
Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery

 , , , , and
, , , , and
Abstract
:1. Introduction
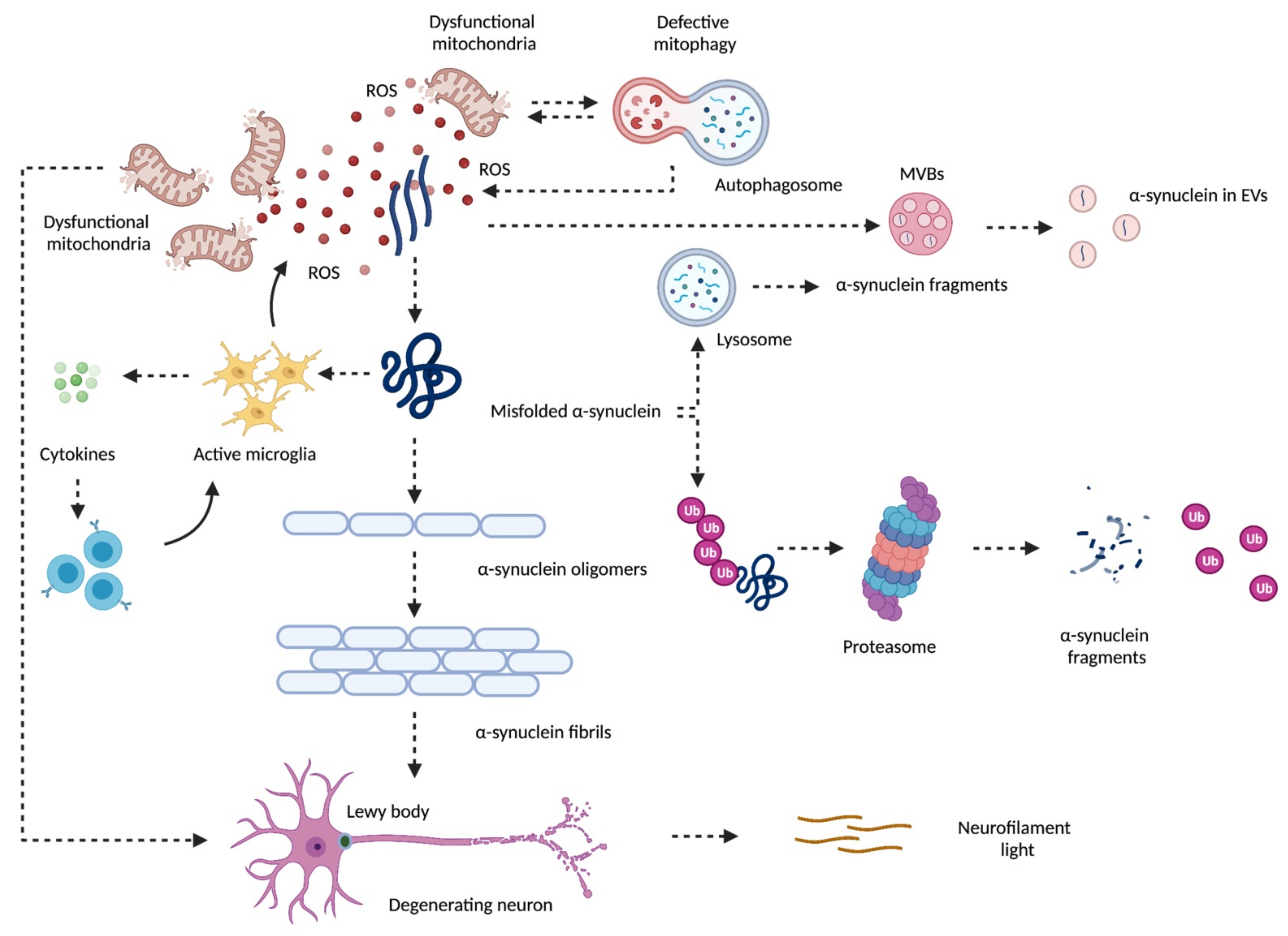
2. Mitochondrial Dysfunction and Neurodegeneration in Parkinson’s Disease
3. α-Synuclein
4. β-Amyloid and p-Tau Pathology
5. Neurofilament Light Chain and Axonal Injury during PD Neurodegeneration
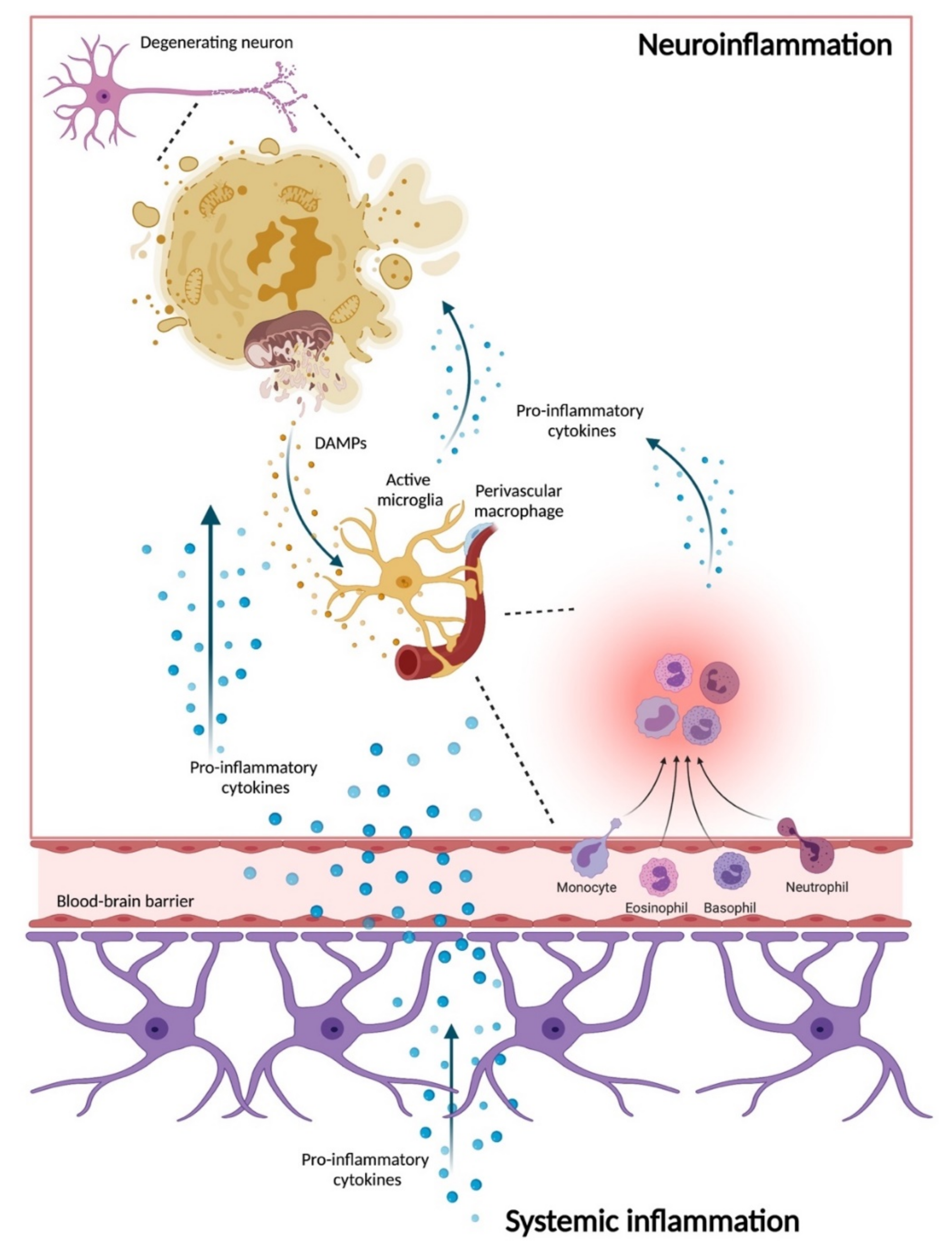
6. Neuroinflammation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ray Dorsey, E.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Dickson, D.W. Neuropathology of Parkinson disease. Park. Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef]
- Parnetti, L.; Gaetani, L.; Eusebi, P.; Paciotti, S.; Hansson, O.; El-Agnaf, O.; Mollenhauer, B.; Blennow, K.; Calabresi, P. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019, 18, 573–586. [Google Scholar] [CrossRef]
- He, R.; Yan, X.; Guo, J.; Xu, Q.; Tang, B.; Sun, Q. Recent advances in biomarkers for Parkinson’s disease. Front. Aging Neurosci. 2018, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Albin, R.; Alcalay, R.; Babcock, D.; Bajaj, V.; Bowman, D.; Buko, A.; Cedarbaum, J.; Chelsky, D.; Cookson, M.R.; et al. Finding useful biomarkers for Parkinson s disease. Sci. Transl. Med. 2018, 10, eaam6003. [Google Scholar] [CrossRef]
- Lewitt, P.A.; Li, J.; Lu, M.; Guo, L.; Auinger, P. Metabolomic biomarkers as strong correlates of Parkinson disease progression. Neurology 2017, 88, 862–869. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi Rastegar, D.; Ho, N.; Halliday, G.M.; Dzamko, N. Parkinson’s progression prediction using machine learning and serum cytokines. NPJ Park. Dis. 2019, 5, 14. [Google Scholar] [CrossRef]
- Picca, A.; Coelho-Junior, H.J.; Cesari, M.; Marini, F.; Miccheli, A.; Gervasoni, J.; Bossola, M.; Landi, F.; Bernabei, R.; Marzetti, E.; et al. The metabolomics side of frailty: Toward personalized medicine for the aged. Exp. Gerontol. 2019, 126, 110692. [Google Scholar] [CrossRef]
- Posavi, M.; Diaz-Ortiz, M.; Liu, B.; Swanson, C.R.; Skrinak, R.T.; Hernandez-Con, P.; Amado, D.A.; Fullard, M.; Rick, J.; Siderowf, A.; et al. Characterization of Parkinson’s disease using blood-based biomarkers: A multicohort proteomic analysis. PLoS Med. 2019, 16, e1002931. [Google Scholar] [CrossRef]
- Calvani, R.; Picca, A.; Landi, G.; Marini, F.; Biancolillo, A.; Coelho-Junior, H.J.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Arcidiacono, A.; et al. A novel multi-marker discovery approach identifies new serum biomarkers for Parkinson’s disease in older people: An EXosomes in PArkiNson Disease (EXPAND) ancillary study. GeroScience 2020, 42, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Landi, G.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Urbani, A.; Bossola, M.; et al. Circulating amino acid signature in older people with Parkinson’s disease: A metabolic complement to the EXosomes in PArkiNson Disease (EXPAND) study. Exp. Gerontol. 2019, 128, 110766. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.; et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Mattison, H.; Liu, C.; Ginghina, C.; Auinger, P.; McDermott, M.; Stewart, T.; Kang, U.; Cain, K.; Shi, M. Longitudinal assessment of tau and amyloid beta in cerebrospinal fluid of Parkinson disease. Acta Neuropathol. 2013, 126, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Cholerton, B.; Shi, M.; Ginghina, C.; Cain, K.; Auinger, P.; Zhang, J. CSF tau and tau/Aβ42 predict cognitive decline in Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Schrag, A.; Siddiqui, U.; Anastasiou, Z.; Weintraub, D.; Schott, J. Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson’s disease: A cohort study. Lancet Neurol. 2017, 16, 66. [Google Scholar] [CrossRef] [Green Version]
- Constantinides, V.; Paraskevas, G.; Emmanouilidou, E.; Petropoulou, O.; Bougea, A.; Vekrellis, K.; Evdokimidis, I.; Stamboulis, E.; Kapaki, E. CSF biomarkers β-amyloid, tau proteins and a-synuclein in the differential diagnosis of Parkinson-plus syndromes. J. Neurol. Sci. 2017, 382, 91–95. [Google Scholar] [CrossRef]
- Bäckström, D.; Eriksson Domellöf, M.; Linder, J.; Olsson, B.; Öhrfelt, A.; Trupp, M.; Zetterberg, H.; Blennow, K.; Forsgren, L. Cerebrospinal Fluid Patterns and the Risk of Future Dementia in Early, Incident Parkinson Disease. JAMA Neurol. 2015, 72, 1175–1182. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.; Öhrfelt, A.; Constantinescu, R.; Andreasson, U.; Surova, Y.; Bostrom, F.; Nilsson, C.; Håkan, W.; Decraemer, H.; Någga, K.; et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch. Neurol. 2012, 69, 1445–1452. [Google Scholar] [CrossRef]
- Aasly, J.; Johansen, K.; Brønstad, G.; Warø, B.; Majbour, N.; Varghese, S.; Alzahmi, F.; Paleologou, K.; Amer, D.; Al-Hayani, A.; et al. Elevated levels of cerebrospinal fluid α-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front. Aging Neurosci. 2014, 6, 248. [Google Scholar] [CrossRef]
- Majbour, N.; Vaikath, N.; Eusebi, P.; Chiasserini, D.; Ardah, M.; Varghese, S.; Haque, M.; Tokuda, T.; Auinger, P.; Calabresi, P.; et al. Longitudinal changes in CSF alpha-synuclein species reflect Parkinson’s disease progression. Mov. Disord. 2016, 31, 1535–1542. [Google Scholar] [CrossRef]
- Delgado-Alvarado, M.; Gago, B.; Gorostidi, A.; Jiménez-Urbieta, H.; Dacosta-Aguayo, R.; Navalpotro-Gómez, I.; Ruiz-Martínez, J.; Bergareche, A.; Martí-Massó, J.; Martínez-Lage, P.; et al. Tau/α-synuclein ratio and inflammatory proteins in Parkinson’s disease: An exploratory study. Mov. Disord. 2017, 32, 1066–1073. [Google Scholar] [CrossRef]
- Herbert, M.; Eeftens, J.; Aerts, M.; Esselink, R.; Bloem, B.; Kuiperij, H.; Verbeek, M. CSF levels of DJ-1 and tau distinguish MSA patients from PD patients and controls. Parkinsonism Relat. Disord. 2014, 20, 112–115. [Google Scholar] [CrossRef]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Malpartida, A.; Williamson, M.; Narendra, D.; Wade-Martins, R.; Ryan, B. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Swatek, K.; Usher, J.; Kueck, A.; Gladkova, C.; Mevissen, T.; Pruneda, J.; Skern, T.; Komander, D. Insights into ubiquitin chain architecture using Ub-clipping. Nature 2019, 572, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Wauer, T.; Simicek, M.; Schubert, A.; Komander, D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 2015, 524, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Gladkova, C.; Maslen, S.; Skehel, J.; Komander, D. Mechanism of parkin activation by PINK1. Nature 2018, 559, 410–414. [Google Scholar] [CrossRef]
- Harper, J.; Ordureau, A.; Heo, J. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Hou, X.; Fiesel, F.; Truban, D.; Castanedes Casey, M.; Lin, W.; Soto, A.; Tacik, P.; Rousseau, L.; Diehl, N.; Heckman, M.; et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 2018, 14, 1404–1418. [Google Scholar] [CrossRef] [Green Version]
- Fiesel, F.; Ando, M.; Hudec, R.; Hill, A.; Castanedes-Casey, M.; Caulfield, T.; Moussaud-Lamodière, E.; Stankowski, J.; Bauer, P.; Lorenzo-Betancor, O.; et al. (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 2015, 16, 1114–1130. [Google Scholar] [CrossRef]
- Hsieh, C.; Shaltouki, A.; Gonzalez, A.; Bettencourt da Cruz, A.; Burbulla, L.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Godena, V.; Brookes-Hocking, N.; Moller, A.; Shaw, G.; Oswald, M.; Sancho, R.; Miller, C.; Whitworth, A.; De Vos, K. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 2014, 5, 5245. [Google Scholar] [CrossRef] [Green Version]
- Bonello, F.; Hassoun, S.; Mouton-Liger, F.; Shin, Y.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef]
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 2020, 16, 203–222. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, X.; Nguyen, D.; Shammas, M.; Wu, B.; Dombi, E.; Springer, D.; Poulton, J.; Sekine, S.; Narendra, D. Loss of CHCHD2 and CHCHD10 activates OMA1 peptidase to disrupt mitochondrial cristae phenocopying patient mutations. Hum. Mol. Genet. 2020, 29, 1547–1567. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, T.; Spinazzi, M.; Martin, S.; Imberechts, D.; Vangheluwe, P.; Bird, M.; De Strooper, B.; Vandenberghe, W. CHCHD2 harboring Parkinson’s disease-linked T61I mutation precipitates inside mitochondria and induces precipitation of wild-type CHCHD2. Hum. Mol. Genet. 2020, 29, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 2017, 8, 15500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, A.; Nishioka, K.; Meng, H.; Takanashi, M.; Hasegawa, I.; Inoshita, T.; Shiba-Fukushima, K.; Li, Y.; Yoshino, H.; Mori, A.; et al. Mutations in CHCHD2 cause α-synuclein aggregation. Hum. Mol. Genet. 2019, 28, 3895–3911. [Google Scholar] [CrossRef] [PubMed]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Tolosa, E.; Wenning, G.; Poewe, W. The diagnosis of Parkinson’s disease. Lancet. Neurol. 2006, 5, 75–86. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.; Bucci, C.; Marzetti, E. Circulating extracellular vesicles: Friends and foes in neurodegeneration. Neural Regen. Res. 2012, 17, 534–542. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, G. Potential of extracellular vesicles in the Parkinson’s disease—Pathological mediators and biomarkers. Neurochem. Int. 2021, 144, 104974. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Romano, R.; Bucci, C.; Marzetti, E. Extracellular Vesicles and Damage-Associated Molecular Patterns: A Pandora’s Box in Health and Disease. Front. Immunol. 2020, 11, 601740. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.; Brotchie, J.; Kalia, L.; Koprich, J.; Tandon, A.; Watts, J.; Lang, A. α-Synuclein-Based Animal Models of Parkinson’s Disease: Challenges and Opportunities in a New Era. Trends Neurosci. 2016, 39, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.; Lee, V. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijaz, B.; Volpicelli-Daley, L. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, P.; Picconi, B.; Tozzi, A.; Ghiglieri, V.; Di Filippo, M. Direct and indirect pathways of basal ganglia: A critical reappraisal. Nat. Neurosci. 2014, 17, 1022–1030. [Google Scholar] [CrossRef]
- Durante, V.; de Iure, A.; Loffredo, V.; Vaikath, N.; De Risi, M.; Paciotti, S.; Quiroga-Varela, A.; Chiasserini, D.; Mellone, M.; Mazzocchetti, P.; et al. Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain 2019, 142, 1365–1385. [Google Scholar] [CrossRef] [Green Version]
- Giordano, N.; Iemolo, A.; Mancini, M.; Cacace, F.; De Risi, M.; Latagliata, E.; Ghiglieri, V.; Bellenchi, G.; Puglisi-Allegra, S.; Calabresi, P.; et al. Motor learning and metaplasticity in striatal neurons: Relevance for Parkinson’s disease. Brain 2018, 141, 505–520. [Google Scholar] [CrossRef] [Green Version]
- El-Agnaf, O.; Salem, S.; Paleologou, K.; Cooper, L.; Fullwood, N.; Gibson, M.; Curran, M.; Court, J.; Mann, D.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Karpowicz, R.; Haney, C.; Mihaila, T.; Sandler, R.; Petersson, E.; Lee, V. Selective imaging of internalized proteopathic α-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J. Biol. Chem. 2017, 292, 13482–13497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chivet, M.; Javalet, C.; Hemming, F.; Pernet-Gallay, K.; Laulagnier, K.; Fraboulet, S.; Sadoul, R. Exosomes as a novel way of interneuronal communication. Biochem. Soc. Trans. 2013, 41, 241–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdinocci, D.; Radford, R.; Siow, S.; Chung, R.; Pountney, D. Potential Modes of Intercellular α-Synuclein Transmission. Int. J. Mol. Sci. 2017, 18, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Li, J. Exosomes in Parkinson’s Disease: Current Perspectives and Future Challenges. ACS Chem. Neurosci. 2019, 10, 964–972. [Google Scholar] [CrossRef]
- Filippini, A.; Gennarelli, M.; Russo, I. α-Synuclein and Glia in Parkinson’s Disease: A Beneficial or a Detrimental Duet for the Endo-Lysosomal System? Cell. Mol. Neurobiol. 2019, 39, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Suk, J.; Patrick, C.; Bae, E.; Cho, J.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.; Zorec, R. Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngolab, J.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Trejo, M.; Masliah, D.; Adame, A.; Masliah, E.; Rissman, R. Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol. Commun. 2017, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.; Kim, J.; Paik, S.; Park, J.; Ahn, Y.; Chung, K. Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. J. Biol. Chem. 2001, 276, 27441–27448. [Google Scholar] [CrossRef] [Green Version]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marie, G.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-synuclein aggregation by exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195. [Google Scholar] [CrossRef]
- Kunadt, M.; Eckermann, K.; Stuendl, A.; Gong, J.; Russo, B.; Strauss, K.; Rai, S.; Kügler, S.; Falomir Lockhart, L.; Schwalbe, M.; et al. Extracellular vesicle sorting of α-Synuclein is regulated by sumoylation. Acta Neuropathol. 2015, 129, 695–713. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.; Goebel, I.; Mubaidin, A.; Wriekat, A.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.; Glauser, L.; Moore, D. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743. [Google Scholar] [CrossRef]
- Gitler, A.; Chesi, A.; Geddie, M.; Strathearn, K.; Hamamichi, S.; Hill, K.; Caldwell, K.; Caldwell, G.; Cooper, A.; Rochet, J.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.; Chan, B.; Park, J.; Hill, K.; Aitken, J.; Cottle, L.; Farghaian, H.; Cole, A.; Lay, P.; Sue, C.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Z.; Shi, M.; Chung, K.; Quinn, J.; Peskind, E.; Galasko, D.; Jankovic, J.; Zabetian, C.; Leverenz, J.; Baird, G.; et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 2010, 133, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, B.; Locascio, J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet. Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- Shi, M.; Bradner, J.; Hancock, A.; Chung, K.; Quinn, J.; Peskind, E.; Galasko, D.; Jankovic, J.; Zabetian, C.; Kim, H.; et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann. Neurol. 2011, 69, 570–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 2016, 139, 481–494. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Liu, C.; Cook, T.; Bullock, K.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Aamodt, E.; Williams, R. Microtubule-associated proteins connect microtubules and neurofilaments in vitro. Biochemistry 1984, 23, 6023–6031. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.; Khan, U.; Hoang, D.; Novikov, D.; Krishnamurthy, P.; Rajamohamed Sait, H.; Little, B.; Sigurdsson, E.; Wadghiri, Y. Non-invasive, in vivo monitoring of neuronal transport impairment in a mouse model of tauopathy using MEMRI. Neuroimage 2013, 64, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Compta, Y.; Revesz, T. Neuropathological and Biomarker Findings in Parkinson’s Disease and Alzheimer’s Disease: From Protein Aggregates to Synaptic Dysfunction. J. Parkinsons Dis. 2021, 11, 107–121. [Google Scholar] [CrossRef]
- Seitz, A.; Kojima, H.; Oiwa, K.; Mandelkow, E.; Song, Y.; Mandelkow, E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002, 21, 4896–4905. [Google Scholar] [CrossRef] [Green Version]
- Kempster, P.; O’Sullivan, S.; Holton, J.; Revesz, T.; Lees, A. Relationships between age and late progression of Parkinson’s disease: A clinico-pathological study. Brain 2010, 133, 1755–1762. [Google Scholar] [CrossRef]
- Duda, J.; Giasson, B.; Mabon, M.; Lee, V.; Trojanowski, J. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann. Neurol. 2002, 52, 205–210. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Uchikado, H.; Dickson, D. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat. Disord. 2007, 13 (Suppl. 3), S221–S224. [Google Scholar] [CrossRef]
- Kalaitzakis, M.; Graeber, M.; Gentleman, S.; Pearce, R. Striatal beta-amyloid deposition in Parkinson disease with dementia. J. Neuropathol. Exp. Neurol. 2008, 67, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Compta, Y.; Parkkinen, L.; O’Sullivan, S.; Vandrovcova, J.; Holton, J.; Collins, C.; Lashley, T.; Kallis, C.; Williams, D.; de Silva, R.; et al. Revesz Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 2011, 134, 1493–1505. [Google Scholar] [CrossRef] [Green Version]
- Harding, A.; Halliday, G. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. 2001, 102, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Kövari, E.; Gold, G.; Herrmann, F.; Canuto, A.; Hof, P.; Bouras, C.; Giannakopoulos, P. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol. 2003, 106, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Morphological substrates of parkinsonism with and without dementia: A retrospective clinico-pathological study. J. Neural Transm. Suppl. 2007, 72, 91–104. [Google Scholar] [CrossRef]
- Jellinger, K.; Seppi, K.; Wenning, G.; Poewe, W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J. Neural Transm. 2002, 109, 329–339. [Google Scholar] [CrossRef]
- Irwin, D.; White, M.; Toledo, J.; Xie, S.; Robinson, J.; Van Deerlin, V.; Lee, V.; Leverenz, J.; Montine, T.; Duda, J.; et al. Neuropathologic substrates of Parkinson disease dementia. Ann. Neurol. 2012, 72, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Oblak, A.; Boeve, B.; Johnson, K.; Dickerson, B.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [Green Version]
- Polanco, J.C.; Li, C.; Durisic, N.; Sullivan, R.; Götz, J. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol. Commun. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polanco, J.; Hand, G.; Briner, A.; Li, C.; Götz, J. Exosomes induce endolysosomal permeabilization as a gateway by which exosomal tau seeds escape into the cytosol. Acta Neuropathol. 2021, 141, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; Surova, Y.; Öhrfelt, A.; Zetterberg, H.; Lindqvist, D.; Hansson, O. CSF biomarkers and clinical progression of Parkinson disease. Neurology 2015, 84, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrelonge, M.; Marder, K.; Weintraub, D.; Alcalay, R. CSF β-Amyloid 1–42 Predicts Progression to Cognitive Impairment in Newly Diagnosed Parkinson Disease. J. Mol. Neurosci. 2016, 58, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latourelle, J.; Beste, M.; Hadzi, T.; Miller, R.; Oppenheim, J.; Valko, M.; Wuest, D.; Church, B.; Khalil, I.; Hayete, B.; et al. Large-scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson’s disease: A longitudinal cohort study and validation. Lancet. Neurol. 2017, 16, 908–916. [Google Scholar] [CrossRef]
- Stav, A.; Aarsland, D.; Johansen, K.; Hessen, E.; Auning, E.; Fladby, T. Amyloid-β and α-synuclein cerebrospinal fluid biomarkers and cognition in early Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 758–764. [Google Scholar] [CrossRef]
- Compta, Y.; Pereira, J.; Ríos, J.; Ibarretxe-Bilbao, N.; Junqué, C.; Bargalló, N.; Cámara, A.; Buongiorno, M.; Fernández, M.; Pont-Sunyer, C.; et al. Combined dementia-risk biomarkers in Parkinson’s disease: A prospective longitudinal study. Parkinsonism Relat. Disord. 2013, 19, 717–724. [Google Scholar] [CrossRef]
- Alves, G.; Lange, J.; Blennow, K.; Zetterberg, H.; Andreasson, U.; Førland, M.; Tysnes, O.; Larsen, J.; Pedersen, K. CSF Aβ42 predicts early-onset dementia in Parkinson disease. Neurology 2014, 82, 1784–1790. [Google Scholar] [CrossRef]
- Ffytche, D.; Pereira, J.; Ballard, C.; Chaudhuri, K.; Weintraub, D.; Aarsland, D. Risk factors for early psychosis in PD: Insights from the Parkinson’s Progression Markers Initiative. J. Neurol. Neurosurg. Psychiatry 2017, 88, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Stewart, T.; Liu, C.; Ginghina, C.; Cain, K.; Auinger, P.; Cholerton, B.; Shi, M.; Zhang, J. Cerebrospinal fluid α-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am. J. Pathol. 2014, 184, 966–975. [Google Scholar] [CrossRef] [Green Version]
- Pagano, G.; De Micco, R.; Yousaf, T.; Wilson, H.; Chandra, A.; Politis, M. REM behavior disorder predicts motor progression and cognitive decline in Parkinson disease. Neurology 2018, 91, e894–e905. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; Surova, Y.; Öhrfelt, A.; Blennow, K.; Zetterberg, H.; Hansson, O. Longitudinal Measurements of Cerebrospinal Fluid Biomarkers in Parkinson’s Disease. Mov. Disord. 2016, 31, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Caspell-Garcia, C.; Simuni, T.; Tosun-Turgut, D.; Wu, I.; Zhang, Y.; Nalls, M.; Singleton, A.; Shaw, L.; Kang, J.; Trojanowski, J.; et al. Multiple modality biomarker prediction of cognitive impairment in prospectively followed de novo Parkinson disease. PLoS ONE 2017, 12, e0175674. [Google Scholar] [CrossRef] [PubMed]
- Førland, M.; Öhrfelt, A.; Dalen, I.; Tysnes, O.; Blennow, K.; Zetterberg, H.; Pedersen, K.; Alves, G.; Lange, J. Evolution of cerebrospinal fluid total α-synuclein in Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 49, 4–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnetti, L.; Farotti, L.; Eusebi, P.; Chiasserini, D.; De Carlo, C.; Giannandrea, D.; Salvadori, N.; Lisetti, V.; Tambasco, N.; Rossi, A.; et al. Differential role of CSF alpha-synuclein species, tau, and Aβ42 in Parkinson’s Disease. Front. Aging Neurosci. 2014, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Rao, M.; Veeranna, N.; Nixon, R. Neurofilaments at a glance. J. Cell Sci. 2012, 125, 3257–3263. [Google Scholar] [CrossRef]
- Gnanapavan, S.; Giovannoni, G. Developing Biomarkers for MS. Curr. Top. Behav. Neurosci. 2015, 26, 179–194. [Google Scholar] [CrossRef]
- Ziemssen, T.; Akgün, K.; Brück, W. Molecular biomarkers in multiple sclerosis. J. Neuroinflamm. 2019, 16, 272. [Google Scholar] [CrossRef] [Green Version]
- Gisslén, M.; Price, R.; Andreasson, U.; Norgren, N.; Nilsson, S.; Hagberg, L.; Fuchs, D.; Spudich, S.; Blennow, K.; Zetterberg, H. Plasma Concentration of the Neurofilament Light Protein (NFL) is a Biomarker of CNS Injury in HIV Infection: A Cross-Sectional Study. EBioMedicine 2015, 3, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Bridel, C.; van Wieringen, W.; Zetterberg, H.; Tijms, B.; Teunissen, C.; Alvarez-Cermeño, J.; Andreasson, U.; Axelsson, M.; Bäckström, D.; Bartos, A.; et al. Diagnostic Value of Cerebrospinal Fluid Neurofilament Light Protein in Neurology: A Systematic Review and Meta-analysis. JAMA Neurol. 2019, 76, 1035–1048. [Google Scholar] [CrossRef]
- Khalil, M.; Pirpamer, L.; Hofer, E.; Voortman, M.M.; Barro, C.; Leppert, D.; Benkert, P.; Ropele, S.; Enzinger, C.; Fazekas, F.; et al. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat. Commun. 2020, 11, 812. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef]
- Lycke, J.; Karlsson, J.; Andersen, O.; Rosengren, L. Neurofilament protein in cerebrospinal fluid: A potential marker of activity in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 1998, 64, 402–404. [Google Scholar] [CrossRef] [Green Version]
- Bergman, J.; Dring, A.; Zetterberg, H.; Blennow, K.; Norgren, N.; Gilthorpe, J.; Bergenheim, T.; Svenningsson, A. Neurofilament light in CSF and serum is a sensitive marker for axonal white matter injury in MS. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disanto, G.; Barro, C.; Benkert, P.; Naegelin, Y.; Schädelin, S.; Giardiello, A.; Zecca, C.; Blennow, K.; Zetterberg, H.; Leppert, D.; et al. Serum Neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann. Neurol. 2017, 81, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Novakova, L.; Axelsson, M.; Khademi, M.; Zetterberg, H.; Blennow, K.; Malmeström, C.; Piehl, F.; Olsson, T.; Lycke, J. Cerebrospinal fluid biomarkers of inflammation and degeneration as measures of fingolimod efficacy in multiple sclerosis. Mult. Scler. 2017, 23, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, I.; Tisell, A.; Cassel, P.; Blennow, K.; Zetterberg, H.; Lundberg, P.; Dahle, C.; Vrethem, M.; Ernerudh, J. Neurofilament levels, disease activity and brain volume during follow-up in multiple sclerosis. J. Neuroinflamm. 2018, 15, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, M.; Teunissen, C.; Otto, M.; Piehl, F.; Sormani, M.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Cantó, E.; Barro, C.; Zhao, C.; Caillier, S.; Michalak, Z.; Bove, R.; Tomic, D.; Santaniello, A.; Häring, D.; Hollenbach, J.; et al. Association Between Serum Neurofilament Light Chain Levels and Long-term Disease Course Among Patients With Multiple Sclerosis Followed up for 12 Years. JAMA Neurol. 2019, 76, 1359–1366. [Google Scholar] [CrossRef]
- Domingues, R.; Fernandes, G.; Leite, F.; Senne, C. Neurofilament light chain in the assessment of patients with multiple sclerosis. Arq. Neuropsiquiatr. 2019, 77, 436–441. [Google Scholar] [CrossRef]
- Varhaug, K.; Torkildsen, Ø.; Myhr, K.; Vedeler, C. Neurofilament Light Chain as a Biomarker in Multiple Sclerosis. Front. Neurol. 2019, 10, 338. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, L.; Eusebi, P.; Mancini, A.; Gentili, L.; Borrelli, A.; Parnetti, L.; Calabresi, P.; Sarchielli, P.; Blennow, K.; Zetterberg, H.; et al. Cerebrospinal fluid neurofilament light chain predicts disease activity after the first demyelinating event suggestive of multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 35, 228–232. [Google Scholar] [CrossRef]
- Zucchi, E.; Bonetto, V.; Sorarù, G.; Martinelli, I.; Parchi, P.; Liguori, R.; Mandrioli, J. Neurofilaments in motor neuron disorders: Towards promising diagnostic and prognostic biomarkers. Mol. Neurodegener. 2020, 15, 58. [Google Scholar] [CrossRef]
- Xu, Z.; Henderson, R.; David, M.; McCombe, P. Neurofilaments as Biomarkers for Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0164625. [Google Scholar] [CrossRef] [Green Version]
- Disanto, G.; Adiutori, R.; Dobson, R.; Martinelli, V.; Dalla Costa, G.; Runia, T.; Evdoshenko, E.; Thouvenot, E.; Trojano, M.; Norgren, N.; et al. Serum neurofilament light chain levels are increased in patients with a clinically isolated syndrome. J. Neurol. Neurosurg. Psychiatry 2016, 87, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Novakova, L.; Zetterberg, H.; Sundström, P.; Axelsson, M.; Khademi, M.; Gunnarsson, M.; Malmeström, C.; Svenningsson, A.; Olsson, T.; Piehl, F.; et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology 2017, 89, 2230–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barro, C.; Benkert, P.; Disanto, G.; Tsagkas, C.; Amann, M.; Naegelin, Y.; Leppert, D.; Gobbi, C.; Granziera, C.; Yaldizli, Ö.; et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain 2018, 141, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Thebault, S.; Abdoli, M.; Fereshtehnejad, S.; Tessier, D.; Tabard-Cossa, V.; Freedman, M. Serum neurofilament light chain predicts long term clinical outcomes in multiple sclerosis. Sci. Rep. 2020, 10, 10381. [Google Scholar] [CrossRef]
- Gunnarsson, M.; Malmeström, C.; Axelsson, M.; Sundström, P.; Dahle, C.; Vrethem, M.; Olsson, T.; Piehl, F.; Norgren, N.; Rosengren, L.; et al. Axonal damage in relapsing multiple sclerosis is markedly reduced by natalizumab. Ann. Neurol. 2011, 69, 83–89. [Google Scholar] [CrossRef]
- Axelsson, M.; Malmeström, C.; Gunnarsson, M.; Zetterberg, H.; Sundström, P.; Lycke, J.; Svenningsson, A. Immunosuppressive therapy reduces axonal damage in progressive multiple sclerosis. Mult. Scler. 2014, 20, 43–50. [Google Scholar] [CrossRef]
- Romme Christensen, J.; Ratzer, R.; Börnsen, L.; Lyksborg, M.; Garde, E.; Dyrby, T.; Siebner, H.; Sorensen, P.; Sellebjerg, F. Natalizumab in progressive MS: Results of an open-label, phase 2A, proof-of-concept trial. Neurology 2014, 82, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Disanto, G.; Lorscheider, J.; Stites, T.; Chen, Y.; Dahlke, F.; Francis, G.; Shrinivasan, A.; Radue, E.; Giovannoni, G.; et al. Fingolimod and CSF neurofilament light chain levels in relapsing-remitting multiple sclerosis. Neurology 2015, 84, 1639–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romme Christensen, J.; Komori, M.; von Essen, M.; Ratzer, R.; Börnsen, L.; Bielekova, B.; Sellebjerg, F. CSF inflammatory biomarkers responsive to treatment in progressive multiple sclerosis capture residual inflammation associated with axonal damage. Mult. Scler. 2019, 25, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Marques, T.; van Rumund, A.; Oeckl, P.; Kuiperij, H.; Esselink, R.; Bloem, B.; Otto, M.; Verbeek, M. Serum NFL discriminates Parkinson disease from atypical parkinsonisms. Neurology 2019, 92, E1479–E1486. [Google Scholar] [CrossRef] [PubMed]
- Sandelius, Å.; Zetterberg, H.; Blennow, K.; Adiutori, R.; Malaspina, A.; Laura, M.; Reilly, M.; Rossor, A. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology 2018, 90, e518–e524. [Google Scholar] [CrossRef] [Green Version]
- Hyun, J.; Kim, Y.; Kim, G.; Kim, S.; Kim, H. Longitudinal analysis of serum neurofilament light chain: A potential therapeutic monitoring biomarker for multiple sclerosis. Mult. Scler. 2020, 26, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Manouchehrinia, A.; Piehl, F.; Hillert, J.; Kuhle, J.; Alfredsson, L.; Olsson, T.; Kockum, I. Confounding effect of blood volume and body mass index on blood neurofilament light chain levels. Ann. Clin. Transl. Neurol. 2020, 7, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Bittner, S.; Steffen, F.; Uphaus, T.; Muthuraman, M.; Fleischer, V.; Salmen, A.; Luessi, F.; Berthele, A.; Klotz, L.; Meuth, S.; et al. Clinical implications of serum neurofilament in newly diagnosed MS patients: A longitudinal multicentre cohort study. EBioMedicine 2020, 56, 102807. [Google Scholar] [CrossRef]
- Hirsch, E.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet. Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- McGeer, P.; Itagaki, S.; Boyes, B.; McGeer, E. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Blum-Degen, D.; Müller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, L.; Perry, V.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Kim, W.; Mohney, R.; Wilson, B.; Jeohn, G.; Liu, B.; Hong, J. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Maatouk, L.; Compagnion, A.; Sauvage, M.; Bemelmans, A.; Leclere-Turbant, S.; Cirotteau, V.; Tohme, M.; Beke, A.; Trichet, M.; Bazin, V.; et al. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat. Commun. 2018, 9, 2450. [Google Scholar] [CrossRef] [PubMed]
- Ros-Bernal, F.; Hunot, S.; Herrero, M.; Parnadeau, S.; Corvol, J.; Lu, L.; Alvarez-Fischer, D.; Carrillo-de Sauvage, M.; Saurini, F.; Coussieu, C.; et al. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc. Natl. Acad. Sci. USA 2011, 108, 6632–6637. [Google Scholar] [CrossRef] [Green Version]
- Marinova-Mutafchieva, L.; Sadeghian, M.; Broom, L.; Davis, J.; Medhurst, A.; Dexter, D. Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: A time course study in a 6-hydroxydopamine model of Parkinson’s disease. J. Neurochem. 2009, 110, 966–975. [Google Scholar] [CrossRef]
- Krashia, P.; Cordella, A.; Nobili, A.; La Barbera, L.; Federici, M.; Leuti, A.; Campanelli, F.; Natale, G.; Marino, G.; Calabrese, V.; et al. Blunting neuroinflammation with resolvin D1 prevents early pathology in a rat model of Parkinson’s disease. Nat. Commun. 2019, 10, 3945. [Google Scholar] [CrossRef] [Green Version]
- La Vitola, P.; Balducci, C.; Baroni, M.; Artioli, L.; Santamaria, G.; Castiglioni, M.; Cerovic, M.; Colombo, L.; Caldinelli, L.; Pollegioni, L.; et al. Peripheral inflammation exacerbates α-synuclein toxicity and neuropathology in Parkinson’s models. Neuropathol. Appl. Neurobiol. 2021, 47, 43–60. [Google Scholar] [CrossRef]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grazioli, S.; Pugin, J. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866I, 165761. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.; Tait, S. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Garaschuk, O. The role of NLRP3 inflammasome for microglial response to peripheral inflammation. Neural Regen. Res. 2021, 16, 294–295. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction and aging: Insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Amir Dache, Z.; Otandault, A.; Tanos, R.; Pastor, B.; Meddeb, R.; Sanchez, C.; Arena, G.; Lasorsa, L.; Bennett, A.; Grange, T.; et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020, 34, 3616–3630. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Biomarkers | Pathogenic Processes | Clinical Trial(s) | References |
|---|---|---|---|
| p-tau/total tau | Post-translational modifications of tau protein and p-tau pathology | − | [16] |
| p-tau/Aβ42 | Post-translational modifications of tau protein, amyloid deposition and p-tau pathology | − | [16,17] |
| Aβ42/total tau, oligomeric α-synuclein/total α-synuclein | Amyloid deposition, α-synuclein, and tau pathology | − | [18,19] |
| Total tau/Aβ42 | Amyloid deposition and p-tau pathology | − | [20] |
| NfL/Aβ42 | Amyloid deposition, neurofilament light chain and axonal injury | − | [21] |
| NfL, Aβ42, p-tau, total tau, and total α-synuclein | Amyloid deposition, α-synuclein and p-tau pathology, and neurofilament light chain and axonal injury | PD01A (α-synuclein, NCT01568099), Nilotinib (α-synuclein and total tau, NCT02281474), MEDI1341 (total α-synuclein, NCT03272165), cerebral dopamine neurotrophic factor (α-synuclein different species, NCT03295786), Glycerol phenylbutyrate (α-synuclein, NCT02046434) | [22] |
| Oligomeric α-synuclein/total α-synuclein, phosphorylated α-synuclein and p-tau | Post-translational modifications (i.e., α-synuclein and tau phosphorylation) | KM-819 (oligomeric α-synuclein, total tau, p-tau, NCT03022799) | [23,24] |
| Total tau/total α-synuclein, p-tau/ α-synuclein, total tau/total α-synuclein with Aβ42, p-tau/total α-synuclein with Aβ42 | Post-translational modifications (i.e., α-synuclein phosphorylation, amyloid deposition, and p-tau pathology) | − | [25] |
| DJ-1 with total tau and p-tau | Mitochondrial dysfunction and post-translational modifications | − | [26] |
| Glucocerebrosidase and β-hexosaminidase, cathepsin D, total α-synuclein, and Aβ42 | Defective autophagy-lysosomal systems, and amyloid and alpha synuclein pathology | Ambroxol (GCase, NCT02941822) | [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Júnior, H.J.; Bucci, C.; Marzetti, E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules 2021, 11, 1508. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11101508
Picca A, Guerra F, Calvani R, Romano R, Coelho-Júnior HJ, Bucci C, Marzetti E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules. 2021; 11(10):1508. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11101508
Chicago/Turabian StylePicca, Anna, Flora Guerra, Riccardo Calvani, Roberta Romano, Hélio José Coelho-Júnior, Cecilia Bucci, and Emanuele Marzetti. 2021. "Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery" Biomolecules 11, no. 10: 1508. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11101508