Untargeted LC-MS Metabolomics Differentiates Between Virulent and Avirulent Clinical Strains of Pseudomonas aeruginosa

 , , , and
, , , and
Abstract
:1. Introduction
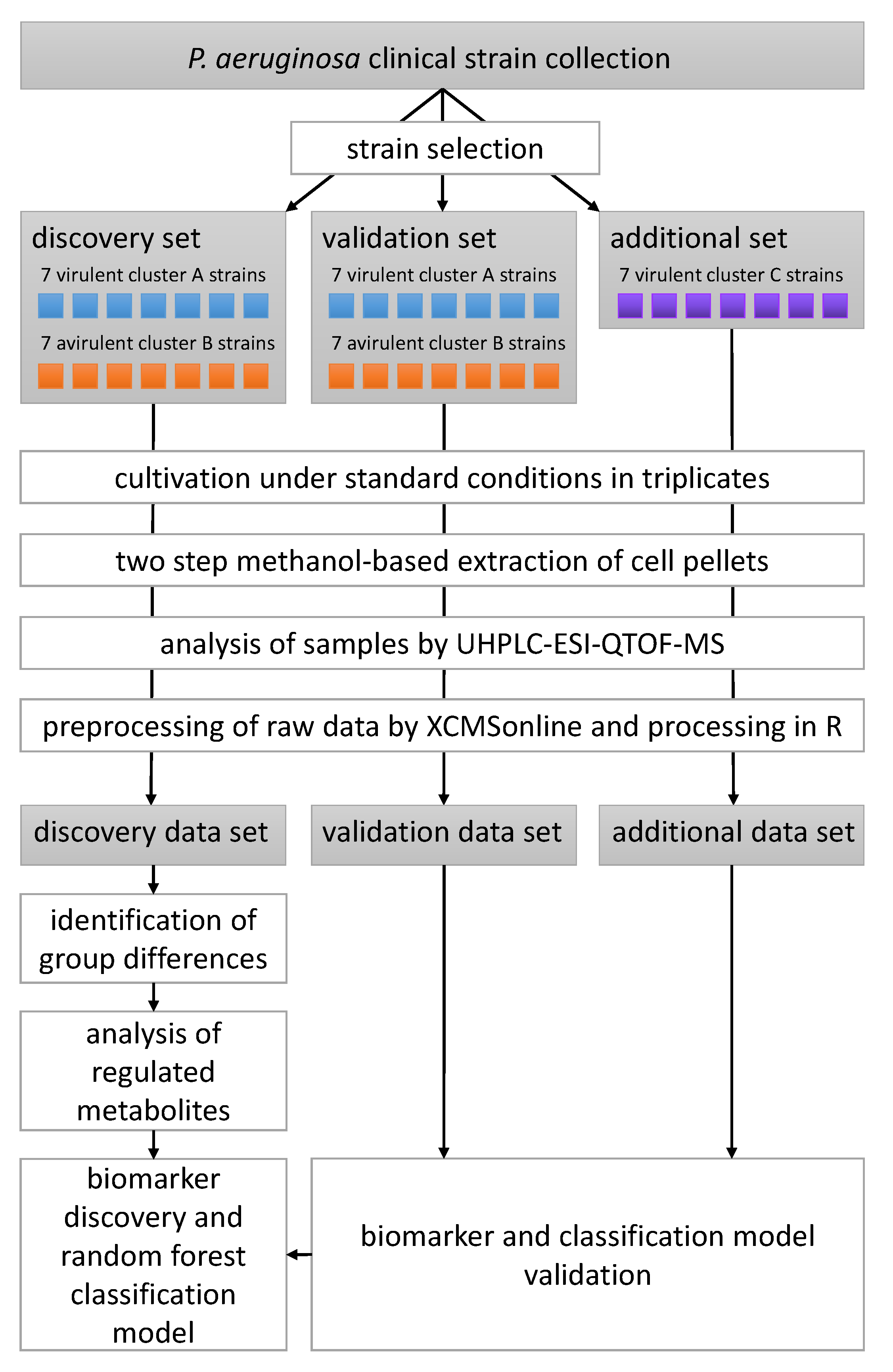
2. Materials and Methods
2.1. Bacterial Strains
2.2. Transcriptomics
2.3. Untargeted Metabolomics
2.4. Data Analysis and Model Building
3. Results
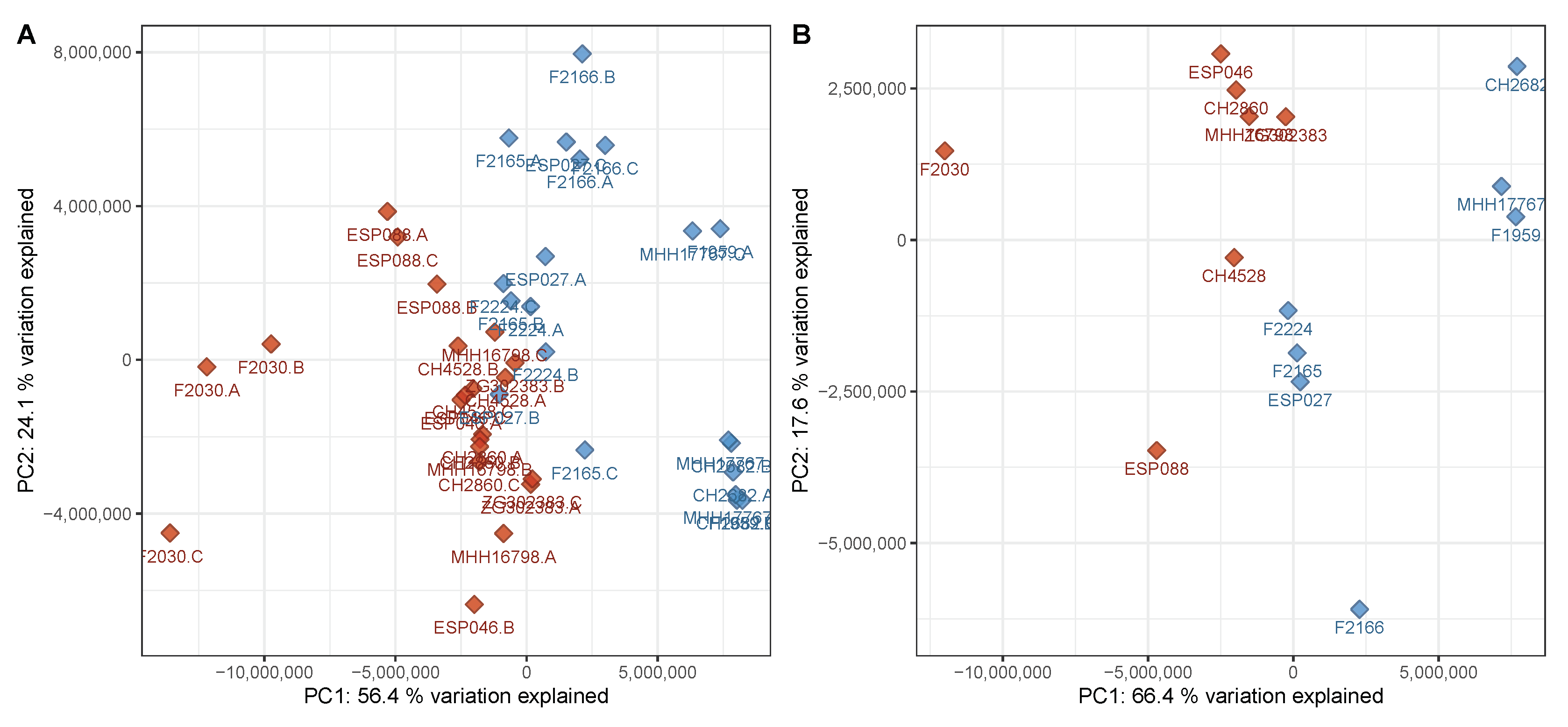
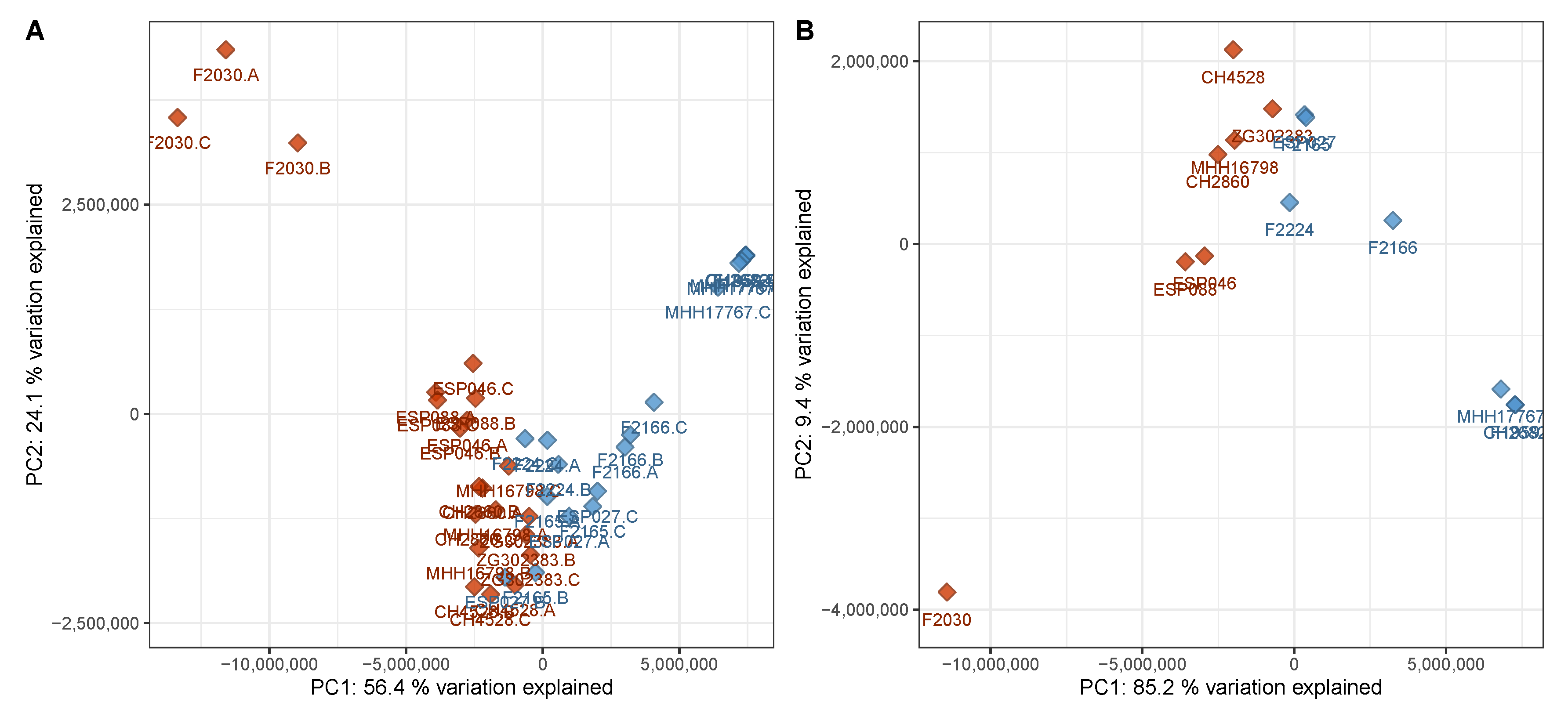
3.1. Virulent Cluster A and Avirulent Cluster B Strains Have Different Metabolic Profiles
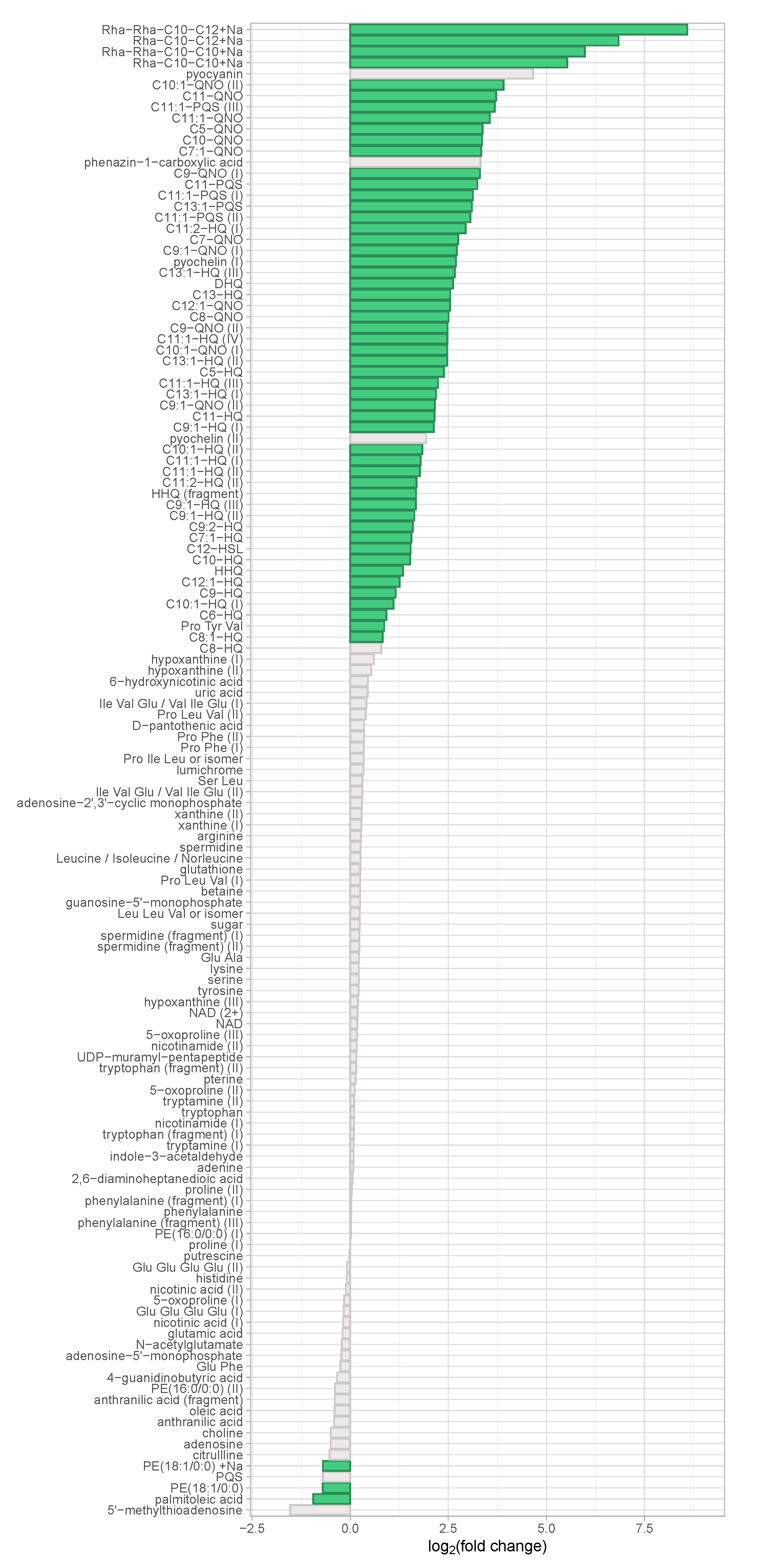
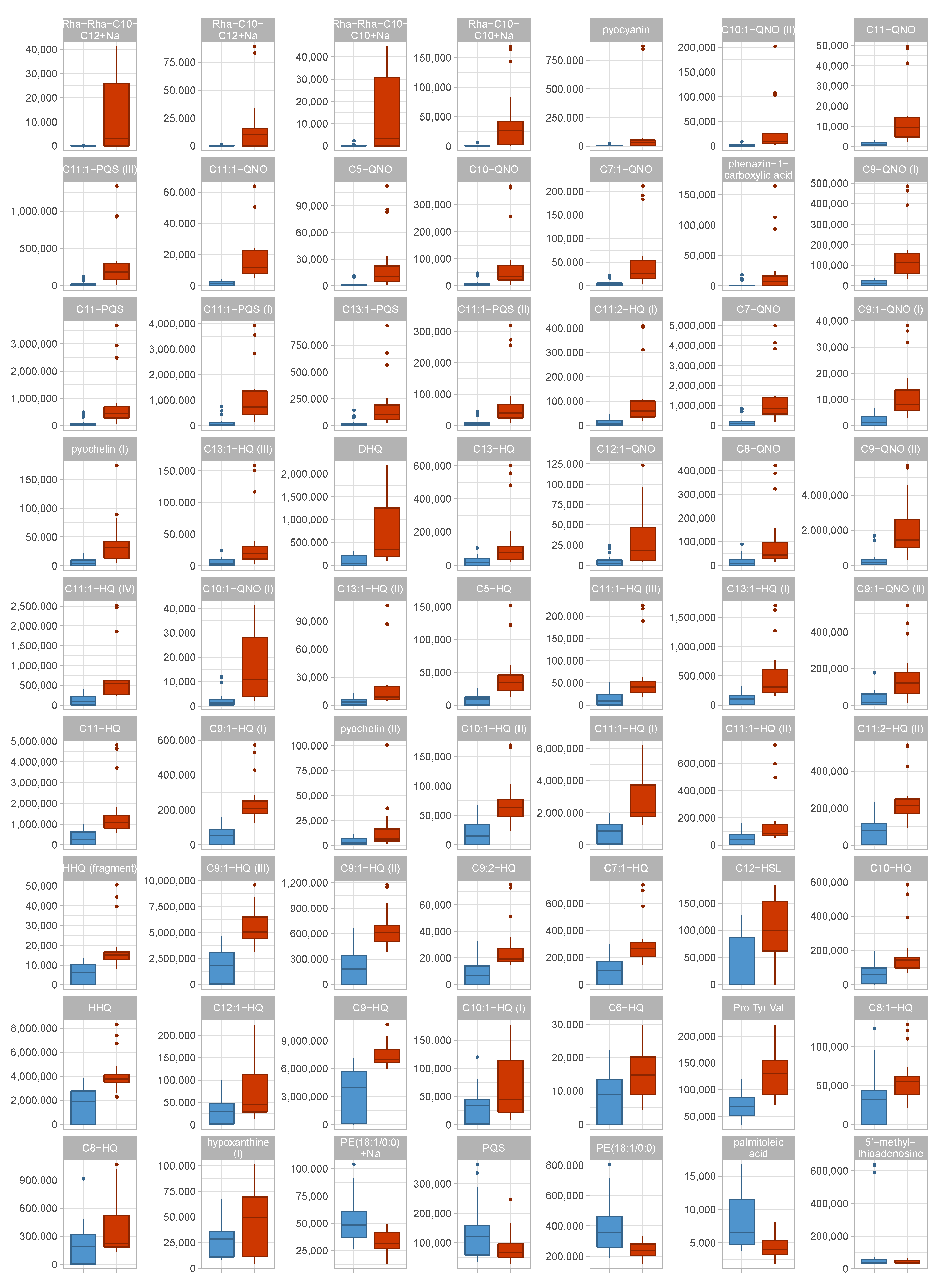
3.2. Metabolic Differences Between Virulent Cluster A and Avirulent Cluster B Strains Manifest in Differential Abundance of Virulence-Associated Secondary Metabolites
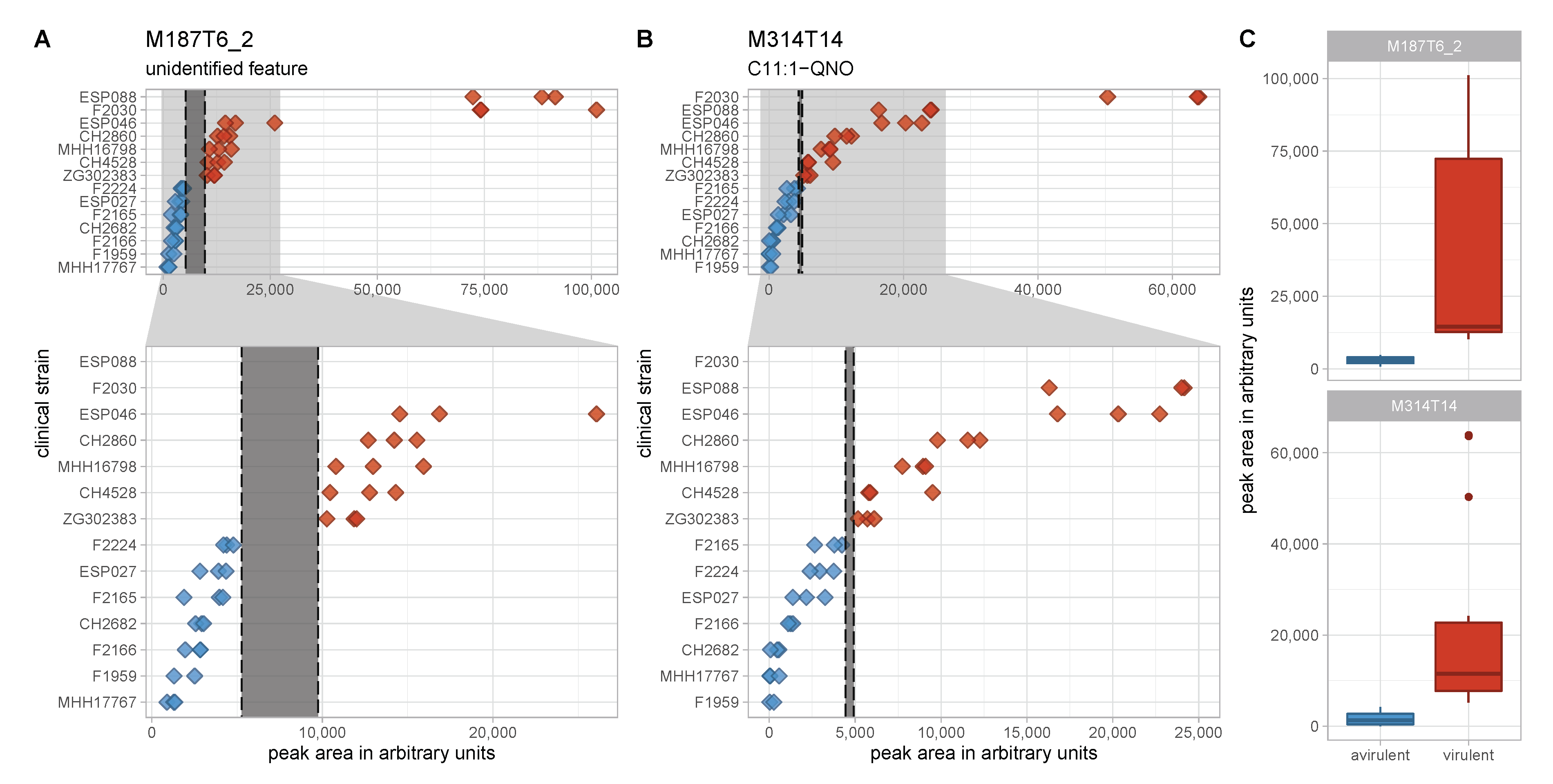
3.3. An Unknown Metabolite Is a Potential Biomarker for Virulent Phenotypes
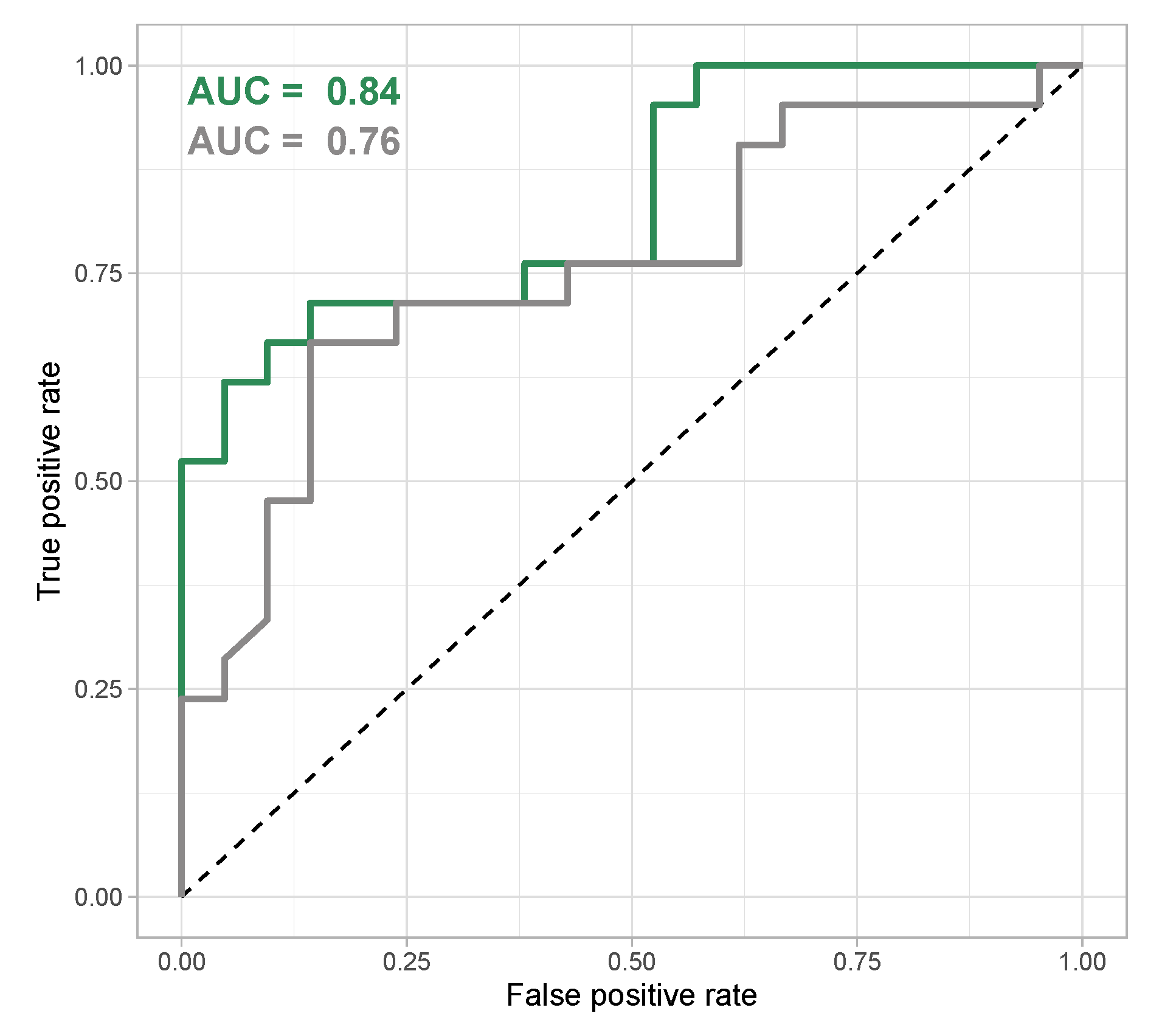
3.4. Virulent and Avirulent Strains with Distinct Biofilm Phenotypes Can Be Differentiated Based on Untargeted Metabolomics Data by Machine Learning
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AQ | alkyl quinolone |
| AUC | area under the curve |
| CF | cystic fibrosis |
| DHQ | 2,4-dihydroxyquinoline |
| ESI-QTOF-MS | electrospray ionisation quadrupole time-of-flight mass spectrometry |
| GNPS | Global Natural Product Social Molecular Networking |
| HSL | homeserine lactone |
| LC-MS | liquid chromatography–mass spectrometry |
| MASST | Mass Spectrometry Search Tool |
| m/z | mass-to-charge ratio |
| nd | not determined |
| nrpg | normalized reads per gene |
| OD600 | optical density at 600 nm |
| padj | adjusted p-value |
| PCA | principal component analysis |
| PE | phosphatidylethanolamine |
| PERMANOVA | Permutational multivariate analysis of variation |
| PQS | Pseudomonas quinolone signal |
| QNO | quinoline-N-oxide |
| Rha | rhamnose, rhamnosyl |
| ROC | receiver operating characteristics |
| VIP | variable importance in projection |
References
- Grosso-Becerra, M.V.; Santos-Medellín, C.; González-Valdez, A.; Méndez, J.L.; Delgado, G.; Morales-Espinosa, R.; Servín-González, L.; Alcaraz, L.D.; Soberón-Chávez, G. Pseudomonas aeruginosa clinical and environmental isolates constitute a single population with high phenotypic diversity. BMC Genom. 2014, 15, 318. [Google Scholar] [CrossRef] [Green Version]
- Gross, J.; Passmore, I.J.; Chung, J.C.S.; Rzhepishevska, O.; Ramstedt, M.; Welch, M. Universal soldier: Pseudomonas aeruginosa—An opportunistic generalist. Front. Biol. 2013, 8, 387–394. [Google Scholar] [CrossRef]
- Stapleton, F.; Dart, J.K.G.; Seal, D.V.; Matheson, M. Epidemiology of Pseudomonas aeruginosa keratitis in contact lens wearers. Epidemiol. Infect. 1995, 114, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.W.; Jung, J.Y.; Pashia, M.E.; Nason, R.; Scholnick, S.; Chole, R.A. Otopathogenic Pseudomonas aeruginosa Strains as Competent Biofilm Formers. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 983. [Google Scholar] [CrossRef] [Green Version]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Driscoll, J.A.; Brody, S.L.; Kollef, M.H. The Epidemiology, Pathogenesis and Treatment of Pseudomonas aeruginosa Infections. Drugs 2007, 67, 351–368. [Google Scholar] [CrossRef] [PubMed]
- de Bentzmann, S.; Plésiat, P. The Pseudomonas aeruginosa opportunistic pathogen and human infections. Environ. Microbiol. 2011, 13, 1655–1665. [Google Scholar] [CrossRef]
- Chastre, J.; Fagon, J.Y. Ventilator-associated Pneumonia. Am. J. Respir. Crit. Care Med. 2002, 165, 867–903. [Google Scholar] [CrossRef]
- Lund-Palau, H.; Turnbull, A.R.; Bush, A.; Bardin, E.; Cameron, L.; Soren, O.; Wierre-Gore, N.; Alton, E.W.F.W.; Bundy, J.G.; Connett, G.; et al. Pseudomonas aeruginosa infection in cystic fibrosis: Pathophysiological mechanisms and therapeutic approaches. Expert Rev. Respir. Med. 2016, 10, 685–697. [Google Scholar] [CrossRef]
- Cornelis, P.; Dingemans, J. Pseudomonas aeruginosa adapts its iron uptake strategies in function of the type of infections. Front. Cell. Infect. Microbiol. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Tümmler, B.; Wiehlmann, L.; Klockgether, J.; Cramer, N. Advances in understanding Pseudomonas. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.C.; Kulasekara, B.R.; Liang, X.; Boyd, D.; Wu, K.; Yang, Q.; Miyada, C.G.; Lory, S. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 8484–8489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, R.S.; Roddam, L.F.; Merritt, A.; Reid, D.W.; Champion, A.C. Virulence gene distribution in clinical, nosocomial and environmental isolates of Pseudomonas aeruginosa. J. Med. Microbiol. 2010, 59, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, J.; Miethke, N.; Kubesch, P.; Bohn, Y.S.; Brockhausen, I.; Cramer, N.; Eberl, L.; Greipel, J.; Herrmann, C.; Herrmann, S.; et al. Intraclonal diversity of the Pseudomonas aeruginosa cystic fibrosis airway isolates TBCF10839 and TBCF121838: Distinct signatures of transcriptome, proteome, metabolome, adherence and pathogenicity despite an almost identical genome sequence. Environ. Microbiol. 2012, 15, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Thöming, J.G.; Tomasch, J.; Preusse, M.; Koska, M.; Grahl, N.; Pohl, S.; Willger, S.D.; Kaever, V.; Müsken, M.; Häussler, S. Parallel evolutionary paths to produce more than one Pseudomonas aeruginosa biofilm phenotype. NPJ Biofilms Microb. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, J.; Tümmler, B. Recent advances in understanding Pseudomonas aeruginosa as a pathogen. F1000Research 2017, 6, 1261. [Google Scholar] [CrossRef]
- Hilker, R.; Munder, A.; Klockgether, J.; Losada, P.M.; Chouvarine, P.; Cramer, N.; Davenport, C.F.; Dethlefsen, S.; Fischer, S.; Peng, H.; et al. Interclonal gradient of virulence in the Pseudomonas aeruginosa pangenome from disease and environment. Environ. Microbiol. 2014, 17, 29–46. [Google Scholar] [CrossRef]
- Bielecki, P.; Komor, U.; Bielecka, A.; Müsken, M.; Puchałka, J.; Pletz, M.W.; Ballmann, M.; dos Santos, V.A.M.; Weiss, S.; Häussler, S. Ex vivotranscriptional profiling reveals a common set of genes important for the adaptation of Pseudomonas aeruginosa to chronically infected host sites. Environ. Microbiol. 2012, 15, 570–587. [Google Scholar] [CrossRef]
- Sousa, A.M.; Monteiro, R.; Pereira, M.O. Unveiling the early events of Pseudomonas aeruginosa adaptation in cystic fibrosis airway environment using a long-term in vitro maintenance. Int. J. Med. Microbiol. 2018, 308, 1053–1064. [Google Scholar] [CrossRef] [Green Version]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The apogee of the omics trilogy. Nature Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Warth, B.; Spilker, M.E.; Siuzdak, G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 2018, 36, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.N.; Koch, G.; Thompson, J.A.; Xavier, K.B.; Cool, R.H.; Quax, W.J. The Multiple Signaling Systems Regulating Virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depke, T.; Franke, R.; Brönstrup, M. Clustering of MS2 spectra using unsupervised methods to aid the identification of secondary metabolites from Pseudomonas aeruginosa. J. Chromatogr. B 2017, 1071, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Ortori, C.A.; Dubern, J.F.; Chhabra, S.R.; Cámara, M.; Hardie, K.; Williams, P.; Barrett, D.A. Simultaneous quantitative profiling of N-acyl-L-homoserine lactone and 2-alkyl-4(1H)-quinolone families of quorum-sensing signaling molecules using LC-MS/MS. Anal. Bioanal. Chem. 2010, 399, 839–850. [Google Scholar] [CrossRef]
- Allegretta, G.; Maurer, C.K.; Eberhard, J.; Maura, D.; Hartmann, R.W.; Rahme, L.; Empting, M. In-depth Profiling of MvfR-Regulated Small Molecules in Pseudomonas aeruginosa after Quorum Sensing Inhibitor Treatment. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Xu, G. Mass-spectrometry-based microbial metabolomics: Recent developments and applications. Ana. Bioanal. Chem. 2014, 407, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Grim, C.M.; Luu, G.T.; Sanchez, L.M. Staring into the void: Demystifying microbial metabolomics. FEMS Microbiol. Lett. 2019, 366. [Google Scholar] [CrossRef]
- Bundy, J.G.; Willey, T.L.; Castell, R.S.; Ellar, D.J.; Brindle, K.M. Discrimination of pathogenic clinical isolates and laboratory strains of Bacillus cereus by NMR-based metabolomic profiling. FEMS Microbiol. Lett. 2005, 242, 127–136. [Google Scholar] [CrossRef] [Green Version]
- t’Kindt, R.; Scheltema, R.A.; Jankevics, A.; Brunker, K.; Rijal, S.; Dujardin, J.C.; Breitling, R.; Watson, D.G.; Coombs, G.H.; Decuypere, S. Metabolomics to Unveil and Understand Phenotypic Diversity between Pathogen Populations. PLoS Negl. Trop. Diseases 2010, 4, e904. [Google Scholar] [CrossRef]
- Maifiah, M.H.M.; Cheah, S.E.; Johnson, M.D.; Han, M.L.; Boyce, J.D.; Thamlikitkul, V.; Forrest, A.; Kaye, K.S.; Hertzog, P.; Purcell, A.W.; et al. Global metabolic analyses identify key differences in metabolite levels between polymyxin-susceptible and polymyxin-resistant Acinetobacter baumannii. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bean, H.D.; Rees, C.A.; Hill, J.E. Comparative analysis of the volatile metabolomes of Pseudomonas aeruginosa clinical isolates. J. Breath Res. 2016, 10, 047102. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K.M.; Ciofu, O.; Molin, S.; Wassermann, T.; Johansen, H.K.; Høiby, N.; Christiansen, L.E. Diversity of metabolic profiles of cystic fibrosis Pseudomonas aeruginosa during the early stages of lung infection. Microbiology 2015, 161, 1447–1462. [Google Scholar] [CrossRef] [Green Version]
- Kordes, A.; Grahl, N.; Koska, M.; Preusse, M.; Arce-Rodriguez, A.; Abraham, W.R.; Kaever, V.; Häussler, S. Establishment of an induced memory response in Pseudomonas aeruginosa during infection of a eukaryotic host. ISME J. 2019, 13, 2018–2030. [Google Scholar] [CrossRef]
- Hornischer, K.; Khaledi, A.; Pohl, S.; Schniederjans, M.; Pezoldt, L.; Casilag, F.; Muthukumarasamy, U.; Bruchmann, S.; Thöming, J.; Kordes, A.; et al. BACTOME—A reference database to explore the sequence- and gene expression-variation landscape of Pseudomonas aeruginosa clinical isolates. Nucleic Acids Res. 2018, 47, D716–D720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaledi, A.; Weimann, A.; Schniederjans, M.; Asgari, E.; Kuo, T.H.; Oliver, A.; Cabot, G.; Kola, A.; Gastmeier, P.; Hogardt, M.; et al. Predicting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef]
- Erdmann, J.; Thöming, J.G.; Pohl, S.; Pich, A.; Lenz, C.; Häussler, S. The Core Proteome of Biofilm-Grown Clinical Pseudomonas aeruginosa Isolates. Cells 2019, 8, 1129. [Google Scholar] [CrossRef] [Green Version]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genom. Res. 2010, 21, 936–939. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genom. Biol. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Mathee, K.; Narasimhan, G.; Valdes, C.; Qiu, X.; Matewish, J.M.; Koehrsen, M.; Rokas, A.; Yandava, C.N.; Engels, R.; Zeng, E.; et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 3100–3105. [Google Scholar] [CrossRef] [Green Version]
- Witzgall, F.; Depke, T.; Hoffmann, M.; Empting, M.; Brönstrup, M.; Müller, R.; Blankenfeldt, W. The Alkylquinolone Repertoire of Pseudomonas aeruginosa is Linked to Structural Flexibility of the FabH-like 2-Heptyl-3-hydroxy-4(1H)-quinolone (PQS) Biosynthesis Enzyme PqsBC. ChemBioChem 2018, 19, 1531–1544. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, Inc.: Boston, MA, USA, 2019. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2011, 84, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahieu, N.G.; Huang, X.; Chen, Y.J.; Patti, G.J. Credentialing Features: A Platform to Benchmark and Optimize Untargeted Metabolomic Methods. Anal. Chem. 2014, 86, 9583–9589. [Google Scholar] [CrossRef] [Green Version]
- Scherber, W. Stable Isotope Labeling to Improve Metabolite Identification in Untargeted Metabolomics of Pathogenic Bacteria. Master’s Thesis, Hochschule Aalen—Technik und Wirtschaft, Aalen, Germany, 2020. [Google Scholar]
- Wang, M.; Jarmusch, A.K.; Vargas, F.; Aksenov, A.A.; Gauglitz, J.M.; Weldon, K.; Petras, D.; da Silva, R.; Quinn, R.; Melnik, A.V.; et al. Mass spectrometry searches using MASST. Nat. Biotechnol. 2020, 38, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package, R package version 2.5-6. 2019.
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef]
- Mavrodi, D.V.; Blankenfeldt, W.; Thomashow, L.S. Phenazine Compounds in Fluorescent Pseudomonas Spp. Biosynthesis and Regulation. Ann. Rev. Phytopathol. 2006, 44, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Guttenberger, N.; Blankenfeldt, W.; Breinbauer, R. Recent developments in the isolation, biological function, biosynthesis, and synthesis of phenazine natural products. Bioorg. Med. Chem. 2017, 25, 6149–6166. [Google Scholar] [CrossRef]
- Boles, B.R.; Thoendel, M.; Singh, P.K. Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol. Microbiol. 2005, 57, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Zulianello, L.; Canard, C.; Köhler, T.; Caille, D.; Lacroix, J.S.; Meda, P. Rhamnolipids Are Virulence Factors That Promote Early Infiltration of Primary Human Airway Epithelia by Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 3134–3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calfee, M.W.; Shelton, J.G.; McCubrey, J.A.; Pesci, E.C. Solubility and Bioactivity of the Pseudomonas Quinolone Signal Are Increased by a Pseudomonas aeruginosa-Produced Surfactant. Infect. Immun. 2005, 73, 878–882. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, K.; Taraz, K.; Budzikiewicz, H. The stereoisomers of pyochelin, a siderophore of Pseudomonas aeruginosa. BioMetals 2004, 17, 409–414. [Google Scholar] [CrossRef]
- Palmer, G.C.; Palmer, K.L.; Jorth, P.A.; Whiteley, M. Characterization of the Pseudomonas aeruginosa Transcriptional Response to Phenylalanine and Tyrosine. J. Bacteriol. 2010, 192, 2722–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, L.; Dong, N.; Yun, Y.; Deng, B.; Ren, D.; Liu, S.; Liang, Y. Chemometric methods in data processing of mass spectrometry-based metabolomics: A review. Anal. Chim. Acta 2016, 914, 17–34. [Google Scholar] [CrossRef]
- Chen, T.; Cao, Y.; Zhang, Y.; Liu, J.; Bao, Y.; Wang, C.; Jia, W.; Zhao, A. Random Forest in Clinical Metabolomics for Phenotypic Discrimination and Biomarker Selection. Evid. Based Complement. Altern. Med. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Frimmersdorf, E.; Horatzek, S.; Pelnikevich, A.; Wiehlmann, L.; Schomburg, D. How Pseudomonas aeruginosa adapts to various environments: A metabolomic approach. Environ. Microbiol. 2010, 12, 1734–1747. [Google Scholar] [CrossRef]
- Bardin, E.E.; Cameron, S.J.S.; Perdones-Montero, A.; Hardiman, K.; Bolt, F.; Alton, E.W.F.W.; Bush, A.; Davies, J.C.; Takáts, Z. Metabolic Phenotyping and Strain Characterisation of Pseudomonas aeruginosa Isolates from Cystic Fibrosis Patients Using Rapid Evaporative Ionisation Mass Spectrometry. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaber, J.A.; Carty, N.L.; McDonald, N.A.; Graham, E.D.; Cheluvappa, R.; Griswold, J.A.; Hamood, A.N. Analysis of quorum sensing-deficient clinical isolates of Pseudomonas aeruginosa. J. Med. Microbiol. 2004, 53, 841–853. [Google Scholar] [CrossRef] [Green Version]
- Davenport, P.W.; Griffin, J.L.; Welch, M. Quorum Sensing Is Accompanied by Global Metabolic Changes in the Opportunistic Human Pathogen Pseudomonas aeruginosa. J. Bacteriol. 2015, 197, 2072–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, R.A.; Phelan, V.V.; Whiteson, K.L.; Garg, N.; Bailey, B.A.; Lim, Y.W.; Conrad, D.J.; Dorrestein, P.C.; Rohwer, F.L. Microbial, host and xenobiotic diversity in the cystic fibrosis sputum metabolome. ISME J. 2015, 10, 1483–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, R.A.; Adem, S.; Mills, R.H.; Comstock, W.; Goldasich, L.D.; Humphrey, G.; Aksenov, A.A.; Melnik, A.V.; da Silva, R.; Ackermann, G.; et al. Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Xu, Y.F.; Lu, W.; Rabinowitz, J.D. Avoiding Misannotation of In-Source Fragmentation Products as Cellular Metabolites in Liquid Chromatography–Mass Spectrometry-Based Metabolomics. Anal. Chem. 2015, 87, 2273–2281. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Gallagher, T.; Zhang, Y.; Elbadawi-Sidhu, M.; Lai, Z.; Fiehn, O.; Whiteson, K.L. Tracking Polymicrobial Metabolism in Cystic Fibrosis Airways: Pseudomonas aeruginosa Metabolism and Physiology Are Influenced by Rothia mucilaginosa-Derived Metabolites. mSphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.; Pereira, M. Pseudomonas aeruginosa Diversification during Infection Development in Cystic Fibrosis Lungs—A Review. Pathogens 2014, 3, 680–703. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Discovery Data Set | |||
| Strain | Biofilm Cluster | GalleriaSurvival (48 h) | Infection/Sampling Site |
| CH2860 | A | 5 | Respiratory tract |
| CH4528 | A | 0 | Respiratory tract |
| ESP046 | A | 0 | nd/other |
| ESP088 | A | 5 | nd/other |
| F2030 | A | 0 | Respiratory tract |
| MHH16798 | A | 20 | Respiratory tract |
| ZG302383 | A | 0 | nd/other |
| CH2682 | B | 95 | Rectal swab |
| ESP027 | B | 100 | nd/other |
| F1959 | B | 100 | Respiratory tract |
| F2165 | B | 100 | Respiratory tract |
| F2166 | B | 100 | Respiratory tract |
| F2224 | B | 95 | nd/other |
| MHH17767 | B | 100 | Respiratory tract |
| Validation Data Set | |||
| Strain | Biofilm Cluster | GalleriaSurvival (48 h) | Infection/Sampling Site |
| CH2690 | A | 0 | Rectal swab |
| ESP058 | A | 0 | nd/other |
| ESP067 | A | 5 | nd/other |
| F1997 | A | 0 | Rectal swab |
| MHH17704 | A | 5 | nd/other |
| Psae1439 | A | 10 | Respiratory tract |
| ZG8038581181 | A | 10 | Respiratory tract |
| CH4681 | B | 90 | Respiratory tract |
| F1764 | B | 95 | Respiratory tract |
| F2020 | B | 95 | Wound infection |
| MHH16050 | B | 60 | nd/other |
| MHH16563 | B | 95 | Respiratory tract |
| MHH17546 | B | 100 | Respiratory tract |
| Psae1837 | B | 75 | nd/other |
| Additional Data Set | |||
| Strain | Biofilm Cluster | GalleriaSurvival (48 h) | Infection/Sampling Site |
| CH2706 | C | 0 | Rectal swab |
| CH4591 | C | 0 | Rectal swab |
| ESP083 | C | 0 | nd/other |
| F1864 | C | 0 | nd/other |
| F2059 | C | 0 | Wound infection |
| ZG316717 | C | 5 | Ear infection |
| ZG8510487 | C | 0 | Urinary tract infection |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Depke, T.; Thöming, J.G.; Kordes, A.; Häussler, S.; Brönstrup, M. Untargeted LC-MS Metabolomics Differentiates Between Virulent and Avirulent Clinical Strains of Pseudomonas aeruginosa. Biomolecules 2020, 10, 1041. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10071041
Depke T, Thöming JG, Kordes A, Häussler S, Brönstrup M. Untargeted LC-MS Metabolomics Differentiates Between Virulent and Avirulent Clinical Strains of Pseudomonas aeruginosa. Biomolecules. 2020; 10(7):1041. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10071041
Chicago/Turabian StyleDepke, Tobias, Janne Gesine Thöming, Adrian Kordes, Susanne Häussler, and Mark Brönstrup. 2020. "Untargeted LC-MS Metabolomics Differentiates Between Virulent and Avirulent Clinical Strains of Pseudomonas aeruginosa" Biomolecules 10, no. 7: 1041. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10071041