288–290. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Sami, N.; Rahman, S.; Kumar, V.; Zaidi, S.; Islam, A.; Ali, S.; Ahmad, F.; Hassan, M.I. Protein aggregation, misfolding and consequential human neurodegenerative diseases. Int. J. Neurosci. 2017, 127, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sami, N.; Kashav, T.; Islam, A.; Ahmad, F.; Hassan, M.I. Protein aggregation and neurodegenerative diseases: From theory to therapy. Eur. J. Med. Chem. 2016, 124, 1105–1120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Islam, A.; Hassan, M.I.; Ahmad, F. Therapeutic progress in amyotrophic lateral sclerosis-beginning to learning. Eur. J. Med. Chem. 2016, 121, 903–917. [Google Scholar] [CrossRef]
- Chokshi, K.; Pancha, I.; Ghosh, T.; Paliwal, C.; Maurya, R.; Ghosh, A.; Mishra, S. Green synthesis, characterization and antioxidant potential of silver nanoparticles biosynthesized from de-oiled biomass of thermotolerant oleaginous microalgae Acutodesmus dimorphus. RSC Adv. 2016, 6, 72269–72274. [Google Scholar] [CrossRef]
- Kumar, V.; Kashav, T.; Islam, A.; Ahmad, F.; Hassan, M.I. Structural insight into C9orf72 hexanucleotide repeat expansions: Towards new therapeutic targets in FTD-ALS. Neurochem. Int. 2016, 100, 11–20. [Google Scholar] [CrossRef]
- Kumar, V.; Prakash, A.; Pandey, P.; Lynn, A.M.; Hassan, M.I. TFE-induced local unfolding and fibrillation of SOD1: Bridging the experiment and simulation studies. Biochem. J. 2018, 475, 1701–1719. [Google Scholar] [CrossRef]
- Kumar, V.; Rahman, S.; Choudhry, H.; Zamzami, M.A.; Sarwar Jamal, M.; Islam, A.; Ahmad, F.; Hassan, M.I. Computing disease-linked SOD1 mutations: Deciphering protein stability and patient-phenotype relations. Sci. Rep. 2017, 7, 4678. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Kumar, V.; Meena, N.K.; Hassan, M.I.; Lynn, A.M. Comparative analysis of thermal unfolding simulations of RNA recognition motifs (RRMs) of TAR DNA-binding protein 43 (TDP-43). J. Biomol. Struct. Dyn. 2019, 37, 178–194. [Google Scholar] [CrossRef]
- Prakash, A.; Kumar, V.; Pandey, P.; Bharti, D.R.; Vishwakarma, P.; Singh, R.; Hassan, M.I.; Lynn, A.M. Solvent sensitivity of protein aggregation in Cu, Zn superoxide dismutase: A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2018, 36, 2605–2617. [Google Scholar] [CrossRef]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef] [Green Version]
- Hertel, J.; Friedrich, N.; Wittfeld, K.; Pietzner, M.; Budde, K.; Van der Auwera, S.; Lohmann, T.; Teumer, A.; Völzke, H.; Nauck, M. Measuring biological age via metabonomics: The metabolic age score. J. Proteome Res. 2015, 15, 400–410. [Google Scholar] [CrossRef]
- Broer, L.; van Duijn, C.M. GWAS and meta-analysis in aging/longevity. In Longevity Genes; Springer: New York, NY, USA, 2015; pp. 107–125. [Google Scholar]
- Vermeij, W.P.; Hoeijmakers, J.H.; Pothof, J. Genome integrity in aging: Human syndromes, mouse models, and therapeutic options. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 427–445. [Google Scholar] [CrossRef]
- McMurray, M.A.; Thorner, J. Septins: Molecular partitioning and the generation of cellular asymmetry. Cell Div. 2009, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M. Lessons on longevity from budding yeast. Nature 2010, 464, 513. [Google Scholar] [CrossRef] [Green Version]
- Ren, R.; Ocampo, A.; Liu, G.-H.; Belmonte, J.C.I. Regulation of stem cell aging by metabolism and epigenetics. Cell Metab. 2017, 26, 460–474. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Shcheprova, Z.; Baldi, S.; Frei, S.B.; Gonnet, G.; Barral, Y. A mechanism for asymmetric segregation of age during yeast budding. Nature 2008, 454, 728. [Google Scholar] [CrossRef]
- Berner, N.; Reutter, K.-R.; Wolf, D.H. Protein quality control of the endoplasmic reticulum and ubiquitin–proteasome-triggered degradation of aberrant proteins: Yeast pioneers the path. Annu. Rev. Biochem. 2018, 87, 751–782. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Yi, D.-G.; Hong, S.; Huh, W.-K. Mitochondrial dysfunction reduces yeast replicative lifespan by elevating RAS-dependent ROS production by the ER-localized NADPH oxidase Yno1. PLoS ONE 2018, 13, e0198619. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; Hu, D.; Kerr, E.O.; Tsuchiya, M.; Westman, E.A.; Dang, N.; Fields, S.; Kennedy, B.K. Increased life span due to calorie restriction in respiratory-deficient yeast. PLoS Genet. 2005, 1, e69. [Google Scholar] [CrossRef]
- Caballero, A.; Ugidos, A.; Liu, B.; Öling, D.; Kvint, K.; Hao, X.; Mignat, C.; Nachin, L.; Molin, M.; Nyström, T. Absence of mitochondrial translation control proteins extends life span by activating sirtuin-dependent silencing. Mol. Cell 2011, 42, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802. [Google Scholar] [CrossRef]
- Greer, E.L.; Maures, T.J.; Hauswirth, A.G.; Green, E.M.; Leeman, D.S.; Maro, G.S.; Han, S.; Banko, M.R.; Gozani, O.; Brunet, A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 2010, 466, 383. [Google Scholar] [CrossRef] [Green Version]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated histone expression promotes life span extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef] [Green Version]
- Oberdoerffer, P.; Sinclair, D.A. The role of nuclear architecture in genomic instability and ageing. Nat. Rev. Mol. Cell Biol. 2007, 8, 692–702. [Google Scholar] [CrossRef]
- McCormick, M.A.; Mason, A.G.; Guyenet, S.J.; Dang, W.; Garza, R.M.; Ting, M.K.; Moller, R.M.; Berger, S.L.; Kaeberlein, M.; Pillus, L. The SAGA histone deubiquitinase module controls yeast replicative lifespan via Sir2 interaction. Cell Rep. 2014, 8, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Kapahi, P.; Chen, D.; Rogers, A.N.; Katewa, S.D.; Li, P.W.-L.; Thomas, E.L.; Kockel, L. With TOR, less is more: A key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010, 11, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Wierman, M.B.; Smith, J.S. Yeast sirtuins and the regulation of aging. FEMS Yeast Res. 2014, 14, 73–88. [Google Scholar] [CrossRef] [Green Version]
- Chiocchetti, A.; Zhou, J.; Zhu, H.; Karl, T.; Haubenreisser, O.; Rinnerthaler, M.; Heeren, G.; Oender, K.; Bauer, J.; Hintner, H. Ribosomal proteins Rpl10 and Rps6 are potent regulators of yeast replicative life span. Exp. Gerontol. 2007, 42, 275–286. [Google Scholar] [CrossRef]
- Ralser, M.; Michel, S.; Breitenbach, M. Sirtuins as regulators of the yeast metabolic network. Front. Pharmacol. 2012, 3, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miceli, M.V.; Jiang, J.C.; Tiwari, A.; Rodriguez-Quiñones, J.F.; Jazwinski, S.M. Loss of mitochondrial membrane potential triggers the retrograde response extending yeast replicative lifespan. Front. Genet. 2012, 2, 102. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Wawryn, J.; Kumara, H.S.; Jazwinski, S. Distinct roles of processes modulated by histone deacetylases Rpd3p, Hda1p, and Sir2p in life extension by caloric restriction in yeast. Exp. Gerontol. 2002, 37, 1023–1030. [Google Scholar] [CrossRef]
- Curran, S.P.; Ruvkun, G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007, 3, e56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.; Wera, S.; Van Dijck, P.; Thevelein, J.M. The PDE1-encoded low-affinity phosphodiesterase in the yeast Saccharomyces cerevisiae has a specific function in controlling agonist-induced cAMP signaling. Mol. Biol. Cell 1999, 10, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVey, M.; Kaeberlein, M.; Tissenbaum, H.A.; Guarente, L. The short life span of Saccharomyces cerevisiae sgs1 and srs2 mutants is a composite of normal aging processes and mitotic arrest due to defective recombination. Genetics 2001, 157, 1531–1542. [Google Scholar]
- Defossez, P.-A.; Prusty, R.; Kaeberlein, M.; Lin, S.-J.; Ferrigno, P.; Silver, P.A.; Keil, R.L.; Guarente, L. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol. Cell 1999, 3, 447–455. [Google Scholar] [CrossRef]
- Liu, J.; Huang, X.; Withers, B.R.; Blalock, E.; Liu, K.; Dickson, R.C. Reducing sphingolipid synthesis orchestrates global changes to extend yeast lifespan. Aging Cell 2013, 12, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jiang, J.C.; Jazwinski, S.M. Gene regulatory changes in yeast during life extension by nutrient limitation. Exp. Gerontol. 2010, 45, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Mirisola, M.G.; Longo, V.D. Conserved role of Ras-GEFs in promoting aging: From yeast to mice. Aging 2011, 3, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazwinski, S.M. Growing old: Metabolic control and yeast aging. Annu. Rev. Microbiol. 2002, 56, 769–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.C.; Stumpferl, S.W.; Tiwari, A.; Qin, Q.; Rodriguez-Quiñones, J.F.; Jazwinski, S.M. Identification of the target of the retrograde response that mediates replicative lifespan extension in Saccharomyces cerevisiae. Genetics 2016, 204, 659–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Hoe, K.L.; Maeng, P.J. Yeast cells lacking the CIT1-encoded mitochondrial citrate synthase are hypersusceptible to heat-or aging-induced apoptosis. Mol. Biol. Cell 2007, 18, 3556–3567. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N.; Guimaraes, J.C.; Gross, T.; Schmidt, A.; Vina-Vilaseca, A.; Nedialkova, D.D.; Aeschimann, F.; Leidel, S.A.; Spang, A.; Zavolan, M. The Gcn4 transcription factor reduces protein synthesis capacity and extends yeast lifespan. Nat. Commun. 2017, 8, 457. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Ohkuni, K.; Couplan, E.; Jazwinski, S.M. The histone acetyltransferase GCN5 modulates the retrograde response and genome stability determining yeast longevity. Biogerontology 2004, 5, 305–316. [Google Scholar] [CrossRef]
- Benjamin, J.J.; Poon, P.P.; Drysdale, J.D.; Wang, X.; Singer, R.A.; Johnston, G.C. Dysregulated Arl1, a regulator of post-Golgi vesicle tethering, can inhibit endosomal transport and cell proliferation in yeast. Mol. Biol. Cell 2011, 22, 2337–2347. [Google Scholar] [CrossRef]
- Kungulovski, G.; Jeltsch, A. Epigenome editing: State of the art, concepts, and perspectives. Trends Genet. 2016, 32, 101–113. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.-H.; Chen, C.-C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Ishimi, Y.; Kojima, M.; Takeuchi, F.; Miyamoto, T.; Yamada, M.-A.; Hanaoka, F. Changes in chromatin structure during aging of human skin fibroblasts. Exp. Cell Res. 1987, 169, 458–467. [Google Scholar] [CrossRef]
- Lund, J.; Tedesco, P.; Duke, K.; Wang, J.; Kim, S.K.; Johnson, T.E. Transcriptional profile of aging in C. elegans. Curr. Biol. 2002, 12, 1566–1573. [Google Scholar] [CrossRef] [Green Version]
- Bennett-Baker, P.E.; Wilkowski, J.; Burke, D.T. Age-associated activation of epigenetically repressed genes in the mouse. Genetics 2003, 165, 2055–2062. [Google Scholar] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Li, J.; Green, C.D.; Yu, X.; Tang, X.; Han, D.; Xian, B.; Wang, D.; Huang, X.; Cao, X. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 2011, 14, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, Y.B.; Kahn, T.G.; Nix, D.A.; Li, X.-Y.; Bourgon, R.; Biggin, M.; Pirrotta, V. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat. Genet. 2006, 38, 700. [Google Scholar] [CrossRef]
- Noer, A.; Lindeman, L.C.; Collas, P. Histone H3 modifications associated with differentiation and long-term culture of mesenchymal adipose stem cells. Stem Cells Dev. 2009, 18, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.C.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef] [Green Version]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.-E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huyen, Y.; Zgheib, O.; DiTullio, R.A., Jr.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406. [Google Scholar] [CrossRef]
- McCord, R.P.; Nazario-Toole, A.; Zhang, H.; Chines, P.S.; Zhan, Y.; Erdos, M.R.; Collins, F.S.; Dekker, J.; Cao, K. Correlated alterations in genome organization, histone methylation, and DNA–lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013, 23, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Sarg, B.; Koutzamani, E.; Helliger, W.; Rundquist, I.; Lindner, H.H. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J. Biol. Chem. 2002, 277, 39195–39201. [Google Scholar] [CrossRef] [Green Version]
- Wilson, V.L.; Jones, P.A. DNA methylation decreases in aging but not in immortal cells. Science 1983, 220, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Goel, N.; Karir, P.; Garg, V.K. Role of DNA methylation in human age prediction. Mech. Ageing Dev. 2017, 166, 33–41. [Google Scholar] [CrossRef]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.-P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarone, F.; Malavolta, M.; Calabrese, R.; Guastafierro, T.; Bacalini, M.G.; Reale, A.; Franceschi, C.; Capri, M.; Hervonen, A.; Hurme, M. Age-dependent expression of DNMT 1 and DNMT 3B in PBMC s from a large E uropean population enrolled in the MARK-AGE study. Aging Cell 2016, 15, 755–765. [Google Scholar] [CrossRef]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, Y.; Yoshino, Y.; Yamazaki, K.; Sao, T.; Mori, Y.; Ochi, S.; Yoshida, T.; Mori, T.; Iga, J.-I.; Ueno, S.-I. DNA methylation changes at TREM2 intron 1 and TREM2 mRNA expression in patients with Alzheimer’s disease. J. Psychiatr. Res. 2017, 92, 74–80. [Google Scholar] [CrossRef]
- Ling, C.; Del Guerra, S.; Lupi, R.; Rönn, T.; Granhall, C.; Luthman, H.; Masiello, P.; Marchetti, P.; Groop, L.; Del Prato, S. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008, 51, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.; Cristofalo, V. Histone acetylation during aging of human cells in culture. Biochem. Biophys. Res. Commun. 1972, 48, 735–742. [Google Scholar] [CrossRef]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Brachmann, C.B.; Sherman, J.M.; Devine, S.E.; Cameron, E.E.; Pillus, L.; Boeke, J.D. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995, 9, 2888–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, P.; Freeman, K.; Coodly, L.; Shore, D. Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 1994, 8, 2257–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotta, M.; Strahl-Bolsinger, S.; Renauld, H.; Laroche, T.; Kennedy, B.K.; Grunstein, M.; Gasser, S.M. Localization of Sir2p: The nucleolus as a compartment for silent information regulators. EMBO J. 1997, 16, 3243–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227. [Google Scholar] [CrossRef]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.-L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef] [Green Version]
- Oppikofer, M.; Kueng, S.; Martino, F.; Soeroes, S.; Hancock, S.M.; Chin, J.W.; Fischle, W.; Gasser, S.M. A dual role of H4K16 acetylation in the establishment of yeast silent chromatin. EMBO J. 2011, 30, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, V.; Chow, M.Z.Y.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef] [Green Version]
- Shia, W.-J.; Osada, S.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Workman, J.L. Characterization of the yeast trimeric-SAS acetyltransferase complex. J. Biol. Chem. 2005, 280, 11987–11994. [Google Scholar] [CrossRef] [Green Version]
- Hecht, A.; Strahl-Bolsinger, S.; Grunstein, M. Spreading of transcriptional represser SIR3 from telomeric heterochromatin. Nature 1996, 383, 92. [Google Scholar] [CrossRef]
- Straight, A.F.; Shou, W.; Dowd, G.J.; Turck, C.W.; Deshaies, R.J.; Johnson, A.D.; Moazed, D. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell 1999, 97, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Saunders, L.; Verdin, E. Sirtuins: Critical regulators at the crossroads between cancer and aging. The Oncogene 2007, 26, 5489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.-I.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Zhou, Z. SIRTain regulators of premature senescence and accelerated aging. Protein Cell 2015, 6, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- Ondracek, C.R.; Frappier, V.; Ringel, A.E.; Wolberger, C.; Guarente, L. Mutations that Allow SIR2 Orthologs to Function in a NAD+-Depleted Environment. Cell Rep. 2017, 18, 2310–2319. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.J.; Martin-Montalvo, A.; Mercken, E.M.; Palacios, H.H.; Ward, T.M.; Abulwerdi, G.; Minor, R.K.; Vlasuk, G.P.; Ellis, J.L.; Sinclair, D.A. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014, 6, 836–843. [Google Scholar] [CrossRef] [Green Version]
- De Magalhães, J.P.; Stevens, M.; Thornton, D. The business of anti-aging science. Trends Biotechnol. 2017, 35, 1062–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61. [Google Scholar] [CrossRef]
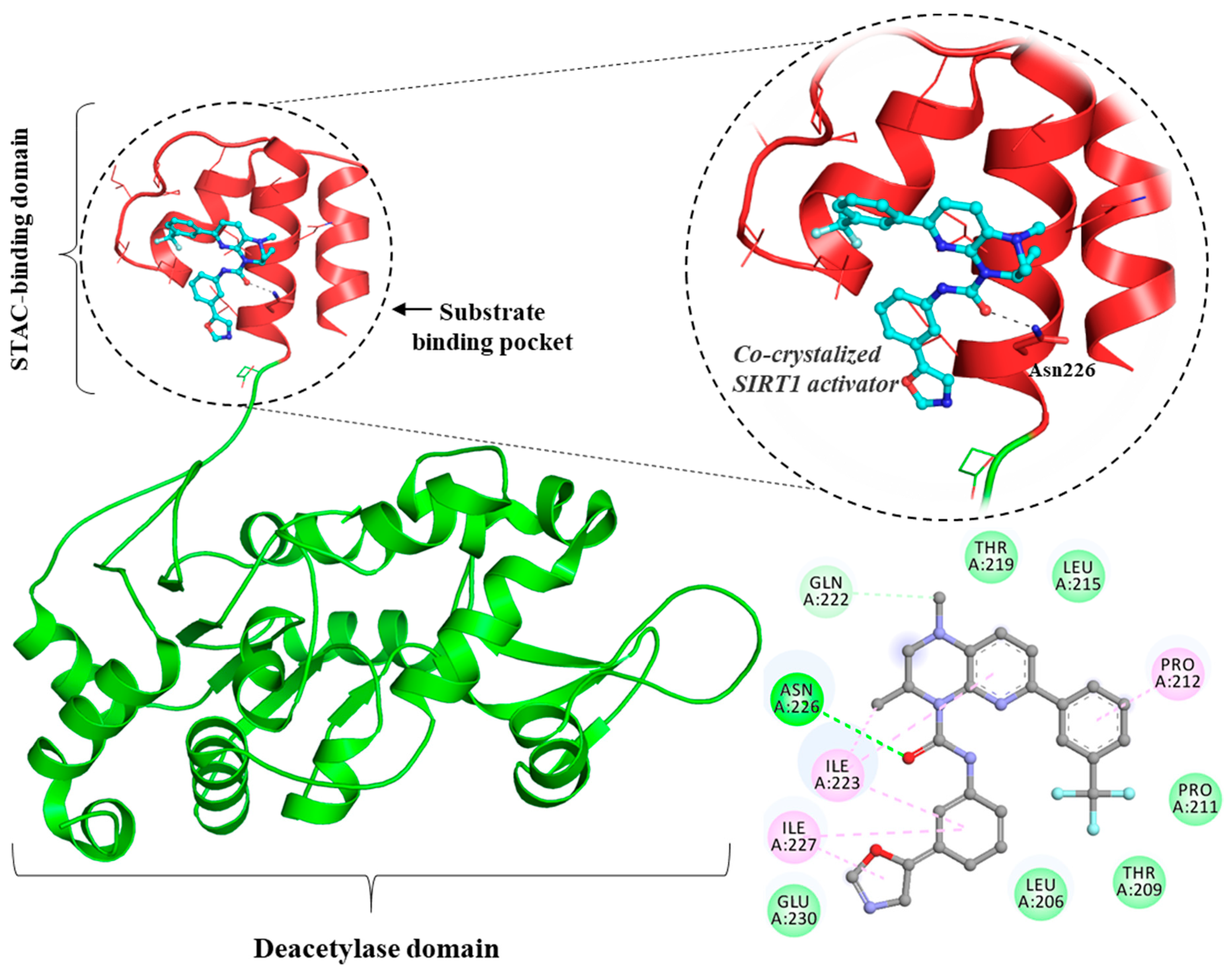
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Gomes, A.P.; Dai, H.; Li, J.; Case, A.W.; Considine, T.; Riera, T.V.; Lee, J.E.; Yen, S.; Lamming, D.W. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013, 339, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. New Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Kolodner, R.D.; Putnam, C.D.; Myung, K. Maintenance of genome stability in Saccharomyces cerevisiae. Science 2002, 297, 552–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, M. A cascade leading to premature aging phenotypes including abnormal tumor profiles in Werner syndrome. Int. J. Mol. Med. 2014, 33, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Gregg, S.Q.; Robinson, A.R.; Niedernhofer, L.J. Physiological consequences of defects in ERCC1–XPF DNA repair endonuclease. DNA Repair 2011, 10, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; Van Leeuwen, W. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038. [Google Scholar] [CrossRef]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillón, J.; Ramírez, M.J.; Pujol, R. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamczyk, J.; Deregowska, A.; Panek, A.; Golec, E.; Lewinska, A.; Wnuk, M. Affected chromosome homeostasis and genomic instability of clonal yeast cultures. Curr. Genet. 2016, 62, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoopes, L.L.M.; Budd, M.; Choe, W.; Weitao, T.; Campbell, J.L. Mutations in DNA replication genes reduce yeast life span. Mol. Cell. Biol. 2002, 22, 4136–4146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesur, I.; Campbell, J.L. The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol. Biol. Cell 2004, 15, 1297–1312. [Google Scholar] [CrossRef] [Green Version]
- Warmerdam, D.O.; Wolthuis, R.M.F. Keeping ribosomal DNA intact: A repeating challenge. Chromosome Res. 2019, 27, 57–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guetg, C.; Lienemann, P.; Sirri, V.; Grummt, I.; Hernandez-Verdun, D.; Hottiger, M.O.; Fussenegger, M.; Santoro, R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J. 2010, 29, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Lu, S.; Gerton, J.L. Roberts syndrome: A deficit in acetylated cohesin leads to nucleolar dysfunction. Rare Dis. 2014, 2, e27743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gard, S.; Light, W.; Xiong, B.; Bose, T.; McNairn, A.J.; Harris, B.; Fleharty, B.; Seidel, C.; Brickner, J.H.; Gerton, J.L. Cohesinopathy mutations disrupt the subnuclear organization of chromatin. J. Cell Biol. 2009, 187, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.-Q.; Petes, T. Genome instability induced by low levels of replicative DNA polymerases in yeast. Genes 2018, 9, 539. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.C.; Pinto, R.M.; Murray, A.W. Heterozygous mutations cause genetic instability in a yeast model of cancer evolution. Nature 2019, 566, 275. [Google Scholar] [CrossRef]
- Sasaki, H.; Yanagi, K.; Ugi, S.; Kobayashi, K.; Ohkubo, K.; Tajiri, Y.; Maegawa, H.; Kashiwagi, A.; Kaname, T. Definitive diagnosis of mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL) syndrome caused by a recurrent de novo mutation in the POLD1 gene. Endocr. J. 2017, EJ17-0287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Defossez, P.-A.; Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, K.A.; Cole, N.H.; Mermier, C.M.; VanDusseldorp, T.A. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876. [Google Scholar] [CrossRef] [Green Version]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size: One figure. J. Nutr. 1935, 10, 63–79. [Google Scholar] [CrossRef]
- Lin, S.-J.; Ford, E.; Haigis, M.; Liszt, G.; Guarente, L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004, 18, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-J.; Kaeberlein, M.; Andalis, A.A.; Sturtz, L.A.; Defossez, P.-A.; Culotta, V.C.; Fink, G.R.; Guarente, L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 2002, 418, 344. [Google Scholar] [CrossRef]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; De Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef]
- Schleit, J.; Wasko, B.M.; Kaeberlein, M. Yeast as a model to understand the interaction between genotype and the response to calorie restriction. FEBS Lett. 2012, 586, 2868–2873. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2004, 2, e296. [Google Scholar] [CrossRef]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Feng, X.; Ennis, K.N.; Behrmann, C.A.; Sarma, P.; Jiang, T.T.; Kofuji, S.; Niu, L.; Stratton, Y.; Thomas, H.E. Pharmacologic targeting of S6K1 in PTEN-deficient neoplasia. Cell Rep. 2017, 18, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Rochon, J.; Bales, C.W.; Ravussin, E.; Redman, L.M.; Holloszy, J.O.; Racette, S.B.; Roberts, S.B.; Das, S.K.; Romashkan, S.; Galan, K.M. Design and conduct of the CALERIE study: Comprehensive assessment of the long-term effects of reducing intake of energy. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2010, 66, 97–108. [Google Scholar] [CrossRef]
- Tamaki, H. Glucose-stimulated cAMP-protein kinase a pathway in yeast Saccharomyces cerevisiae. J. Biosci. Bioeng. 2007, 104, 245–250. [Google Scholar] [CrossRef]
- Sun, J.; Kale, S.P.; Childress, A.M.; Pinswasdi, C.; Jazwinski, S.M. Divergent roles of RAS1 and RAS2 in yeast longevity. J. Biol. Chem. 1994, 269, 18638–18645. [Google Scholar]
- Kang, W.K.; Kim, Y.H.; Kang, H.A.; Kwon, K.-S.; Kim, J.-Y. Sir2 phosphorylation through cAMP-PKA and CK2 signaling inhibits the lifespan extension activity of Sir2 in yeast. Elife 2015, 4, e09709. [Google Scholar] [CrossRef] [PubMed]
- Enns, L.C.; Morton, J.F.; Treuting, P.R.; Emond, M.J.; Wolf, N.S.; McKnight, G.; Rabinovitch, P.S.; Ladiges, W.C. Disruption of protein kinase A in mice enhances healthy aging. PLoS ONE 2009, 4, e5963. [Google Scholar] [CrossRef] [Green Version]
- Burkewitz, K.; Zhang, Y.; Mair, W.B. AMPK at the nexus of energetics and aging. Cell Metab. 2014, 20, 10–25. [Google Scholar] [CrossRef] [Green Version]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.; Soulard, A.; Huber, A.; Lippman, S.; Mukhopadhyay, D.; Deloche, O.; Wanke, V.; Anrather, D.; Ammerer, G.; Riezman, H. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 2007, 26, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Chen, D.; Riddle, D.L. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 2004, 131, 3897–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Passtoors, W.M.; Beekman, M.; Deelen, J.; van der Breggen, R.; Maier, A.B.; Guigas, B.; Derhovanessian, E.; van Heemst, D.; de Craen, A.J.; Gunn, D.A. Gene expression analysis of mTOR pathway: Association with human longevity. Aging Cell 2013, 12, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Basu, B.; Dean, E.; Puglisi, M.; Greystoke, A.; Ong, M.; Burke, W.; Cavallin, M.; Bigley, G.; Womack, C.; Harrington, E.A. First-in-human pharmacokinetic and pharmacodynamic study of the dual m-TORC 1/2 inhibitor AZD2014. Clin. Cancer Res. 2015, 21, 3412–3419. [Google Scholar] [CrossRef] [Green Version]
- Shum, M.; Bellmann, K.; St-Pierre, P.; Marette, A. Pharmacological inhibition of S6K1 increases glucose metabolism and Akt signalling in vitro and in diet-induced obese mice. Diabetologia 2016, 59, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Pachler, K.; Karl, T.; Kolmann, K.; Mehlmer, N.; Eder, M.; Loeffler, M.; Oender, K.; Hochleitner, E.O.; Lottspeich, F.; Bresgen, N. Functional interaction in establishment of ribosomal integrity between small subunit protein rpS6 and translational regulator rpL10/Grc5p. FEMS Yeast Res. 2004, 5, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Steffen, K.K.; MacKay, V.L.; Kerr, E.O.; Tsuchiya, M.; Hu, D.; Fox, L.A.; Dang, N.; Johnston, E.D.; Oakes, J.A.; Tchao, B.N. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell 2008, 133, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Heeren, G.; Rinnerthaler, M.; Laun, P.; von Seyerl, P.; Kössler, S.; Klinger, H.; Jarolim, S.; Simon-Nobbe, B.; Hager, M.; Schüller, C. The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1. Aging 2009, 1, 622. [Google Scholar] [CrossRef] [Green Version]
- Saez, I.; Vilchez, D. The mechanistic links between proteasome activity, aging and agerelated diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the aging proteome in health and disease. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2014, 69, S33–S38. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Cheon, S.Y.; Kim, H.; Rubinsztein, D.C.; Lee, J.E. Autophagy, Cellular Aging and Age-related Human Diseases. Exp. Neurobiol. 2019, 28, 643. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chondrogianni, N.; Georgila, K.; Kourtis, N.; Tavernarakis, N.; Gonos, E.S. 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans. FASEB J. 2014, 29, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyo, J.-O.; Yoo, S.-M.; Ahn, H.-H.; Nah, J.; Hong, S.-H.; Kam, T.-I.; Jung, S.; Jung, Y.-K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [Green Version]
- Kruegel, U.; Robison, B.; Dange, T.; Kahlert, G.; Delaney, J.R.; Kotireddy, S.; Tsuchiya, M.; Tsuchiyama, S.; Murakami, C.J.; Schleit, J. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 2011, 7, e1002253. [Google Scholar] [CrossRef]
- Reverter-Branchat, G.; Cabiscol, E.; Tamarit, J.; Ros, J. Oxidative damage to specific proteins in replicative and chronological-aged Saccharomyces cerevisiae common targets and prevention by calorie restriction. J. Biol. Chem. 2004, 279, 31983–31989. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.L.; Timney, B.L.; Rout, M.P.; Wente, S.R. Altering nuclear pore complex function impacts longevity and mitochondrial function in S. cerevisiae. J. Cell Biol. 2015, 208, 729–744. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Hidalgo, M.; Santos-Ocaña, C.; Padilla, S.; Villalba, J.M.; López-Lluch, G.; Martín-Montalvo, A.; Minor, R.K.; Sinclair, D.A.; De Cabo, R.; Navas, P. NQR1 controls lifespan by regulating the promotion of respiratory metabolism in yeast. Aging Cell 2009, 8, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, S.; Arimoto, A.; Kuramoto, Y.; Kozako, T.; Honda, S.-I.; Shimeno, H.; Soeda, S. Fasting promotes the expression of SIRT1, an NAD+-dependent protein deacetylase, via activation of PPARα in mice. Mol. Cell. Biochem. 2010, 339, 285–292. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Adler, J.; Adamovich, Y.; Asher, G.; Reuven, N.; Shaul, Y. NQO1 binds and supports SIRT1 function. BioRxiv 2017, 139907. [Google Scholar]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Adamovich, Y.; Shlomai, A.; Tsvetkov, P.; Umansky, K.B.; Reuven, N.; Estall, J.L.; Spiegelman, B.M.; Shaul, Y. The protein level of PGC-1α, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell. Biol. 2013, 33, 2603–2613. [Google Scholar] [CrossRef] [Green Version]
- Sieverling, L.; Hong, C.; Koser, S.D.; Ginsbach, P.; Kleinheinz, K.; Hutter, B.; Braun, D.M.; Cortés-Ciriano, I.; Xi, R.; Kabbe, R.; et al. Genomic footprints of activated telomere maintenance mechanisms in cancer. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Crabbe, L.; Cesare, A.J.; Kasuboski, J.M.; Fitzpatrick, J.A.; Karlseder, J. Human telomeres are tethered to the nuclear envelope during postmitotic nuclear assembly. Cell Rep. 2012, 2, 1521–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Mello, N.P.; Jazwinski, S.M. Telomere length constancy during aging of Saccharomyces cerevisiae. J. Bacteriol. 1991, 173, 6709–6713. [Google Scholar] [CrossRef] [Green Version]
- Austriaco, N.R.; Guarente, L.P. Changes of telomere length cause reciprocal changes in the lifespan of mother cells in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 9768–9772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Jay, K.A.; Smith, D.L.; Zhang, Y.; Liu, Z.; Zheng, J.; Tian, R.; Li, H.; Blackburn, E.H. Early telomerase inactivation accelerates aging independently of telomere length. Cell 2015, 160, 928–939. [Google Scholar] [CrossRef] [Green Version]
- Smeal, T.; Claus, J.; Kennedy, B.; Cole, F.; Guarente, L. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 1996, 84, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Villeponteau, B.; Jazwinski, S.M. Effect of replicative age on transcriptional silencing near telomeres in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1996, 219, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, K.; Capell, B.C. The senescence-associated secretory phenotype: Critical effector in skin cancer and aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef] [Green Version]
- Hackett, J.A.; Greider, C.W. End resection initiates genomic instability in the absence of telomerase. Mol. Cell. Biol. 2003, 23, 8450–8461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, J.A.; Feldser, D.M.; Greider, C.W. Telomere dysfunction increases mutation rate and genomic instability. Cell 2001, 106, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Abdallah, P.; Luciano, P.; Runge, K.W.; Lisby, M.; Géli, V.; Gilson, E.; Teixeira, M.T. A two-step model for senescence triggered by a single critically short telomere. Nat. Cell Biol. 2009, 11, 988–993. [Google Scholar] [CrossRef] [Green Version]
- Khadaroo, B.; Teixeira, M.T.; Luciano, P.; Eckert-Boulet, N.; Germann, S.M.; Simon, M.N.; Gallina, I.; Abdallah, P.; Gilson, E.; Géli, V.; et al. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat. Cell Biol. 2009, 11, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Wang, Z.; Liu, J.-P. Roles of telomere biology in cell senescence, replicative and chronological ageing. Cells 2019, 8, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, T.S.; Taggart, A.K.; Zakian, V.A. Cell cycle-dependent regulation of yeast telomerase by Ku. Nat. Struct. Mol. Biol. 2004, 11, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Vogel, H.; Holcomb, V.B.; Gu, Y.; Hasty, P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol. Cell. Biol. 2007, 27, 8205–8214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.-J.; Lee, K.-H.; Park, J.-E.; Yi, Y.-S.; Yun, M.-Y.; Ham, Y.-H.; Kim, T.-J.; Choi, H.M.; Han, G.J.; Lee, J.; et al. Decreased expression of DNA repair proteins Ku70 and Mre11 is associated with aging and may contribute to the cellular senescence. Exp. Mol. Med. 2006, 38, 686–693. [Google Scholar] [CrossRef]
- Fontana, G.A.; Reinert, J.K.; Thomä, N.H.; Rass, U. Shepherding DNA ends: Rif1 protects telomeres and chromosome breaks. Microb. Cell 2018, 5, 327. [Google Scholar] [CrossRef]
- Wotton, D.; Shore, D. A novel Rap1p-interacting factor, Rif2p, cooperates with Rif1p to regulate telomere length in Saccharomyces cerevisiae. Genes Dev. 1997, 11, 748–760. [Google Scholar] [CrossRef] [Green Version]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [Green Version]
- Rice, C.; Skordalakes, E. Structure and function of the telomeric CST complex. Comput. Struct. Biotechnol. J. 2016, 14, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Vulliamy, T.; Marrone, A.; Dokal, I.; Mason, P.J. Association between aplastic anaemia and mutations in telomerase RNA. The Lancet 2002, 359, 2168–2170. [Google Scholar] [CrossRef]
- Calado, R.T.; Young, N.S. Telomere diseases. New Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. γH2AX as a molecular marker of aging and disease. Epigenetics 2010, 5, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Name Description | Cellular Functions | Impact of Null Phenotype on Lifespan | References |
|---|---|---|---|---|
| TOR1, SCH9 | Ser/Thr protein kinase involved in the signaling of the target of rapamycin | A protein kinase subunit of TOR complex which controls growth in response to nutrients by regulating translation | Increased | [10,40] |
| SIR2 | Silent information regulator | NAD-dependent Histone deacetylase. Plays important roles in silencing at HML, HMR, telomeres, and rDNA | Decreased | [41] |
| RPL9A, RPL6B, RPL19A | Ribosomal 60S subunit protein L9A | Structural constituent of the large 60S subunit of Ribosome, involved in translation. | Increased | [9,42] |
| ADH1 | Alcohol dehydrogenase enzyme | Reduces acetaldehyde to ethanol during fermentation; involved in NADH oxidation. | Increased | [43] |
| COX4 | Cytochrome c oxidase | Subunit IV of cytochrome c oxidase; functions in mitochondrial inner membrane ETC. | Increased | [44] |
| RPD3 | Reduced potassium Dependency | Histone deacetylase, a component of both the Rpd3S and Rpd3L complexes and regulates transcription. | Increased | [45] |
| DBP3 | Dead box protein | RNA-dependent ATPase, involved in rRNA processing. | Decreased | [46] |
| SGF73 | SAGA-associated factor 73 | DUB module subunit of SAGA and SLIK; contributes to de-ubiquitination activity. | Increased | [39] |
| PDE1 | Phosphodiesterase | Low-affinity cAMP phosphodiesterase. | Decreased | [47] |
| SGS1 | Slow growth suppressor | ATP-dependent DNA helicase. | Decreased | [48] |
| FOB1 | Fork blocking less | Nucleolar protein that binds to the rDNA replication fork barrier site; required for replication fork blocking. | Increased | [49] |
| PKH2, HXK1, HXK2 | Pkb-kinase homolog | Serine/threonine-protein kinase; involved in signaling cascade; involved in glucose metabolism, endocytosis, and cell wall integrity. | Increased | [50,51] |
| CDC25 | Cell division cycle | Membrane-bound guanine nucleotide exchange factor (GEF); regulates adenylate cyclase. | Increased | [52] |
| GPR1, GPA2, CYR1 | G-Protein-coupled receptor | Senses and integrates nutritional signals and decides cell fate via PKA and cAMP synthesis. | Increased | [53] |
| PHO84, CIT2 | PHOsphate metabolism and CITrate synthase | Effector of retrograde response in the extension of longevity. | Decreased | [54,55] |
| SOV1 | Synthesis of var | Member of the yeast mitochondrial translation control (MTC) module. | Increased | [34] |
| GCN4, GCN5 | General control nonderepressible | Roles in transcriptional activation. | Decreased | [56,57] |
| ASF1 | Anti-silencing function | Role in H3K56 acetylation; involved in chromatin assembly and disassembly. | Decreased | [37] |
| YPT6 | Yeast protein two | Rab family GTPase, required for retrograde transport. | Increased | [58] |
| YNO1/AIM14 | Yeast NADPH oxidase 1/altered inheritance rate of mitochondria | Endoplasmic reticulum localized NADPH oxidase. | Increased | [32] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahiya, R.; Mohammad, T.; Alajmi, M.F.; Rehman, M.T.; Hasan, G.M.; Hussain, A.; Hassan, M.I. Insights into the Conserved Regulatory Mechanisms of Human and Yeast Aging. Biomolecules 2020, 10, 882. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060882
Dahiya R, Mohammad T, Alajmi MF, Rehman MT, Hasan GM, Hussain A, Hassan MI. Insights into the Conserved Regulatory Mechanisms of Human and Yeast Aging. Biomolecules. 2020; 10(6):882. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060882
Chicago/Turabian StyleDahiya, Rashmi, Taj Mohammad, Mohamed F. Alajmi, Md. Tabish Rehman, Gulam Mustafa Hasan, Afzal Hussain, and Md. Imtaiyaz Hassan. 2020. "Insights into the Conserved Regulatory Mechanisms of Human and Yeast Aging" Biomolecules 10, no. 6: 882. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10060882