Glycyl-L-Prolyl-L-Glutamate Pseudotripeptides for Treatment of Alzheimer’s Disease

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Cellular Model of AD
2.2. Treatments
2.3. MTT Assay
2.4. LDH Assay
2.5. Determination of AChE Activity
2.6. Determination of β-Secretase and α-Secretase Activities
2.7. TAC and TOS Analysis
2.8. Apoptosis Detection by Hoechst 33258 Staining
2.9. Apoptosis-Necrosis Assay
2.10. Molecular Genetic Analysis
2.10.1. Total RNA Isolation
2.10.2. cDNA Synthesis
2.10.3. PCR Array
2.11. Statistical Analyses
3. Results and Discussion
3.1. Aβ1-42 Treatments Induced Cell Death in Differentiated SH-SY5Y Cells
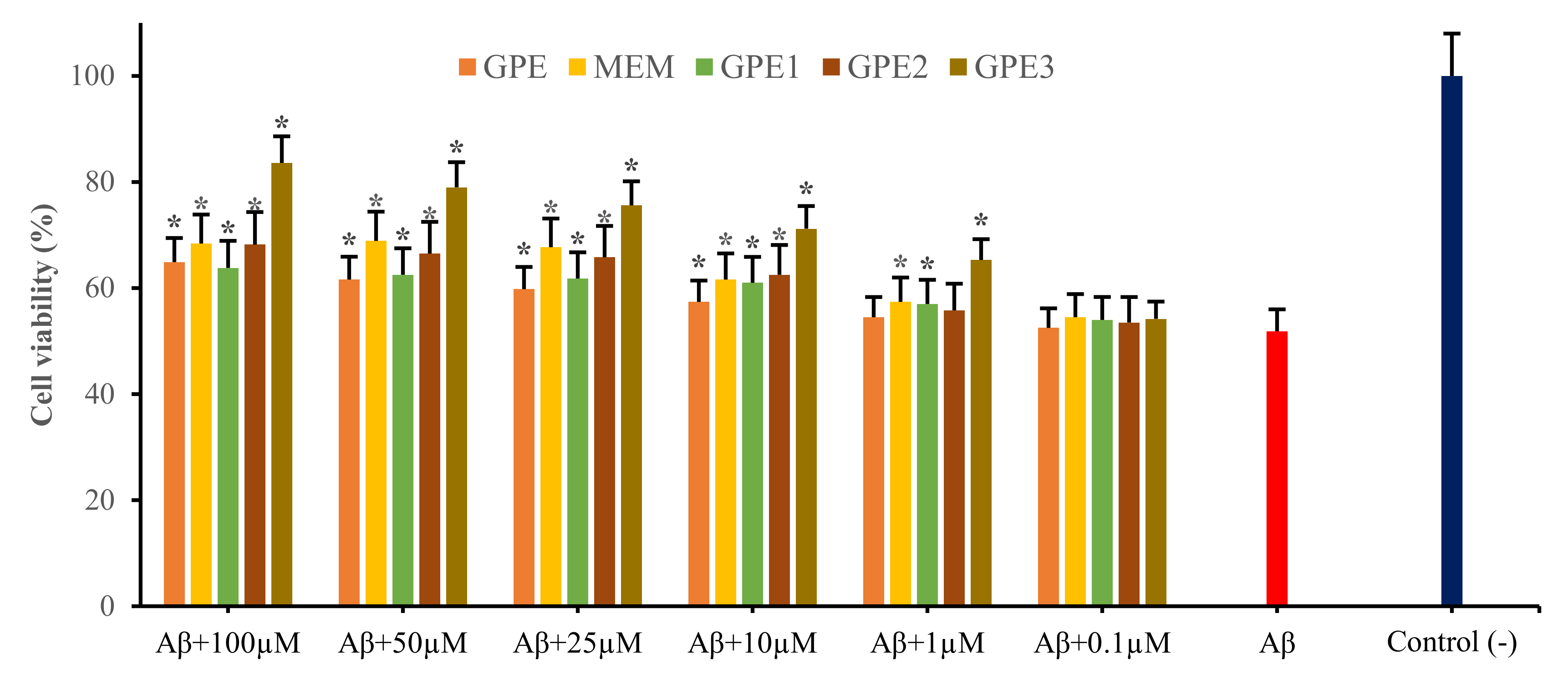
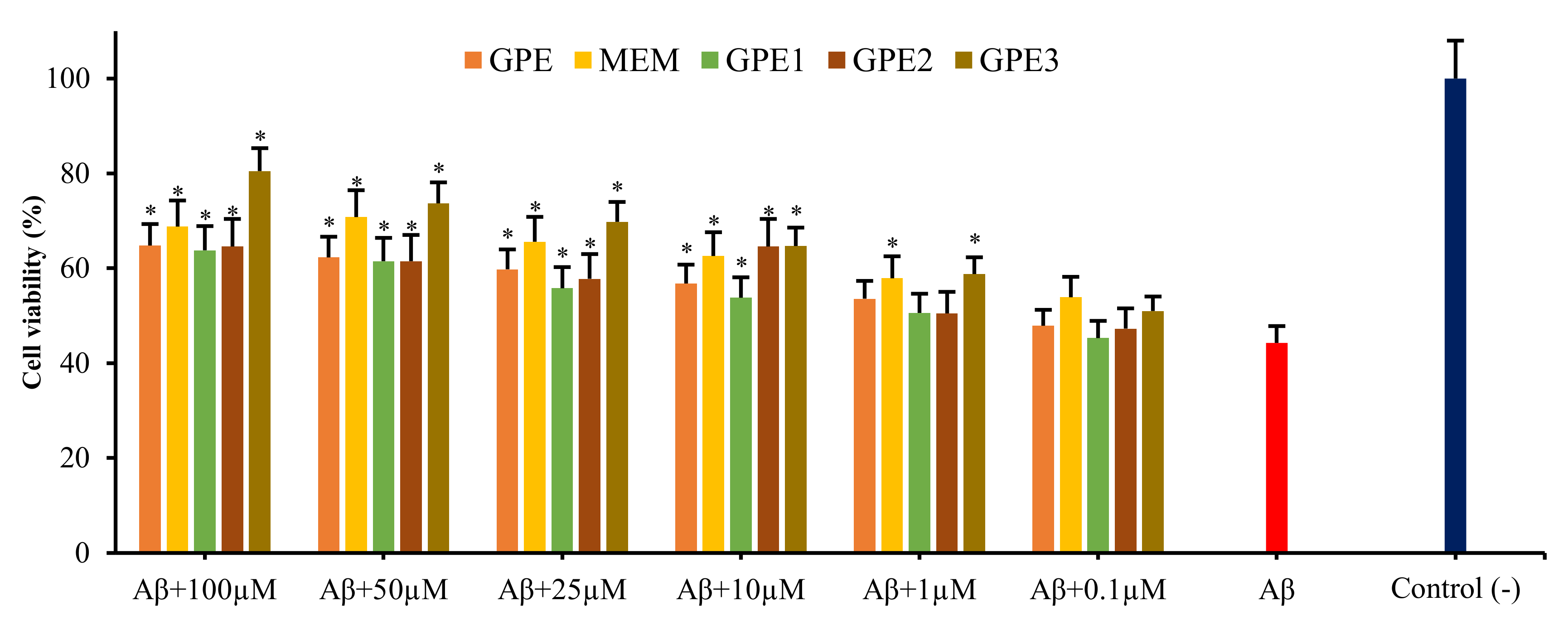
3.2. Neuroprotective Potentials of Novel GPEs on Aβ1-42-Induced Cytotoxicity in In Vitro Cellular AD Model
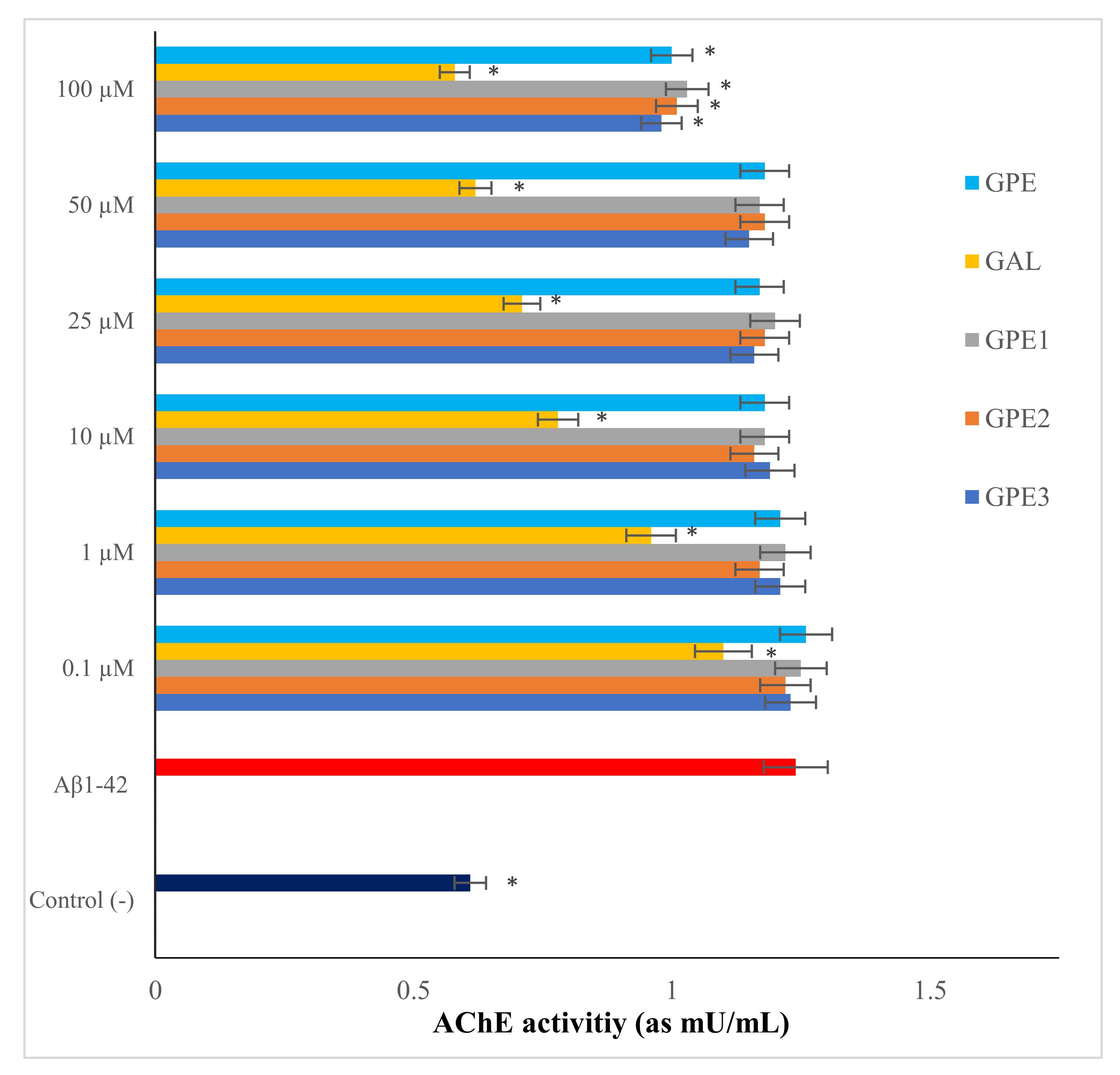
3.3. The In Vitro Effects of Novel GPEs on AChE Activity
3.4. The In Vitro Effects of Novel GPEs on β-Secretase and α-Secretase Activities
3.5. Effects of Applications with Novel GPEs on TAC and TOS Levels in the Cellular Model of AD
3.6. Effects of Novels GPEs on Apoptosis and Necrosis in the Cellular Model of AD
3.7. Molecular Genetic Responses to GPE3 in Cellular AD Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamamoto, H.; Murphy, L.J. Enzymatic conversion of IGF-I to des(1–3)IGF-I in rat serum and tissues: A further potential site of growth hormone regulation of IGF-I action. J. Endocrinol. 1995, 146, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Sara, V.R.; Carlsson-Skwirut, C.; Bergman, T.; Jörnvall, H.; Roberts, P.J.; Crawford, M.; Håkansson, L.N.; Civalero, I.; Nordberg, A. Identification of Gly-Pro-Glu (GPE), the aminoterminal tripeptide of insulin-like growth factor 1 which is truncated in brain, as a novel neuroactive peptide. Biochem. Biophys. Res. Commun. 1989, 165, 766–771. [Google Scholar] [CrossRef]
- Burgos-Ramos, E.; Martos-Moreno, G.Á.; López, M.G.; Herranz, R.; Aguado-Llera, D.; Egea, J. The N-terminal tripeptide of insulin-like growth factor-I protects against β-amyloid-induced somatostatin depletion by calcium and glycogen synthase kinase 3β modulation. J. Neurochem. 2009, 109, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Sizonenko, S.V.; Sirimanne, E.S.; Williams, C.E.; Gluckman, P.D. Neuroprotective effects of the N-terminal tripeptide of IGF-1, glycine-proline-glutamate, in the immature rat brain after hypoxic–ischemic injury. Brain Res. 2001, 922, 42–50. [Google Scholar] [CrossRef]
- Guan, J.; Thomas, G.B.; Lin, H.; Mathai, S.; Bachelor, D.C.; George, S. Neuroprotective effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate (GPE) following intravenous infusion in hypoxic–ischemic adult rats. Neuropharmacology 2004, 47, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Aguado-Llera, D.; Martín-Martínez, M.; García-López, M.T.; Arilla-Ferreiro, E.; Barrios, V. Gly-Pro-Glu protects β-amyloid-induced somatostatin depletion in the rat cortex. Neuroreport 2004, 15, 1979–1982. [Google Scholar] [CrossRef]
- Minelli, A.; Conte, C.; Cacciatore, I.; Cornacchia, C.; Pinnen, F. Molecular mechanism underlying the cerebral effect of Gly-Pro-Glu tripeptide bound to l-dopa in a Parkinson’s animal model. Amino Acids 2012, 43, 1359–1367. [Google Scholar] [CrossRef]
- Guan, J.; Krishnamurthi, R.; Waldvogel, H.J.; Faull, R.L.M.; Clark, R.; Gluckman, P. N-terminal tripeptide of IGF-1 (GPE) prevents the loss of TH positive neurons after 6-OHDA induced nigral lesion in rats. Brain Res. 2000, 859, 286–292. [Google Scholar] [CrossRef]
- Stepanova, P.; Srinivasan, V.; Lindholm, D.; Voutilainen, M.H. Cerebral dopamine neurotrophic factor (CDNF) protects against quinolinic acid-induced toxicity in in vitro and in vivo models of Huntington’s disease. Sci. Rep. 2020, 10, 19045. [Google Scholar] [CrossRef]
- Dong, X.; Wang, Y.; Qin, Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Aguado-Llera, D.; Canelles, S.; Fernández-Mendívil, C.; Frago, L.M.; Argente, J.; Arilla-Ferreiro, E. Improvement in inflammation is associated with the protective effect of Gly-Pro-Glu and cycloprolylglycine against Aβ-induced depletion of the hippocampal somatostatinergic system. Neuropharmacology 2019, 151, 112–126. [Google Scholar] [CrossRef]
- Marinelli, L.; Fornasari, E.; Di Stefano, A.; Turkez, H.; Genovese, S.; Epifano, F. Synthesis and biological evaluation of novel analogues of Gly-l-Pro-l-Glu (GPE) as neuroprotective agents. Bioorg. Med. Chem. Lett. 2019, 29, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Alonso De Diego, S.A.; Gutiérrez-Rodríguez, M.; Pérez De Vega, M.J.; Casabona, D.; Cativiela, C.; González-Muñiz, R. New Gly-Pro-Glu (GPE) analogues: Expedite solid-phase synthesis and biological activity. Bioorganic Med. Chem. Lett. 2006, 16, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Ceña, V. Sequential Treatment of SH-SY5Y Cells with Retinoic Acid and Brain-Derived Neurotrophic Factor Gives Rise to Fully Differentiated, Neurotrophic Factor-Dependent, Human Neuron-Like Cells. J. Neurochem. 2002, 75, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Shan, X.; Li, X.; Fan, W.; Zhang, S.Q.; Zhang, J. Toxoplasma gondii rhoptry protein ROP16 mediates partially SH-SY5Y cells apoptosis and cell cycle arrest by directing ser15/37 phosphorylation of p53. Int. J. Biol. Sci. 2015, 11, 1215–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkez, H.; Cacciatore, I.; Arslan, M.E.; Fornasari, E.; Marinelli, L.; Di Stefano, A. Histidyl-proline diketopiperazine isomers as multipotent anti-alzheimer drug candidates. Biomolecules 2020, 10, 737. [Google Scholar] [CrossRef]
- Kucinska, M.; Giron, M.D.; Piotrowska, H.; Lisiak, N.; Granig, W.H.; Lopez-Jaramillo, F.J. Novel promising estrogenic receptor modulators: Cytotoxic and estrogenic activity of benzanilides and dithiobenzanilides. PLoS ONE 2016, 11, e0145615. [Google Scholar] [CrossRef]
- Küçükdoğru, R.; Türkez, H.; Arslan, M.E.; Tozlu, Ö.Ö.; Sönmez, E.; Mardinoğlu, A. Neuroprotective effects of boron nitride nanoparticles in the experimental Parkinson’s disease model against MPP+ induced apoptosis. Metab. Brain Dis. 2020, 35, 947–957. [Google Scholar] [CrossRef]
- Emsen, B.; Aslan, A.; Togar, B.; Turkez, H. In vitro antitumor activities of the lichen compounds olivetoric, physodic and psoromic acid in rat neuron and glioblastoma cells. Pharm. Biol. 2016, 54, 1748–1762. [Google Scholar] [CrossRef] [Green Version]
- Alak, G.; Özgeriş, F.B.; Yeltekin, A.Ç.; Parlak, V.; Ucar, A.; Caglar, O. Hematological and Hepatic Effects of Ulexite in Zebrafish. Environ. Toxicol. Pharmacol. 2020, 80, 103496. [Google Scholar] [CrossRef]
- Turkez, H.; Togar, B.; Tatar, A.; Geyıkoglu, F.; Hacımuftuoglu, A. Cytotoxic and cytogenetic effects of α-copaene on rat neuron and N2a neuroblastoma cell lines. Biologia 2014, 69, 936–942. [Google Scholar] [CrossRef]
- Togar, B.; Türkez, H.; Stefano, A.; Tatar, A.; Cetin, D. Zingiberene attenuates hydrogen peroxide-induced toxicity in neuronal cells. Hum. Exp. Toxicol. 2015, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhao, Q.; Xiang, H.; Liu, M.; Zhang, Q.; Xue, W. Antiproliferative activity and apoptosis-inducing mechanism of constituents from Toona sinensis on human cancer cells. Cancer Cell Int. 2013, 13, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majstorović, I.; Vučević, D.; Pavlović, B.; Vasilijić, S.; Čolić, M. An anti-DEC-205 monoclonal antibody stimulates binding of thymocytes to rat thymic dendritic cells and promotes apoptosis of thymocytes. Cent. Eur. J. Immunol. 2014, 39, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Aydın, E.; Türkez, H.; Hacımüftüoğlu, F.; Tatar, A.; Geyikoğlu, F. Molecular genetic and biochemical responses in human airway epithelial cell cultures exposed to titanium nanoparticles in vitro. J. Biomed. Mater. Res. A 2017, 105, 2056–2064. [Google Scholar] [CrossRef]
- Laserra, S.; Basit, A.; Sozio, P.; Marinelli, L.; Fornasari, E.; Cacciatore, I. Solid lipid nanoparticles loaded with lipoyl-memantine codrug: Preparation and characterization. Int. J. Pharm. 2015, 485, 183–191. [Google Scholar] [CrossRef]
- Jeong, J.H.; Jeong, H.R.; Jo, Y.N.; Kim, H.J.; Shin, J.H.; Heo, H.J. Ameliorating effects of aged garlic extracts against Aβ-induced neurotoxicity and cognitive impairment. BMC Complement Altern. Med. 2013, 13, 268. [Google Scholar] [CrossRef] [Green Version]
- Korecka, J.A.; van Kesteren, R.E.; Blaas, E.; Spitzer, S.O.; Kamstra, J.H.; Smit, A.B. Phenotypic Characterization of Retinoic Acid Differentiated SH-SY5Y Cells by Transcriptional Profiling. PLoS ONE 2013, 8, e63862. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Gray, N.W.; Brimijoin, S. Amyloid-beta increases acetylcholinesterase expression in neuroblastoma cells by reducing enzyme degradation. J. Neurochem. 2004, 86, 470–478. [Google Scholar] [CrossRef]
- Nilsson-Hakansson, L.; Civalero, I.; Zhang, X.; Carlsson-Skwirut, C.; Sara, V.R.; Nordberg, A. Effects of IGF-1, truncated IGF-1 and the tripeptide Gly-Pro-Glu on acetylcholine release from parietal cortex of rat brain. Neuroreport 1993, 4, 1111–1114. [Google Scholar]
- Mori, T.; Koyama, N.; Yokoo, T.; Segawa, T.; Maeda, M.; Sawmiller, D. Gallic acid is a dual α/β-secretase modulator that reverses cognitive impairment and remediates pathology in Alzheimer mice. J. Biol. Chem. 2020, 295, 16251–16266. [Google Scholar] [CrossRef] [PubMed]
- Sennvik, K.; Fastbom, J.; Blomberg, M.; Wahlund, L.-O.; Winblad, B.; Benedikz, E. Levels of α- and β-secretase cleaved amyloid precursor protein in the cerebrospinal fluid of Alzheimer’s disease patients. Neurosci. Lett. 2000, 278, 169–172. [Google Scholar] [CrossRef]
- Famer, D.; Crisby, M. Rosuvastatin reduces caspase-3 activity and up-regulates α-secretase in human neuroblastoma SH-SY5Y cells exposed to Aβ. Neurosci. Lett. 2004, 371, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Marambaud, P. Identification and biology of α-secretase. J. Neurochem. 2012, 120, 34–45. [Google Scholar] [CrossRef]
- Evin, G.; Hince, C. BACE1 as a Therapeutic Target in Alzheimer’s Disease: Rationale and Current Status. Drugs Aging 2013, 30, 755–764. [Google Scholar] [CrossRef]
- Banerjee, P.; Sahoo, A.; Anand, S.; Ganguly, A.; Righi, G.; Bovicelli, P. Multiple Mechanisms of Iron-Induced Amyloid Beta-Peptide Accumulation in SHSY5Y Cells: Protective Action of Negletein. Neuromol. Med. 2014, 16, 787–798. [Google Scholar] [CrossRef]
- Yamamori, H.; Tanaka, T.; Kudo, T.; Takeda, M. Amyloid-beta down-regulates XIAP expression in human SH-SY5Y neuroblastoma cells. Neuroreport 2004, 15, 851–854. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Amyloid β-Peptide(1-42) Contributes to the Oxidative Stress and Neurodegeneration Found in Alzheimer Disease Brain. Brain Pathol. 2006, 14, 426–432. [Google Scholar] [CrossRef]
- Cioanca, O.; Hritcu, L.; Mihasan, M.; Trifan, A.; Hancianu, M. Inhalation of coriander volatile oil increased anxiolytic–antidepressant-like behaviors and decreased oxidative status in beta-amyloid (1-42) rat model of Alzheimer’s disease. Physiol. Behav. 2014, 131, 68–74. [Google Scholar] [CrossRef]
- Yu, L.; Wang, S.; Chen, X.; Yang, H.; Li, X.; Xu, Y. Orientin alleviates cognitive deficits and oxidative stress in Aβ1–42-induced mouse model of Alzheimer’s disease. Life Sci. 2015, 121, 104–109. [Google Scholar] [CrossRef]
- Hacioglu, C.; Kar, F.; Kar, E.; Kara, Y.; Kanbak, G. Effects of Curcumin and Boric Acid Against Neurodegenerative Damage Induced by Amyloid Beta (1–42). Biol. Trace Elem. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Saeed, K.; Shah, S.A.; Ullah, R.; Alam, S.I.; Park, J.S.; Saleem, S. Quinovic Acid Impedes Cholesterol Dyshomeostasis, Oxidative Stress, and Neurodegeneration in an Amyloid-β-Induced Mouse Model. Oxid. Med. Cell Longev. 2020, 2020, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E. The therapeutic importance of understanding mechanisms of neuronal cell death in neurodegenerative disease. Mol. Neurodegener. 2009, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Sutton, E.T.; Hellermann, G.R.; Thomas, T. β-Amyloid-induced endothelial necrosis and inhibition of nitric oxide production. Exp. Cell Res. 1997, 230, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Cacciatore, I.; Fornasari, E.; Marinelli, L.; Eusepi, P.; Ciulla, M.; Ozdemir, O. Memantine-derived drugs as potential antitumor agents for the treatment of glioblastoma. Eur. J. Pharm. Sci. 2017, 109, 402–411. [Google Scholar] [CrossRef]
- Ivins, K.J.; Thornton, P.L.; Rohn, T.T.; Cotman, C.W. Neuronal Apoptosis Induced by β-Amyloid Is Mediated by Caspase-8. Neurobiol. Dis. 1999, 6, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Wang, X.; Kusiak, J.W. Signaling events in amyloid β-peptide-induced neuronal death and insulin-like growth factor I protection. J. Biol. Chem. 2002, 277, 17649–17656. [Google Scholar] [CrossRef] [Green Version]
- Pike, C.J. Estrogen Modulates Neuronal Bcl-xl Expression and β-Amyloid-Induced Apoptosis. J. Neurochem. 2001, 72, 1552–1563. [Google Scholar] [CrossRef]
- Su, J.H.; Anderson, A.J.; Cribbs, D.H.; Tu, C.; Tong, L.; Kesslack, P. Fas and Fas Ligand are associated with neuritic degeneration in the AD brain and participate in β-amyloid-induced neuronal death. Neurobiol. Dis. 2003, 12, 182–193. [Google Scholar] [CrossRef]
- Clarimón, J.; Bertranpetit, J.; Boada, M.; Tàrraga, L.; Comas, D. HSP70-2 (HSPA1B) is Associated with Noncognitive Symptoms in Late-Onset Alzheimer’s Disease. J. Geriatr. Psychiatr. Neurol. 2003, 16, 146–150. [Google Scholar] [CrossRef]
- Chi, L.-M.; Wang, X.; Nan, G.-X. In silico analyses for molecular genetic mechanism and candidate genes in patients with Alzheimer’s disease. Acta Neurol. Belg. 2016, 116, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J. Heme oxygenase-1 protects against Alzheimer’s amyloid-β1-42-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoni, E.; Serafini, M.M.; Caporaso, R.; Marchetti, C.; Racchi, M.; Minarini, A. Targeting the Nrf2/Amyloid-Beta Liaison in Alzheimer’s Disease: A Rational Approach. ACS Chem. Neurosci. 2017, 8, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, L.; Leissring, M.A. Insulin-degrading enzyme is exported via an unconventional protein secretion pathway. Mol. Neurodegener. 2009, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Ponzoni, M. Antiangiogenic strategies in neuroblastoma. Cancer Treat Rev. 2005, 31, 27–34. [Google Scholar] [CrossRef]
- Mann, A.; Tyndale, R.F. Cytochrome P450 2D6 enzyme neuroprotects against 1-methyl-4-phenylpyridinium toxicity in SH-SY5Y neuronal cells. Eur. J. Neurosci. 2010, 31, 1185–1193. [Google Scholar] [CrossRef] [Green Version]
- Koliasnikova, K.N.; Povarnina, P.Y.; Tallerova, A.V.; Firsova, Y.N.; Nikolaev, S.V.; Antipova, T.A. Glyproline Pro-Ampakine with Neuroprotective Activity. Neuroprotection—New Approaches Prospect IntechOpen 2020. [Google Scholar] [CrossRef] [Green Version]
- Jameson, E.; Walter, J.H. Medium-chain acyl-CoA dehydrogenase deficiency. Paediatr. Child Health 2019, 29, 123–126. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Cell Viability (as %) | |
|---|---|---|
| Control | 100 | |
| Aβ1-42 concentrations (as μM) | 1.25 | 81.26 ± 5.06 |
| 2.5 | 73.42 ± 5.24 | |
| 5 | 66.93 ± 6.55 | |
| 10 | 61.41 ± 4.87 | |
| 20 | 51.84 ± 5.21 | |
| 40 | 43.65 ± 4.38 | |
| 80 | 32.11 ± 4.22 | |
| 160 | 11.22 ± 2.78 | |
| Group | α-Secretase Activity | β-Secretase Activity |
|---|---|---|
| Control (-) | 18.8 ± 3.6 c | 2854.5 ± 308.6 b |
| Control (+) | 27.6 ± 4.7 d | 1635.6 ± 211.8 a |
| 20 µM Aβ1-42 | 6.2 ± 1.3 a | 6884.8 ± 545.0 c |
| Aβ +0.1 µM GPE | 6.1 ± 1.1 a | 6774.5 ± 478.2 c |
| Aβ +1 µM GPE | 6.4 ± 1.0 a | 6841.6 ± 396.5 c |
| Aβ +10 µM GPE | 6.8 ± 1.2 a | 6692.0 ± 383.4 c |
| Aβ +25 µM GPE | 7.4 ± 1.4 a | 6735.5 ± 411.7 c |
| Aβ +50 µM GPE | 10.6 ± 1.4 b | 6689.2 ± 549.4 c |
| Aβ +100 µM GPE | 12.5 ± 1.7 b | 6714.4 ± 661.0 c |
| Aβ +0.1 µM GPE1 | 6.0 ± 0.8 a | 7028.7 ± 486.6 c |
| Aβ +1 µM GPE1 | 6.3 ± 1.1 a | 6894.4 ± 463.1 c |
| Aβ +10 µM GPE1 | 6.7 ± 1.0 a | 6838.5 ± 471.4 c |
| Aβ +25 µM GPE1 | 6.9 ± 1.4 a | 6845.4 ± 504.5 c |
| Aβ +50 µM GPE1 | 7.1 ± 1.2 a | 6771.2 ± 522.2 c |
| Aβ +100 µM GPE1 | 7.4 ± 1.4 a | 6768.4 ± 478.2 c |
| Aβ +0.1 µM GPE2 | 6.1 ± 1.0 a | 7041.4 ± 506.0 c |
| Aβ +1 µM GPE2 | 6.4 ± 1.1 a | 7012.0 ± 543.6 c |
| Aβ +10 µM GPE2 | 6.4 ± 1.3 a | 6861.4 ± 431.5 c |
| Aβ +25 µM GPE2 | 6.7 ± 1.4 a | 6844.5 ± 447.7 c |
| Aβ +50 µM GPE2 | 7.0 ± 1.5 a | 6864.2 ± 391.2 c |
| Aβ +100 µM GPE2 | 7.2 ± 1.3 a | 6785.0 ± 488.0 c |
| Aβ +0.1 µM GPE3 | 6.1 ± 1.7 a | 6993.0 ± 512.6 c |
| Aβ +1 µM GPE3 | 6.4 ± 1.3 a | 6981.4 ± 455.2 c |
| Aβ +10 µM GPE3 | 6.9 ± 1.5 a | 6873.5 ± 476.6 c |
| Aβ +25 µM GPE3 | 7.3 ± 1.3 a | 6865.6 ± 442.0 c |
| Aβ +50 µM GPE3 | 10.5 ± 1.6 b | 6756.2 ± 465.6 c |
| Aβ +100 µM GPE3 | 12.8 ± 1.8 b | 6776.0 ± 511.8 c |
| Group | TAC Level (mmolTrolox Equiv./L) | TOS Level (µmol H2O2 Equiv./L) | |
|---|---|---|---|
| Control (-) | 5.08 ± 0.62 a | 1.86 ± 0.24 a | |
| Control (+) | 13.96 ± 1.14 e | 5.45 ± 0.42 b | |
| GPE | 0.1 µM | 5.22 ± 0.41 a | 1.77 ± 0.27 a |
| 1 µM | 5.49 ± 0.57 a | 1.81 ± 0.21 a | |
| 10 µM | 6.38 ± 0.48 b | 1.88 ± 0.14 a | |
| 25 µM | 8.69 ± 0.63 bc | 1.83 ± 0.20 a | |
| 50 µM | 10.91 ± 0.57 c | 1.87 ± 0.22 a | |
| 100 µM | 12.08 ± 0.96 c | 1.90 ± 0.18 a | |
| GPE1 | 0.1 µM | 5.06 ± 0.35 a | 1.84 ± 0.20 a |
| 1 µM | 5.38 ± 0.51 a | 1.73 ± 0.16 a | |
| 10 µM | 6.79 ± 0.39 b | 1.82 ± 0.18 a | |
| 25 µM | 8.25 ± 0.70 bc | 1.68 ± 0.20 a | |
| 50 µM | 9.78 ± 0.61 c | 1.83 ± 0.22 a | |
| 100 µM | 11.02 ± 0.94 c | 1.95 ± 0.22 a | |
| GPE2 | 0.1 µM | 5.08 ± 0.40 a | 1.73 ± 0.26 a |
| 1 µM | 5.21 ± 0.61 a | 1.85 ± 0.21 a | |
| 10 µM | 5.93 ± 0.71 a | 1.91 ± 0.26 a | |
| 25 µM | 7.44 ± 0.58 b | 1.87 ± 0.18 a | |
| 50 µM | 8.68 ± 0.56 bc | 1.93 ± 0.20 a | |
| 100 µM | 10.04 ± 0.83 c | 1.98 ± 0.28 a | |
| GPE3 | 0.1 µM | 5.25 ± 0.30 a | 1.66 ± 0.22 a |
| 1 µM | 5.86 ± 0.43 a | 1.64 ± 0.18 a | |
| 10 µM | 7.73 ± 0.54 b | 1.74 ± 0.16 a | |
| 25 µM | 9.18 ± 0.66 c | 1.88 ± 0.25 a | |
| 50 µM | 10.94 ± 0.64 c | 1.81 ± 0.21 a | |
| 100 µM | 12.74 ± 0.87 d | 1.89 ± 0.17 a | |
| Groups | TAC Level (mmolTrolox Equiv./L) | TOS Level (µmol H2O2 Equiv./L) |
|---|---|---|
| Control (-) | 5.08 ± 0.62 b | 1.86 ± 0.24 a |
| Control (+) | 13.96 ± 1.14 f | 5.45 ± 0.42 d |
| 20 µM Aβ1-42 | 3.22 ± 0.58 a | 4.37 ± 0.40 c |
| Aβ +0.1 µM GPE | 3.89 ± 0.46 a | 4.09 ± 0.36 bc |
| Aβ +1 µM GPE | 4.95 ± 0.61 b | 3.85 ± 0.34 b |
| Aβ +10 µM GPE | 6.24 ± 0.54 bc | 3.27 ± 0.40 b |
| Aβ +25 µM GPE | 7.12 ± 0.63 c | 2.89 ± 0.33 b |
| Aβ +50 µM GPE | 9.65 ± 0.72 d | 2.60 ± 0.20 b |
| Aβ +100 µM GPE | 10.04 ± 0.79 d | 2.34 ± 0.28 b |
| Aβ +0.1 µM GPE1 | 3.41 ± 0.52 a | 4.13 ± 0.26 c |
| Aβ +1 µM GPE1 | 4.35 ± 0.54 ab | 3.96 ± 0.32 bc |
| Aβ +10 µM GPE1 | 5.12 ± 0.47 b | 3.63 ± 0.34 b |
| Aβ +25 µM GPE1 | 5.34 ± 0.45 b | 3.17 ± 0.35 b |
| Aβ +50 µM GPE1 | 5.71 ± 0.50 b | 2.87 ± 0.21 b |
| Aβ +100 µM GPE1 | 6.33 ± 0.55 bc | 2.66 ± 0.23 b |
| Aβ +0.1 µM GPE2 | 3.88 ± 0.42 a | 4.02 ± 0.30 bc |
| Aβ +1 µM GPE2 | 4.22 ± 0.51 a | 3.84 ± 0.42 b |
| Aβ +10 µM GPE2 | 4.41 ± 0.39 a | 3.56 ± 0.33 b |
| Aβ +25 µM GPE2 | 4.49 ± 0.53 a | 3.29 ± 0.25 b |
| Aβ +50 µM GPE2 | 4.83 ± 0.51 b | 3.08 ± 0.26 b |
| Aβ +100 µM GPE2 | 6.38 ± 0.55 bc | 2.93 ± 0.30 b |
| Aβ +0.1 µM GPE3 | 4.15 ± 0.35 a | 4.06 ± 0.28 bc |
| Aβ +1 µM GPE3 | 4.78 ± 0.47 b | 3.66 ± 0.36 b |
| Aβ +10 µM GPE3 | 5.34 ± 0.45 b | 3.15 ± 0.27 b |
| Aβ +25 µM GPE3 | 7.66 ± 0.81 c | 2.71 ± 0.32 ab |
| Aβ +50 µM GPE3 | 9.86 ± 0.73 d | 2.55 ± 0.18 ab |
| Aβ +100 µM GPE3 | 10.93 ± 0.88 e | 2.17 ± 0.26 a |
| Gene | Aβ 1-42 | Aβ 1-42 Plus GPE3 |
|---|---|---|
| ACADVL | −0.71 ± 0.05 | 5.65 ± 1.25 |
| ADM2 | −0.38 ± 0.12 | 2.90 ± 0.28 |
| AKT1 | 0.15 ± 0.02 | 0.09 ± 0.03 |
| BCL2 | −0.86 ± 0.07 | 0.34 ± 0.01 |
| BCL2L1 | −0.91 ± 0.11 | 0.72 ± 0.03 |
| BRCA1 | −0.18 ± 0.02 | 6.92 ± 0.48 |
| CASP8 | 3.27 ± 0.21 | 0.29 ± 0.02 |
| CASP9 | 2.87 ± 0.35 | 0.54 ± 0.05 |
| CYP2D6 | −0.22 ± 0.21 | 3.94 ± 0.16 |
| DNAJB9 | −0.83 ± 0.18 | 16.95 ± 1.80 |
| FASLG | 0.94 ± 0.04 | 0.35 ± 0.03 |
| HMOX1 | −0.47 ± 0.12 | 8.95 ± 1.55 |
| HSPA1A | 3.80 ± 0.21 | 1.15 ± 0.45 |
| METAP2 | −0.66 ± 0.03 | 3.35 ± 0.90 |
| MK167 | −0.26 ± 0.01 | 3.20 ± 0.84 |
| NQO1 | −0.65 ± 0.06 | 5.66 ± 1.02 |
| SLC7A11 | −0.33 ± 0.02 | 6.96 ± 2.05 |
| UHRF1 | −0.81 ± 0.05 | 8.55 ± 1.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turkez, H.; Cacciatore, I.; Marinelli, L.; Fornasari, E.; Aslan, M.E.; Cadirci, K.; Kahraman, C.Y.; Caglar, O.; Tatar, A.; Di Biase, G.; et al. Glycyl-L-Prolyl-L-Glutamate Pseudotripeptides for Treatment of Alzheimer’s Disease. Biomolecules 2021, 11, 126. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11010126
Turkez H, Cacciatore I, Marinelli L, Fornasari E, Aslan ME, Cadirci K, Kahraman CY, Caglar O, Tatar A, Di Biase G, et al. Glycyl-L-Prolyl-L-Glutamate Pseudotripeptides for Treatment of Alzheimer’s Disease. Biomolecules. 2021; 11(1):126. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11010126
Chicago/Turabian StyleTurkez, Hasan, Ivana Cacciatore, Lisa Marinelli, Erika Fornasari, Mehmet Enes Aslan, Kenan Cadirci, Cigdem Yuce Kahraman, Ozge Caglar, Abdulgani Tatar, Giuseppe Di Biase, and et al. 2021. "Glycyl-L-Prolyl-L-Glutamate Pseudotripeptides for Treatment of Alzheimer’s Disease" Biomolecules 11, no. 1: 126. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11010126