
Non-Antibody-Based Binders for the Enrichment of Proteins for Analysis by Mass Spectrometry


Abstract
:1. Introduction
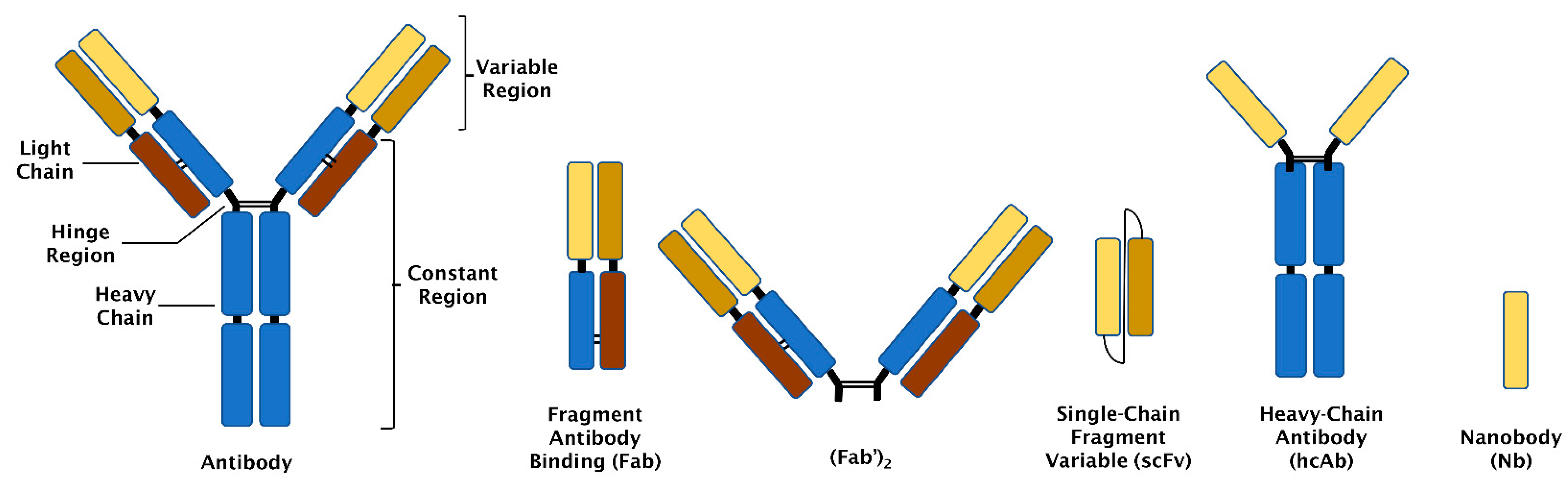
2. Antibody-Based Binders
2.1. Antibodies
2.2. Fragment Antibody Binding (Fab)
2.3. Single Chain Fragment Variable (scFv)
2.4. Heavy Chain Antibodies (hcAbs)
2.5. Nanobodies (Nbs)
3. Non-Antibody-Based Binders
3.1. Aptamers
3.2. DARPins (Designed Ankyrin Repeat Proteins)
3.3. Affimers
3.4. Knottins
3.5. Avimers
3.6. Monobodies
3.7. Anticalins
3.8. Fynomers
3.9. Affibodies
4. Phage Display
5. Immobilisation Approaches
5.1. Physical Adsorption
5.2. Covalent Immobilisation
5.3. Affinity Immobilisation
6. Non-Antibody-Based Affinity Enrichment of Proteins in Combination with Mass Spectrometry
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Luque-Garcia, J.L.; Neubert, T.A. Sample preparation for serum/plasma profiling and biomarker identification by mass spectrometry. J. Chromatogr. A 2007, 1153, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Hortin, G.L.; Sviridov, D. The dynamic range problem in the analysis of the plasma proteome. J. Proteom. 2010, 73, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Bellei, E.; Bergamini, S.; Monari, E.; Fantoni, L.; Cuoghi, A.; Ozben, T.; Tomasi, A. High-abundance proteins depletion for serum proteomic analysis: Concomitant removal of non-targeted proteins. Amino Acids 2011, 40, 145–156. [Google Scholar] [CrossRef]
- Whiteaker, J.R.; Zhao, L.; Schoenherr, R.M.; Kennedy, J.J.; Ivey, R.G.; Paulovich, A.G. Peptide Immunoaffinity Enrichment with Targeted Mass Spectrometry: Application to Quantification of ATM Kinase Phospho-Signaling. Methods Mol. Biol. 2017, 1599, 197–213. [Google Scholar]
- Ezan, E.; Dubois, M.; Becher, F. Bioanalysis of recombinant proteins and antibodies by mass spectrometry. Analyst 2009, 134, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.N.; Bryan, P.; Kozhich, A. Techniques for quantitative LC–MS/MS analysis of protein therapeutics: Advances in enzyme digestion and immunocapture. Bioanalysis 2016, 8, 847–856. [Google Scholar] [CrossRef]
- Yu, X.; Yang, Y.-P.; Dikici, E.; Deo, S.K.; Daunert, S. Beyond Antibodies as Binding Partners: The Role of Antibody Mimetics in Bioanalysis. Annu. Rev. Anal. Chem. 2017, 10, 293–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilffert, D.; Bischoff, R.; Merbel, N.C.v.d. Antibody-free workflows for protein quantification by LC–MS/MS. Bioanalysis 2015, 7, 763–779. [Google Scholar] [CrossRef]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.L. Recombinant monoclonal antibody technology. Transfus. Clin. Biol. 2002, 9, 15–22. [Google Scholar] [CrossRef]
- Horwitz, A.H.; Chang, C.P.; Better, M.; Hellstrom, K.E.; Robinson, R.R. Secretion of functional antibody and Fab fragment from yeast cells. Proc. Natl. Acad. Sci. USA 1988, 85, 8678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Gutshall, L.; Jiang, H.; Baker, A.; Beil, E.; Obmolova, G.; Carton, J.; Taudte, S.; Amegadzie, B. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expr. Purif. 2009, 67, 182–189. [Google Scholar] [CrossRef]
- Akesson, P.; Moritz, L.; Truedsson, M.; Christensson, B.; von Pawel-Rammingen, U. IdeS, a highly specific immunoglobulin G (IgG)-cleaving enzyme from Streptococcus pyogenes, is inhibited by specific IgG antibodies generated during infection. Infect. Immun. 2006, 74, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Sereno, A.J.; Huang, F.; Lewis, S.M.; Lieu, R.L.; Weldon, C.; Torres, C.; Fine, C.; Batt, M.A.; Fitchett, J.R.; et al. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. mAbs 2015, 7, 470–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.M.; Hamid, M. scFv Antibody: Principles and Clinical Application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef]
- Miller, B.R.; Demarest, S.J.; Lugovskoy, A.; Huang, F.; Wu, X.; Snyder, W.B.; Croner, L.J.; Wang, N.; Amatucci, A.; Michaelson, J.S.; et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng. Des. Sel. 2010, 23, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Weaver-Feldhaus, J.; Gray, S.A.; Siegel, R.W.; Feldhaus, M.J. Production, purification, and characterization of human scFv antibodies expressed in Saccharomyces cerevisiae, Pichia pastoris, and Escherichia coli. Protein Expr. Purif. 2005, 42, 255–267. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Drabek, D.; Janssens, R.; de Boer, E.; Rademaker, R.; Kloess, J.; Skehel, J.; Grosveld, F. Expression Cloning and Production of Human Heavy-Chain-Only Antibodies from Murine Transgenic Plasma Cells. Front. Immunol. 2016, 7, 619. [Google Scholar] [CrossRef] [Green Version]
- Crivianu-Gaita, V.; Thompson, M. Aptamers, antibody scFv, and antibody Fab’ fragments: An overview and comparison of three of the most versatile biosensor biorecognition elements. Biosens. Bioelectron. 2016, 85, 32–45. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Historical Perspective and Future Outlook. Front. Immunol. 2017, 8, 1589. [Google Scholar] [CrossRef] [Green Version]
- Song, K.-M.; Lee, S.; Ban, C. Aptamers and Their Biological Applications. Sensors 2012, 12, 612–631. [Google Scholar] [CrossRef] [Green Version]
- Chai, C.; Xie, Z.; Grotewold, E. SELEX (Systematic Evolution of Ligands by EXponential Enrichment), as a powerful tool for deciphering the protein-DNA interaction space. Methods Mol. Biol. 2011, 754, 249–258. [Google Scholar] [PubMed]
- Reverdatto, S.; Rai, V.; Xue, J.; Burz, D.S.; Schmidt, A.M.; Shekhtman, A. Combinatorial library of improved peptide aptamers, CLIPs to inhibit RAGE signal transduction in mammalian cells. PLoS ONE 2013, 8, e65180. [Google Scholar] [CrossRef] [Green Version]
- Geyer, C.R.; Brent, R. [13]Selection of genetic agents from random peptide aptamer expression libraries. In Methods in Enzymology; Thorner, J., Emr, S.D., Abelson, J.N., Eds.; Academic Press: Cambridge, MA, USA, 2000; Volume 328, pp. 171–208. [Google Scholar]
- Kratschmer, C.; Levy, M. Effect of Chemical Modifications on Aptamer Stability in Serum. Nucleic Acid Ther. 2017, 27, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical Modifications of Nucleic Acid Aptamers for Therapeutic Purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Shilova, O.N.; Deyev, S.M. DARPins: Promising Scaffolds for Theranostics. Acta Nat. 2019, 11, 42–53. [Google Scholar] [CrossRef]
- Plückthun, A. Designed Ankyrin Repeat Proteins (DARPins): Binding Proteins for Research, Diagnostics, and Therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef]
- Stefan, N.; Martin-Killias, P.; Wyss-Stoeckle, S.; Honegger, A.; Zangemeister-Wittke, U.; Plückthun, A. DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J. Mol. Biol. 2011, 413, 826–843. [Google Scholar] [CrossRef]
- Zahnd, C.; Pecorari, F.; Straumann, N.; Wyler, E.; Plückthun, A. Selection and characterization of Her2 binding-designed ankyrin repeat proteins. J. Biol. Chem. 2006, 281, 35167–35175. [Google Scholar] [CrossRef] [Green Version]
- Shamsuddin, S.H.; Jayne, D.G.; Tomlinson, D.C.; McPherson, M.J.; Millner, P.A. Selection and characterisation of Affimers specific for CEA recognition. Sci. Rep. 2021, 11, 744. [Google Scholar] [CrossRef] [PubMed]
- Gelly, J.C.; Gracy, J.; Kaas, Q.; Le-Nguyen, D.; Heitz, A.; Chiche, L. The KNOTTIN website and database: A new information system dedicated to the knottin scaffold. Nucleic Acids Res. 2004, 32 (Suppl. 1), D156–D159. [Google Scholar] [CrossRef] [Green Version]
- Postic, G.; Gracy, J.; Périn, C.; Chiche, L.; Gelly, J.-C. KNOTTIN: The database of inhibitor cystine knot scaffold after 10 years, toward a systematic structure modeling. Nucleic Acids Res. 2018, 46, D454–D458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeon, R.; Chen, Z. In vitro-engineered non-antibody protein therapeutics. Protein Cell 2018, 9, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherf, G.M.; Cochran, J.R. Applications of Yeast Surface Display for Protein Engineering. Methods Mol. Biol. 2015, 1319, 155–175. [Google Scholar] [PubMed] [Green Version]
- Silverman, J.; Lu, Q.; Bakker, A.; To, W.; Duguay, A.; Alba, B.M.; Smith, R.; Rivas, A.; Li, P.; Le, H.; et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 2005, 23, 1556–1561. [Google Scholar] [CrossRef]
- Jeong, K.J.; Mabry, R.; Georgiou, G. Avimers hold their own. Nat. Biotechnol. 2005, 23, 1493–1494. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef]
- Schmit, N.E.; Neopane, K.; Hantschel, O. Targeted Protein Degradation through Cytosolic Delivery of Monobody Binders Using Bacterial Toxins. ACS Chem. Biol. 2019, 14, 916–924. [Google Scholar] [CrossRef]
- Skerra, A. Alternative binding proteins: Anticalins—Harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities. FEBS J. 2008, 275, 2677–2683. [Google Scholar] [CrossRef]
- Rothe, C.; Skerra, A. Anticalin(®) Proteins as Therapeutic Agents in Human Diseases. BioDrugs 2018, 32, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Barinka, C.; Ptacek, J.; Richter, A.; Novakova, Z.; Morath, V.; Skerra, A. Selection and characterization of Anticalins targeting human prostate-specific membrane antigen (PSMA). Protein Eng. Des. Sel. 2016, 29, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlatter, D.; Brack, S.; Banner, D.W.; Batey, S.; Benz, J.; Bertschinger, J.; Huber, W.; Joseph, C.; Rufer, A.; van der Klooster, A.; et al. Generation, characterization and structural data of chymase binding proteins based on the human Fyn kinase SH3 domain. mAbs 2012, 4, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, A.L. Antibody fragments: Hope and hype. mAbs 2010, 2, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moelleken, J.; Endesfelder, M.; Gassner, C.; Lingke, S.; Tomaschek, S.; Tyshchuk, O.; Lorenz, S.; Reiff, U.; Mølhøj, M. GingisKHAN™ protease cleavage allows a high-throughput antibody to Fab conversion enabling direct functional assessment during lead identification of human monoclonal and bispecific IgG1 antibodies. mAbs 2017, 9, 1076–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, H.; Takekoshi, M. Production of Antibody Fab Fragments in Escherichia coli. Antib. Expr. Prod. 2011, 7, 165–178. [Google Scholar]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronemeyer, P.; Ditz, R.; Strube, J. Trends in Upstream and Downstream Process Development for Antibody Manufacturing. Bioengineering 2014, 1, 188–212. [Google Scholar] [CrossRef] [Green Version]
- Leenaars, M.; Hendriksen, C.F.M. Critical Steps in the Production of Polyclonal and Monoclonal Antibodies: Evaluation and Recommendations. ILAR J. 2005, 46, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Strom, M.; Hammond, D.S.; Shigdar, S. Anything You Can Do, I Can Do Better: Can Aptamers Replace Antibodies in Clinical Diagnostic Applications? Molecules 2019, 24, 4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voskuil, J. Commercial antibodies and their validation. F1000Research 2014, 3, 232. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Rusling, J.; Dixit, C.K. Site-selective orientated immobilization of antibodies and conjugates for immunodiagnostics development. Methods 2017, 116, 95–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, A.; Power, C.A. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Quintero-Hernández, V.; Juárez-González, V.R.; Ortíz-León, M.; Sánchez, R.; Possani, L.D.; Becerril, B. The change of the scFv into the Fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol. Immunol. 2007, 44, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Kumar Kulabhusan, P.; Hussain, B.; Yüce, M. Current Perspectives on Aptamers as Diagnostic Tools and Therapeutic Agents. Pharmaceutics 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- SELEX. Encyclopedia of Cancer; Schwab, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; p. 3359. [Google Scholar]
- Borghouts, C.; Kunz, C.; Delis, N.; Groner, B. Monomeric recombinant peptide aptamers are required for efficient intracellular uptake and target inhibition. Mol. Cancer Res. 2008, 6, 267–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, R.; Sosabowski, J.; Livanos, M.; Leyton, J.; Vigor, K.; Bhavsar, G.; Nagy-Davidescu, G.; Rashid, M.; Miranda, E.; Yeung, J.; et al. Development of the designed ankyrin repeat protein (DARPin) G3 for HER2 molecular imaging. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 288–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puard, V.; Tan, K.-Y.; Baldwin, K.; Carrol, E.; Pattison, R.; Beynon, R.; Ko Ferrigno, P. Affimer®, Novel Affinity Reagent and Targeting Tools for Life Sciences; [Paper presentation] RPPA Workshop 2015; George Mason University: Manassas, VA, USA, 2015. [Google Scholar]
- Kyle, S. Affimer Proteins: Theranostics of the Future? Trends Biochem. Sci. 2018, 43, 230–232. [Google Scholar] [CrossRef]
- Thangsunan, P. Affimer-Based Impedimetric Biosensors: The New Analytical Platform for Biorecognition Applications. Ph.D. Thesis, University of Leeds, Leeds, UK, 2018. [Google Scholar]
- Raina, M.; Sharma, R.; Deacon, S.E.; Tiede, C.; Tomlinson, D.; Davies, A.G.; McPherson, M.J.; Wälti, C. Antibody mimetic receptor proteins for label-free biosensors. Analyst 2015, 140, 803–810. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.J.; Cochran, J.R. Chapter nine—Engineering Knottins as Novel Binding Agents. In Methods in Enzymology; Wittrup, K.D., Verdine, G.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 503, pp. 223–251. [Google Scholar]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Moore, S.J.; Leung, C.L.; Cochran, J.R. Knottins: Disulfide-bonded therapeutic and diagnostic peptides. Drug Discov. Today: Technol. 2012, 9, e3–e11. [Google Scholar] [CrossRef]
- Hantschel, O. Monobodies as possible next-generation protein therapeutics—A perspective. Swiss Med. Wkly. 2017, 147, w14545. [Google Scholar]
- Silacci, M.; Baenziger-Tobler, N.; Lembke, W.; Zha, W.; Batey, S.; Bertschinger, J.; Grabulovski, D. Linker length matters, fynomer-Fc fusion with an optimized linker displaying picomolar IL-17A inhibition potency. J. Biol. Chem. 2014, 289, 14392–14398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlberg, E.; Lendel, C.; Helgstrand, M.; Allard, P.; Dincbas-Renqvist, V.; Hedqvist, A.; Berglund, H.; Nygren, P.-Å.; Härd, T. An affibody in complex with a target protein: Structure and coupled folding. Proc. Natl. Acad. Sci. USA 2003, 100, 3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arap, M.A. Phage display technology—Applications and innovations. Genet. Mol. Biol. 2005, 28. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-H.; Liu, I.J.; Lu, R.-M.; Wu, H.-C. Advancement and applications of peptide phage display technology in biomedical science. J. Biomed. Sci. 2016, 23, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusmini, F.; Zhong, Z.; Feijen, J. Protein immobilization strategies for protein biochips. Biomacromolecules 2007, 8, 1775–1789. [Google Scholar] [CrossRef]
- Lee, J.E.; Seo, J.H.; Kim, C.S.; Kwon, Y.; Ha, J.H.; Choi, S.S.; Cha, H.J. A comparative study on antibody immobilization strategies onto solid surface. Korean J. Chem. Eng. 2013, 30, 1934–1938. [Google Scholar] [CrossRef]
- Kausaite-Minkstimiene, A.; Ramanaviciene, A.; Kirlyte, J.; Ramanavicius, A. Comparative study of random and oriented antibody immobilization techniques on the binding capacity of immunosensor. Anal. Chem. 2010, 82, 6401–6408. [Google Scholar] [CrossRef]
- de la Torre, D.; Chin, J.W. Reprogramming the genetic code. Nat. Rev. Genet. 2021, 22, 169–184. [Google Scholar] [CrossRef]
- Camarero, J.A. Recent developments in the site-specific immobilization of proteins onto solid supports. Biopolymers 2008, 90, 450–458. [Google Scholar] [CrossRef]
- Jonkheijm, P.; Weinrich, D.; Schröder, H.; Niemeyer, C.M.; Waldmann, H. Chemical strategies for generating protein biochips. Angew. Chem. Int. Ed. Engl. 2008, 47, 9618–9647. [Google Scholar] [CrossRef]
- Seo, M.H.; Han, J.; Jin, Z.; Lee, D.W.; Park, H.S.; Kim, H.S. Controlled and oriented immobilization of protein by site-specific incorporation of unnatural amino acid. Anal. Chem. 2011, 83, 2841–2845. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D. Applications of reversible covalent chemistry in analytical sample preparation. Analyst 2012, 137, 5457–5482. [Google Scholar] [CrossRef] [PubMed]
- Tans, R.; van Rijswijck, D.M.H.; Davidson, A.; Hannam, R.; Ricketts, B.; Tack, C.J.; Wessels, H.J.C.T.; Gloerich, J.; van Gool, A.J. Affimers as an alternative to antibodies for protein biomarker enrichment. Protein Expr. Purif. 2020, 174, 105677. [Google Scholar] [CrossRef]
- Klont, F.; Hadderingh, M.; Horvatovich, P.; Ten Hacken, N.H.T.; Bischoff, R. Affimers as an Alternative to Antibodies in an Affinity LC-MS Assay for Quantification of the Soluble Receptor of Advanced Glycation End-Products (sRAGE) in Human Serum. J. Proteome Res. 2018, 17, 2892–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klont, F.; Pouwels, S.D.; Hermans, J.; van de Merbel, N.C.; Horvatovich, P.; ten Hacken, N.H.T.; Bischoff, R. A fully validated liquid chromatography-mass spectrometry method for the quantification of the soluble receptor of advanced glycation end-products (sRAGE) in serum using immunopurification in a 96-well plate format. Talanta 2018, 182, 414–421. [Google Scholar] [CrossRef]
- Radko, S.; Ptitsyn, K.; Novikova, S.; Kiseleva, Y.; Moysa, A.; Kurbatov, L.; Mannanova, M.; Zgoda, V.; Ponomarenko, E.; Lisitsa, A.; et al. Evaluation of Aptamers as Affinity Reagents for an Enhancement of SRM-Based Detection of Low-Abundance Proteins in Blood Plasma. Biomedicines 2020, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-H.; Chang, K.-W.; Cheng, H.-W.; Liu, C.-J. SMAD4 Somatic Mutations in Head and Neck Carcinoma Are Associated With Tumor Progression. Front. Oncol. 2019, 9, 1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodford-Richens, K.L.; Rowan, A.J.; Gorman, P.; Halford, S.; Bicknell, D.C.; Wasan, H.S.; Roylance, R.R.; Bodmer, W.F.; Tomlinson, I.P. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 9719–9723. [Google Scholar] [CrossRef] [Green Version]
- Ngo, D.; Sinha, S.; Shen, D.; Kuhn, E.W.; Keyes, M.J.; Shi, X.; Benson, M.D.; O’Sullivan, J.F.; Keshishian, H.; Farrell, L.A.; et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation 2016, 134, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Lassman, M.E.; McAvoy, T.; Lee, A.Y.; Chappell, D.L.; Laterza, O.F. An evaluation of an aptamer for use as an affinity reagent with MS: PCSK9 as an example protein. Bioanalysis 2016, 8, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.S.; Fong, L.G.; Young, S.G. PCSK9 function and physiology. J. Lipid Res. 2008, 49, 1152–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
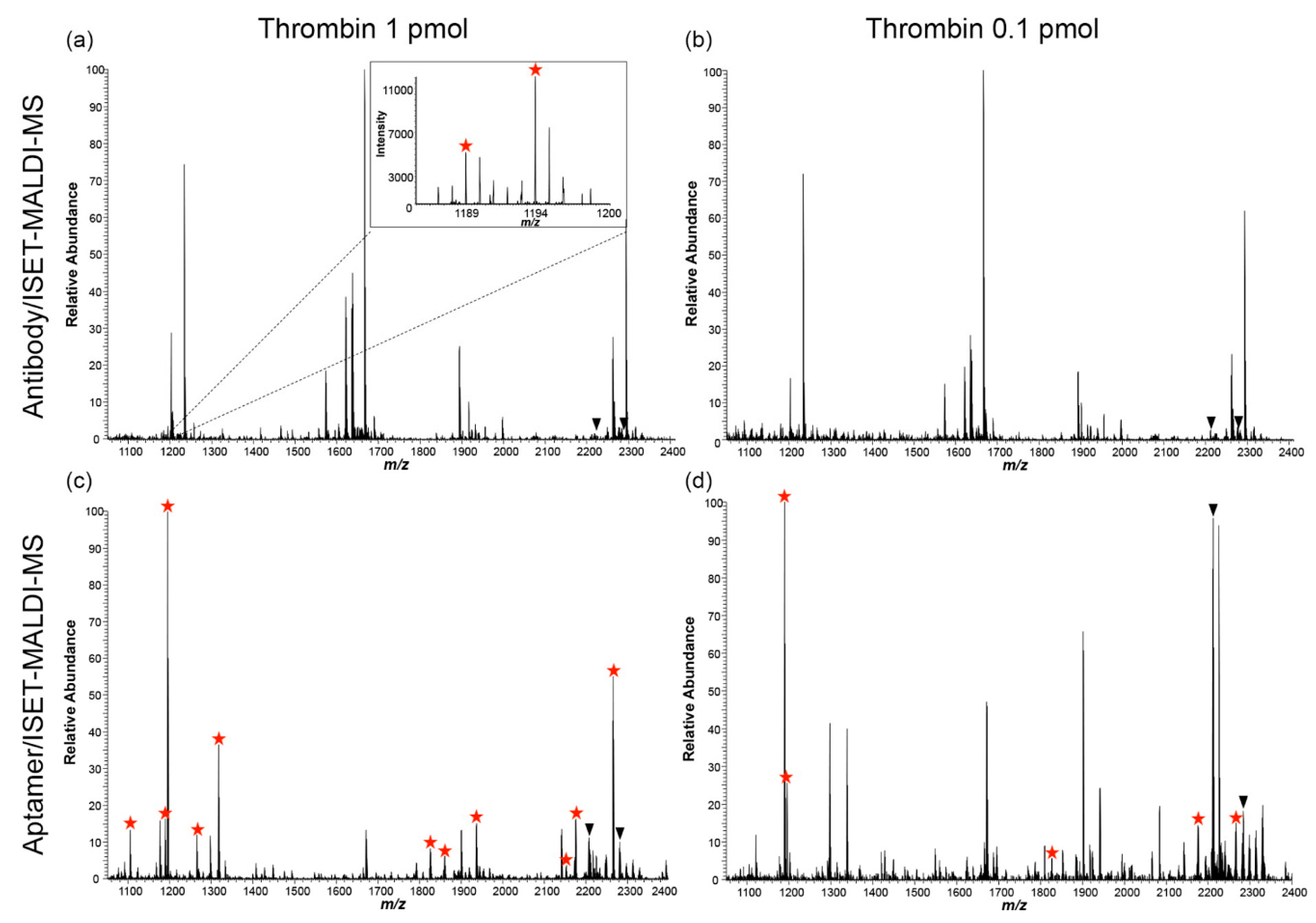
- Lee, S.J.; Adler, B.; Ekström, S.; Rezeli, M.; Végvári, Á.; Park, J.W.; Malm, J.; Laurell, T. Aptamer/ISET-MS: A new affinity-based MALDI technique for improved detection of biomarkers. Anal. Chem. 2014, 86, 7627–7634. [Google Scholar] [CrossRef] [PubMed]
- Harmansa, S.; Affolter, M. Protein binders and their applications in developmental biology. Development 2018, 145, dev148874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015, 33, 408–418. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody-Based Binder | Size | Production | Refs |
|---|---|---|---|
| Antibody | ~150 kDa | Hybridoma or recombinant DNA technology and mammalian cell expression | [8,9] |
| Fragment Antibody Binding (Fab) | ~50 kDa | Proteolysis (e.g., with papain, IdeS, or GingisKHAN™) or recombinant DNA technology and mammalian, yeast, or bacterial cell expression | [10,11,12,13] |
| Single-Chain Fragment Variable (scFv) | ~25 kDa | Recombinant DNA technology and yeast or bacterial cell expression | [14,15,16] |
| Heavy Chain Antibodies | ~75 kDa | Hybridoma or recombinant DNA technology and mammalian cell expression | [17,18] |
| Nanobodies | ~15 kDa | Recombinant DNA technology and plant, mammalian, or bacterial cell expression | [19] |
| Non-Antibody-Based Binder | Scaffold | Size | Production | Refs |
|---|---|---|---|---|
| Aptamers | Oligonucleotide/Protein scaffolds | 5–30 kDa | Chemical synthesis as part of the Systematic Evolution of Ligands by Exponential Enrichment (SELEX) procedure/Phage display and bacterial expression | [20,21,22,23,24,25] |
| DARPins | Ankyrin repeats | 14–18 kDa | Phage or ribosome display and bacterial expression | [26,27,28,29,30] |
| Affimers | Human stefin A or phytocystatin | 12–14 kDa | Phage display and bacterial expression | [31,32,33] |
| Knottins | Inhibitor cysteine knot | ~4 kDa | Chemical synthesis or yeast display and yeast expression | [34,35,36] |
| Avimers | A-domain region of cells | ~4 kDa | Phage display and bacterial expression | [35,37] |
| Monobodies | Human fibronectin type III domain | ~10 kDa | Phage or yeast display and bacterial expression | [38,39,40] |
| Anticalins | Lipocalins | ~20 kDa | Phage display and bacterial expression | [41,42] |
| Fynomers | Human tyrosine kinase Src Homology 3 domain | ~7 kDa | Phage display and bacterial expression | [43,44] |
| Affibodies | S. aureus Protein A | ~7 kDa | Phage display and bacterial expression | [36,44,45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olaleye, O.; Govorukhina, N.; van de Merbel, N.C.; Bischoff, R. Non-Antibody-Based Binders for the Enrichment of Proteins for Analysis by Mass Spectrometry. Biomolecules 2021, 11, 1791. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11121791
Olaleye O, Govorukhina N, van de Merbel NC, Bischoff R. Non-Antibody-Based Binders for the Enrichment of Proteins for Analysis by Mass Spectrometry. Biomolecules. 2021; 11(12):1791. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11121791
Chicago/Turabian StyleOlaleye, Oladapo, Natalia Govorukhina, Nico C. van de Merbel, and Rainer Bischoff. 2021. "Non-Antibody-Based Binders for the Enrichment of Proteins for Analysis by Mass Spectrometry" Biomolecules 11, no. 12: 1791. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11121791