
Effect of Pirfenidone on TGF-β1-Induced Myofibroblast Differentiation and Extracellular Matrix Homeostasis of Human Orbital Fibroblasts in Graves’ Ophthalmopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Culture of Orbital Fibroblasts
2.2. Chemicals and Antibodies
2.3. Determination of Cell Viability
2.4. Western Blot Analysis
2.5. MMP-2/-9 Enzyme Activity Assay
2.6. Statistical Analysis
3. Results
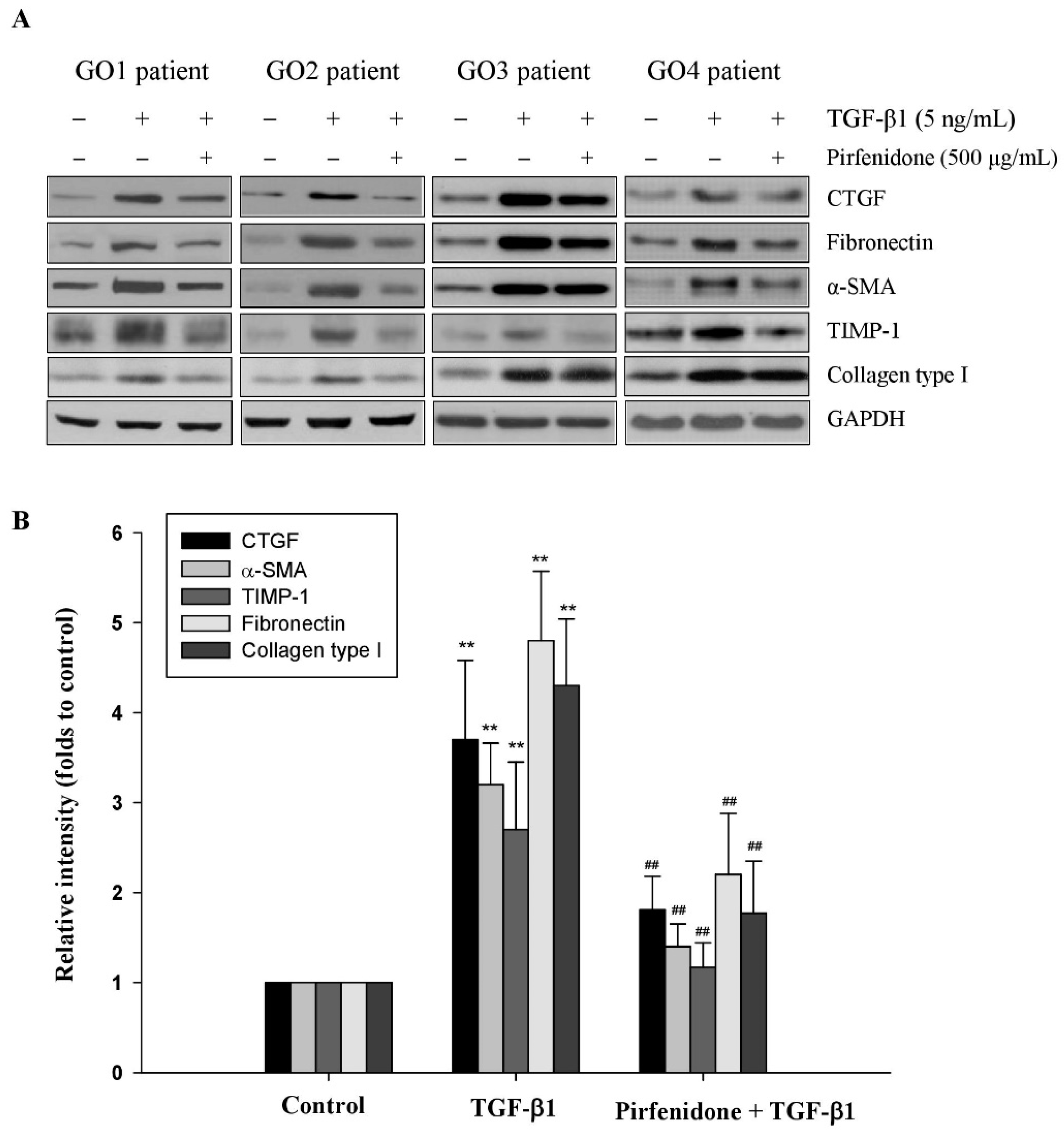
3.1. Pirfenidone Inhibited TGF-β1-Induced Fibrotic Protein Expression in GO Orbital Fibroblasts
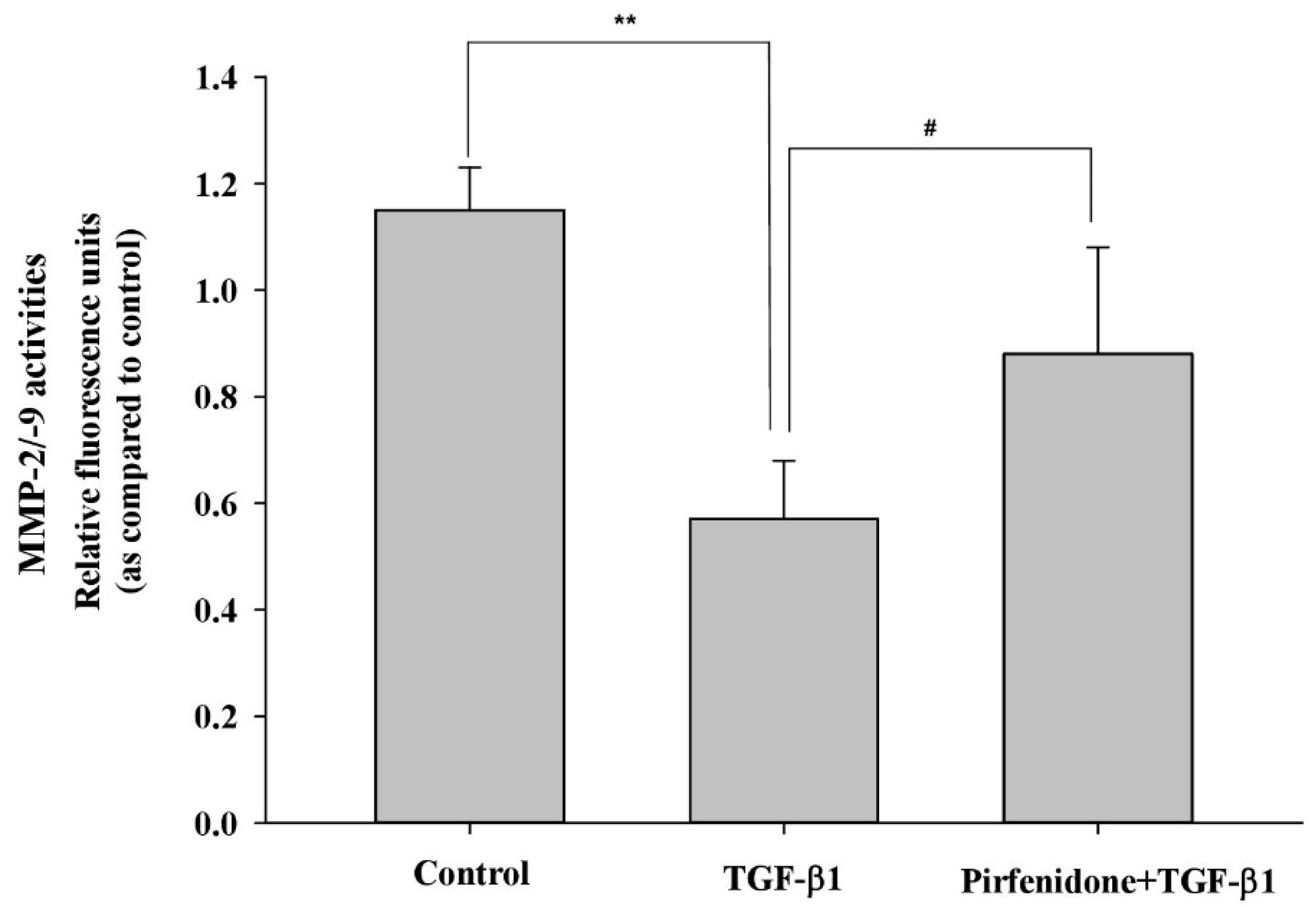
3.2. Pirfenidone Diminished TGF-β1-Mediated ECM Metabolism in GO Orbital Fibroblasts
3.3. Pirfenidone Abolished TGF-β1-Induced p38 and JNK Phosphorylation in GO Orbital Fibroblasts
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hiromatsu, Y.; Eguchi, H.; Tani, J.; Kasaoka, M.; Teshima, Y. Graves’ ophthalmopathy: Epidemiology and natural history. Intern. Med. 2014, 53, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khong, J.J.; McNab, A.A.; Ebeling, P.R.; Craig, J.E.; Selva, D. Pathogenesis of thyroid eye disease: Review and update on molecular mechanisms. Br. J. Ophthalmol. 2016, 100, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dik, W.A.; Virakul, S.; van Steensel, L. Current perspectives on the role of orbital fibroblasts in the pathogenesis of Graves’ ophthalmopathy. Exp. Eye Res. 2016, 142, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Smith, T.J. Current concepts in the molecular pathogenesis of thyroid-associated ophthalmopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1735–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Nikolic–Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, P.; Reszec, J.; Eckstein, A.; Johnson, K.; Grzybowski, A.; Chyczewski, L.; Mysliwiec, J. Markers of inflammation and fibrosis in the orbital fat/connective tissue of patients with Graves’ orbitopathy: Clinical implications. Mediat. Inflamm. 2014, 2014, 412158. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Wu, S.B.; Kao, S.C.; Kau, H.C.; Lee, F.; Wei, Y.H. The protective effect of antioxidants on orbital fibroblasts from patients with Graves’ ophthalmopathy in response to oxidative stress. Mol. Vis. 2013, 19, 927–934. [Google Scholar] [PubMed]
- Tsai, C.C.; Wu, S.B.; Kau, H.C.; Wei, Y.H. Essential role of connective tissue growth factor (CTGF) in transforming growth factor-β1 (TGF-β1)-induced myofibroblast transdifferentiation from Graves’ orbital fibroblasts. Sci. Rep. 2018, 8, 7276. [Google Scholar] [CrossRef] [PubMed]
- Koumas, L.; Smith, T.J.; Feldon, S.; Blumberg, N.; Phipps, R.P. Thy-1 expression in human fibroblast subsets defines myofibroblastic or lipofibroblastic phenotypes. Am. J. Pathol. 2003, 163, 1291–1300. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.Y.; Wu, S.B.; Kau, H.C.; Tsai, C.C. JNK and p38 inhibitors prevent transforming growth factor-β1-induced myofibroblast transdifferentiation in human Graves’ orbital fibroblasts. Int. J. Mol. Sci. 2021, 22, 2952. [Google Scholar] [CrossRef]
- Richeldi, L.; Yasothan, U.; Kirkpatrick, P. Pirfenidone. Nat. Rev. Drug Discov. 2011, 10, 489–490. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Bradford, W.Z.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Glasscock, K.F.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.H.; Nathan, S.D.; et al. Sensitivity analyses of the change in FVC in a Phase 3 trial of pirfenidone for idiopathic pulmonary fibrosis. Chest 2015, 148, 196–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Westra, I.M.; Oosterhuis, D.; Groothuis, G.M.; Olinga, P. The effect of antifibrotic drugs in rat precision-cut fibrotic liver slices. PLoS ONE 2014, 9, e95462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagami, K.; Oka, T.; Wang, Q.; Ishizu, T.; Lee, J.K.; Miwa, K.; Akazawa, H.; Naito, A.T.; Sakata, Y.; Komuro, I. Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts. Am. J. Physiol Heart Circ. Physiol. 2015, 309, H512–H522. [Google Scholar] [CrossRef] [Green Version]
- Tampe, D.; Zeisberg, M. Potential approaches to reverse or repair renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 226–237. [Google Scholar] [CrossRef]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Zhang, G.F.; Liao, X.P.; Li, X.J.; Zhang, J.; Lin, H.; Chen, Z.; Zhang, X. Anti-fibrotic effects of pirfenidone by interference with the hedgehog signalling pathway in patients with systemic sclerosis-associated interstitial lung disease. Int. J. Rheum. Dis. 2018, 21, 477–486. [Google Scholar] [CrossRef]
- Wells, A.R.; Leung, K.P. Pirfenidone attenuates the profibrotic contractile phenotype of differentiated human dermal myofibroblasts. Biochem. Biophys. Res. Commun. 2020, 521, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.A.; Jeon, B.K.; Choi, Y.H.; Back, K.O.; Lee, J.B.; Kook, K.H. Pirfenidone attenuates the IL-1β-induced hyaluronic acid increase in orbital fibroblasts from patients with thyroid-associated ophthalmopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2276–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Chen, Q.; Zhao, C.; Yang, X.; Cun, Q.; Yang, W.; Zhang, Y.; Zhu, Y.; Zhong, H. The in vitro anti-fibrotic effect of Pirfenidone on human pterygium fibroblasts is associated with down-regulation of autocrine TGF-beta and MMP-1. Int. J. Med. Sci. 2020, 17, 734–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.; Lee, K.; Ryu, S.W.; Im, M.; Kook, K.H.; Choi, C. Pirfenidone inhibits transforming growth factor-β1-induced fibrogenesis by blocking nuclear translocation of Smads in human retinal pigment epithelial cell line ARPE-19. Mol. Vis. 2012, 18, 1010–1020. [Google Scholar] [PubMed]
- Roztocil, E.; Hammond, C.L.; Gonzalez, M.O.; Feldon, S.E.; Woeller, C.F. The aryl hydrocarbon receptor pathway controls matrix metalloproteinase-1 and collagen levels in human orbital fibroblasts. Sci. Rep. 2020, 10, 8477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Q.; Wang, J.; Xu, C.; Huang, X.; Ruan, Z.; Dai, Y. Pirfenidone alleviates pulmonary fibrosis in vitro and in vivo through regulating Wnt/GSK-3beta/beta-catenin and TGF-beta1/Smad2/3 signaling pathways. Mol. Med. 2020, 26, 49. [Google Scholar] [CrossRef]
- Li, G.; Ren, J.; Hu, Q.; Deng, Y.; Chen, G.; Guo, K.; Li, R.; Li, Y.; Wu, L.; Wang, G.; et al. Oral pirfenidone protects against fibrosis by inhibiting fibroblast proliferation and TGF-β signaling in a murine colitis model. Biochem. Pharmacol. 2016, 117, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, X.; Wang, B.; Nie, Y.; Wen, J.; Wang, Q.; Gu, C. Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology (Carlton) 2017, 22, 589–597. [Google Scholar] [CrossRef]
- Guo, X.; Yang, Y.; Liu, L.; Liu, X.; Xu, J.; Wu, K.; Yu, M. Pirfenidone induces G1 arrest in human Tenon’s fibroblasts in vitro involving AKT and MAPK signaling pathways. J. Ocul. Pharmacol. Ther. 2017, 33, 366–374. [Google Scholar] [CrossRef]
- Hall, C.L.; Wells, A.R.; Leung, K.P. Pirfenidone reduces profibrotic responses in human dermal myofibroblasts, in vitro. Lab. Investig. 2018, 98, 640–655. [Google Scholar] [CrossRef]
- Shi, K.; Wang, F.; Xia, J.; Zuo, B.; Wang, Z.; Cao, X. Pirfenidone inhibits epidural scar fibroblast proliferation and differentiation by regulating TGF-β1-induced Smad-dependent and -independent pathways. Am. J. Transl. Res. 2019, 11, 1593–1604. [Google Scholar] [PubMed]
- Kim, H.; Choi, Y.H.; Park, S.J.; Lee, S.Y.; Kim, S.J.; Jou, I.; Kook, K.H. Antifibrotic effect of Pirfenidone on orbital fibroblasts of patients with thyroid-associated ophthalmopathy by decreasing TIMP-1 and collagen levels. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3061–3066. [Google Scholar] [CrossRef] [PubMed]
- Douglas, R.S.; Kahaly, G.J.; Patel, A.; Sile, S.; Thompson, E.H.Z.; Perdok, R.; Fleming, J.C.; Fowler, B.T.; Marcocci, C.; Marinò, M.; et al. Teprotumumab for the Treatment of Active Thyroid Eye Disease. N. Engl. J. Med. 2020, 382, 341–352. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.-B.; Hou, T.-Y.; Kau, H.-C.; Tsai, C.-C. Effect of Pirfenidone on TGF-β1-Induced Myofibroblast Differentiation and Extracellular Matrix Homeostasis of Human Orbital Fibroblasts in Graves’ Ophthalmopathy. Biomolecules 2021, 11, 1424. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11101424
Wu S-B, Hou T-Y, Kau H-C, Tsai C-C. Effect of Pirfenidone on TGF-β1-Induced Myofibroblast Differentiation and Extracellular Matrix Homeostasis of Human Orbital Fibroblasts in Graves’ Ophthalmopathy. Biomolecules. 2021; 11(10):1424. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11101424
Chicago/Turabian StyleWu, Shi-Bei, Tzu-Yu Hou, Hui-Chuan Kau, and Chieh-Chih Tsai. 2021. "Effect of Pirfenidone on TGF-β1-Induced Myofibroblast Differentiation and Extracellular Matrix Homeostasis of Human Orbital Fibroblasts in Graves’ Ophthalmopathy" Biomolecules 11, no. 10: 1424. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11101424