
Glycosaminoglycans: Carriers and Targets for Tailored Anti-Cancer Therapy


 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Focus on GAGs’ Structure and Roles
2.1. Heparin and Heparan Sulfate
2.2. Chondroitin Sulfate/Dermatan Sulfate
2.3. Keratan Sulfate
2.4. Hyaluronan
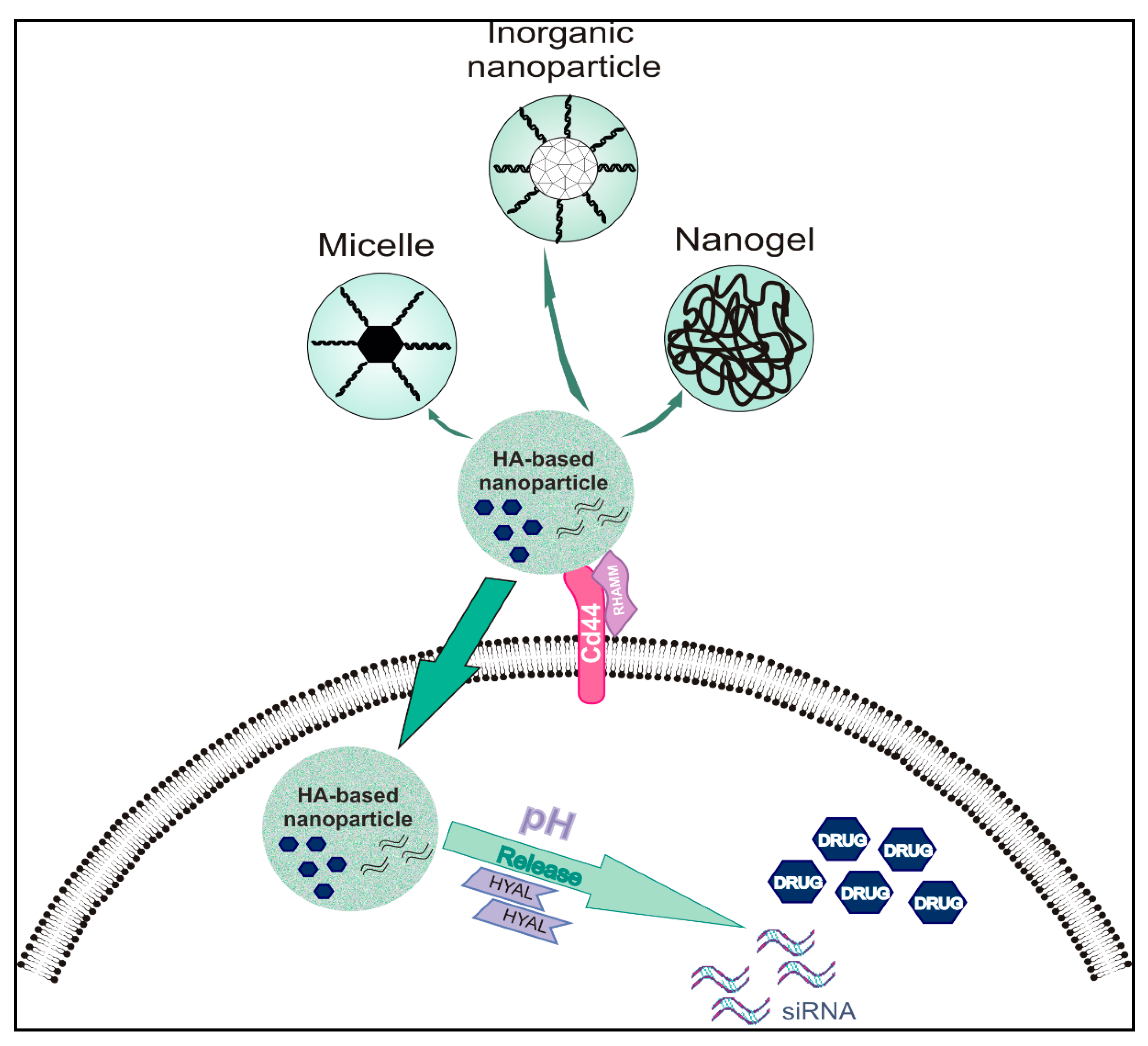
3. Types of Nanoparticles and Materials Utilized for Targeted Drug Delivery—Focus on GAG-Based Nanoparticles

{kind=link}
{kind=link}
| Nanoparticle System | Material | Nanocarriers Type | Examples of Carried Agents | Reference |
|---|---|---|---|---|
| Lipids | Phospholipids | Liposomes, solid lipid particles | RGD peptide, apatinib | [89] |
| Synthetic polymers | Poly(N-isopropylacrylamide, poly-N-vinylpyrrolidone, poly(lactic-co-glycolic acid) | Micelles, nanoparticles, | Doxorubicin, curcumin, indocyanine green | [85,88,93,94,104,112] |
| Natural polymers | HA, alginate, chitosan, heparosan, carboxymethyl starch, CS, Hep | Microcapsules nanospheres, nanoparticles, nanogel, micelles | Doxorubicin, BSA, tirapazamine, cisplatin | [84,85,86,87,92,101,106,111] Section 1 |
| Dendrimer | Polyester, Polyacetal/polyketal | Micelles | Camptothecin, methotrexate | [90] |
| Silica | Mesoporous silica | Nanoparticles | Doxorubicin, fluorescein isothiocyanate | [91] |
| Metal | Gold | Nanoparticles, nanorods | Doxorubicin, bleomycin | [105] |
3.1. Heparin and Heparan Sulfate for Anticancer Drug Delivery
3.1.1. Micellar Heparin Nanoparticles
3.1.2. Heparin-Coated Metal NanoParticles
3.1.3. Heparin Nanogels
3.1.4. Summary
3.2. Chondroitin Sulfate and Dermatan Sulfate-Based Nanoparticles as Drug Delivery Systems
Summary
3.3. Keratan Sulfate in Anticancer Drug Delivery
Summary
3.4. Hyaluronic Acid-Based Nanoparticles for Controlled Drug Release in Cancer
3.4.1. Hyaluronic Acid-Based Micelles
3.4.2. Hyaluronic Acid-Based Nanogels
3.4.3. Inorganic Hyaluronic Acid-Based Nanoparticles
3.4.4. Clinical Trials Implementing Hyaluronic Acid-Based Nanoparticles
4. Targeting GAGs in Cancer—New Prospective
4.1. Targeting Heparan Sulfate/Heparin
4.2. Enzymatic Modulation of HS–Protein Interactions
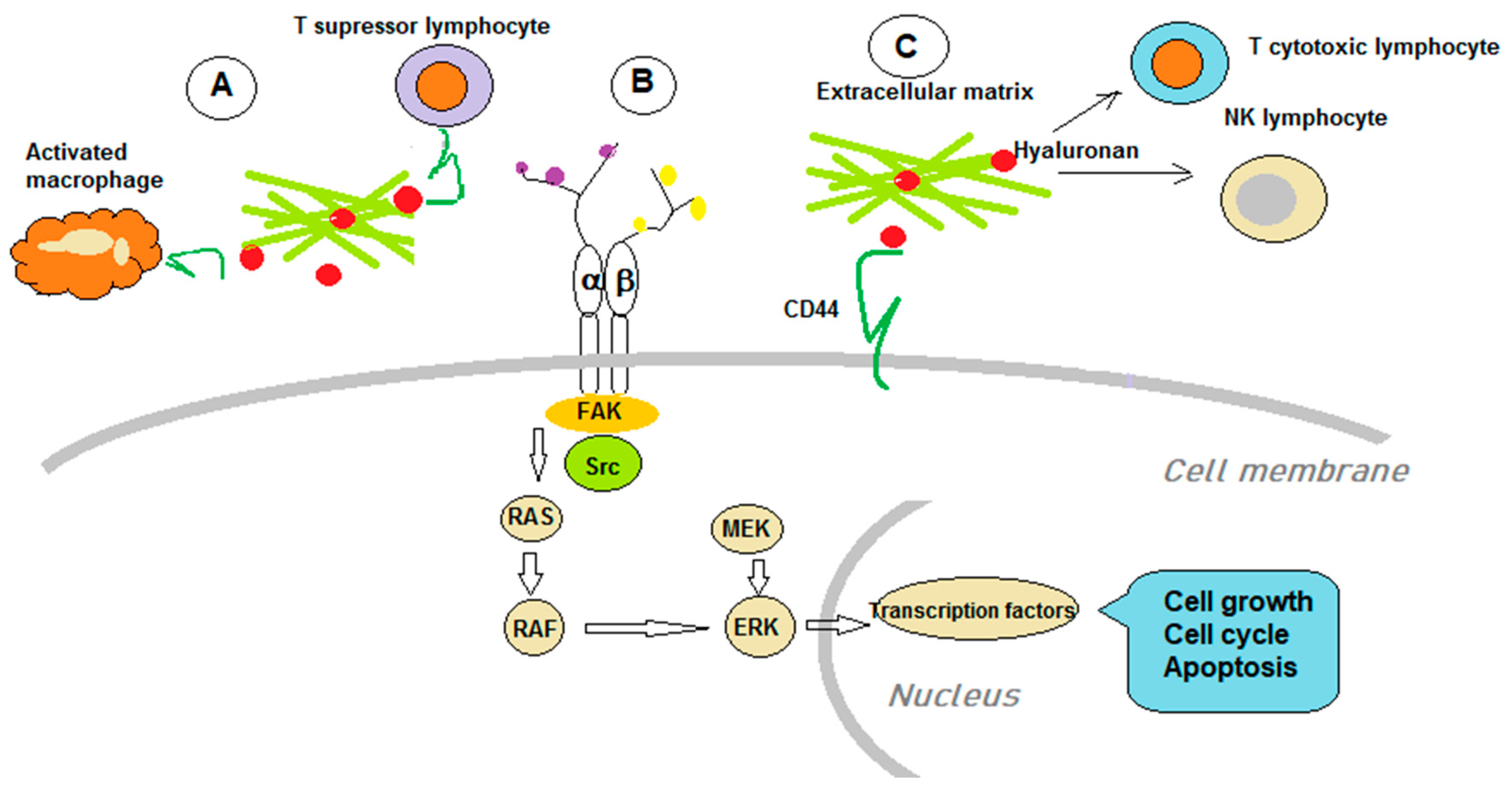
5. GAGs and Immunological Aspects of Cancer Therapy
5.1. GAGs Roles in Tumor Immunology
5.2. GAGs as Immunotherapy Targets
6. GAGs as Potential Cancer Therapy Response Biomarkers
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tzanakakis, G.; Giatagana, E.M.; Kuskov, A.; Berdiaki, A.; Tsatsakis, A.M.; Neagu, M.; Nikitovic, D. Proteoglycans in the Pathogenesis of Hormone-Dependent Cancers: Mediators and Effectors. Cancers 2020, 12, 2401. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Neagu, M.; Tsatsakis, A.; Nikitovic, D. Proteoglycans and Immunobiology of Cancer-Therapeutic Implications. Front. Immunol. 2019, 10, 875. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Piperigkou, Z.; Theocharis, A.D.; Watanabe, H.; Franchi, M.; Baud, S.; Brezillon, S.; Gotte, M.; Passi, A.; Vigetti, D.; et al. Proteoglycan Chemical Diversity Drives Multifunctional Cell Regulation and Therapeutics. Chem. Rev. 2018, 118, 9152–9232. [Google Scholar] [CrossRef]
- Nikitovic, D.; Papoutsidakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican affects tumor cell functions, tumor-ECM interactions, angiogenesis and inflammatory response. Matrix Biol. 2014, 35, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavao, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key players in cancer cell biology and treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Tzardi, M.; Berdiaki, A.; Tsatsakis, A.; Tzanakakis, G.N. Cancer microenvironment and inflammation: Role of hyaluronan. Front. Immunol. 2015, 6, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitovic, D.; Berdiaki, A.; Spyridaki, I.; Krasanakis, T.; Tsatsakis, A.; Tzanakakis, G.N. Proteoglycans-Biomarkers and Targets in Cancer Therapy. Front. Endocrinol. 2018, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karangelis, D.E.; Kanakis, I.; Asimakopoulou, A.P.; Karousou, E.; Passi, A.; Theocharis, A.D.; Triposkiadis, F.; Tsilimingas, N.B.; Karamanos, N.K. Glycosaminoglycans as key molecules in atherosclerosis: The role of versican and hyaluronan. Curr. Med. Chem. 2010, 17, 4018–4026. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Cavdarli, S.; Uchimura, K.; Allain, F.; Delannoy, P. Glycosylation changes in inflammatory diseases. Adv. Protein Chem. Struct. Biol. 2020, 119, 111–156. [Google Scholar] [PubMed]
- Kouvidi, K.; Berdiaki, A.; Nikitovic, D.; Katonis, P.; Afratis, N.; Hascall, V.C.; Karamanos, N.K.; Tzanakakis, G.N. Role of receptor for hyaluronic acid-mediated motility (RHAMM) in low molecular weight hyaluronan (LMWHA)-mediated fibrosarcoma cell adhesion. J. Biol. Chem. 2011, 286, 38509–38520. [Google Scholar] [CrossRef] [Green Version]
- Schwertfeger, K.L.; Cowman, M.K.; Telmer, P.G.; Turley, E.A.; McCarthy, J.B. Hyaluronan, Inflammation, and Breast Cancer Progression. Front. Immunol. 2015, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, P.; Presto, J.; Spillmann, D.; Lindahl, U.; Kjellen, L. Heparin/heparan sulfate biosynthesis: Processive formation of N-sulfated domains. J. Biol. Chem. 2008, 283, 20008–20014. [Google Scholar] [CrossRef] [Green Version]
- Vigetti, D.; Karousou, E.; Viola, M.; Deleonibus, S.; De Luca, G.; Passi, A. Hyaluronan: Biosynthesis and signaling. Biochim. Biophys. Acta 2014, 1840, 2452–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikami, T.; Kitagawa, H. Biosynthesis and function of chondroitin sulfate. Biochim. Biophys. Acta 2013, 1830, 4719–4733. [Google Scholar] [CrossRef]
- Pomin, V.H. Keratan sulfate: An up-to-date review. Int. J. Biol. Macromol. 2015, 72, 282–289. [Google Scholar] [CrossRef]
- Habuchi, H.; Habuchi, O.; Kimata, K. Sulfation pattern in glycosaminoglycan: Does it have a code? Glycoconj. J. 2004, 21, 47–52. [Google Scholar] [CrossRef]
- Sarkar, A.; Desai, U.R. A Simple Method for Discovering Druggable, Specific Glycosaminoglycan-Protein Systems. Elucidation of Key Principles from Heparin/Heparan Sulfate-Binding Proteins. PLoS ONE 2015, 10, e0141127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef]
- Kjellen, L.; Lindahl, U. Specificity of glycosaminoglycan-protein interactions. Curr. Opin. Struct. Biol. 2018, 50, 101–108. [Google Scholar] [CrossRef]
- Morla, S. Glycosaminoglycans and Glycosaminoglycan Mimetics in Cancer and Inflammation. Int. J. Mol. Sci. 2019, 20, 1963. [Google Scholar] [CrossRef] [Green Version]
- Sasisekharan, R.; Shriver, Z.; Venkataraman, G.; Narayanasami, U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat. Rev. Cancer 2002, 2, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, J.; Matsumoto, T.; Vanwildemeersch, M.; Sasaki, T.; Timpl, R.; Claesson-Welsh, L.; Spillmann, D.; Lindahl, U. Role of heparan sulfate domain organization in endostatin inhibition of endothelial cell function. EMBO J. 2002, 21, 6303–6311. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Yu, G.; Sun, L.; Zhang, Z.; Zhao, X.; Chai, W. Elevate level of glycosaminoglycans and altered sulfation pattern of chondroitin sulfate are associated with differentiation status and histological type of human primary hepatic carcinoma. Oncology 2007, 72, 347–356. [Google Scholar] [CrossRef]
- Pudelko, A.; Wisowski, G.; Olczyk, K.; Kozma, E.M. The dual role of the glycosaminoglycan chondroitin-6-sulfate in the development, progression and metastasis of cancer. FEBS J. 2019, 286, 1815–1837. [Google Scholar] [CrossRef] [PubMed]
- Fuster, M.M.; Wang, L.; Castagnola, J.; Sikora, L.; Reddi, K.; Lee, P.H.; Radek, K.A.; Schuksz, M.; Bishop, J.R.; Gallo, R.L.; et al. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J. Cell Biol. 2007, 177, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waaijer, C.J.; de Andrea, C.E.; Hamilton, A.; van Oosterwijk, J.G.; Stringer, S.E.; Bovee, J.V. Cartilage tumour progression is characterized by an increased expression of heparan sulphate 6O-sulphation-modifying enzymes. Virchows Arch. 2012, 461, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Hatabe, S.; Kimura, H.; Arao, T.; Kato, H.; Hayashi, H.; Nagai, T.; Matsumoto, K.; De Velasco, M.; Fujita, Y.; Yamanouchi, G.; et al. Overexpression of heparan sulfate 6-O-sulfotransferase-2 in colorectal cancer. Mol. Clin. Oncol. 2013, 1, 845–850. [Google Scholar] [CrossRef] [Green Version]
- Jeney, A.; Timar, J.; Pogany, G.; Paku, S.; Moczar, E.; Mareel, M.; Otvos, L.; Kopper, L.; Lapis, K. Glycosaminoglycans as novel target in antitumor therapy. Tokai J. Exp. Clin. Med. 1990, 15, 167–177. [Google Scholar]
- Kowitsch, A.; Zhou, G.; Groth, T. Medical application of glycosaminoglycans: A review. J. Tissue Eng. Regen. Med. 2018, 12, e23–e41. [Google Scholar] [CrossRef] [PubMed]
- Prydz, K.; Dalen, K.T. Synthesis and sorting of proteoglycans. J. Cell Sci. 2000, 113 Pt 2, 193–205. [Google Scholar]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Vanky, P.; Tzanakakis, G.N.; Tsegenidis, T.; Hjerpe, A. Ion-pair high-performance liquid chromatography for determining disaccharide composition in heparin and heparan sulphate. J. Chromatogr. A 1997, 765, 169–179. [Google Scholar] [CrossRef]
- Nikitovic, D.; Tsatsakis, A.M.; Karamanos, N.K.; Tzanakakis, G.N. The effects of genistein on the synthesis and distribution of glycosaminoglycans/proteoglycans by two osteosarcoma cell lines depends on tyrosine kinase and the estrogen receptor density. Anticancer Res. 2003, 23, 459–464. [Google Scholar] [PubMed]
- Duchez, S.; Pascal, V.; Cogne, N.; Jayat-Vignoles, C.; Julien, R.; Cogne, M. Glycotranscriptome study reveals an enzymatic switch modulating glycosaminoglycan synthesis during B-cell development and activation. Eur. J. Immunol. 2011, 41, 3632–3644. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Kitagawa, H. Heparin and heparan sulfate biosynthesis. IUBMB Life 2002, 54, 163–175. [Google Scholar] [CrossRef]
- Feyerabend, T.B.; Li, J.P.; Lindahl, U.; Rodewald, H.R. Heparan sulfate C5-epimerase is essential for heparin biosynthesis in mast cells. Nat. Chem. Biol. 2006, 2, 195–196. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, L.S.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular Cell Biology, 4th ed.; Freeman, W.H.: New York, NY, USA, 2000. [Google Scholar]
- Lindahl, U.; Kusche-Gullberg, M.; Kjellen, L. Regulated diversity of heparan sulfate. J. Biol. Chem. 1998, 273, 24979–24982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casu, B.; Lindahl, U. Structure and biological interactions of heparin and heparan sulfate. Adv. Carbohydr. Chem. Biochem. 2001, 57, 159–206. [Google Scholar]
- Powell, A.K.; Yates, E.A.; Fernig, D.G.; Turnbull, J.E. Interactions of heparin/heparan sulfate with proteins: Appraisal of structural factors and experimental approaches. Glycobiology 2004, 14, 17R–30R. [Google Scholar] [CrossRef] [Green Version]
- Nagamine, S.; Tamba, M.; Ishimine, H.; Araki, K.; Shiomi, K.; Okada, T.; Ohto, T.; Kunita, S.; Takahashi, S.; Wismans, R.G.; et al. Organ-specific sulfation patterns of heparan sulfate generated by extracellular sulfatases Sulf1 and Sulf2 in mice. J. Biol. Chem. 2012, 287, 9579–9590. [Google Scholar] [CrossRef] [Green Version]
- Gallangher, J.T. Heparan sulfate: A heparin in miniature. In Handbook Experimental Pharmacology; Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 207, pp. 347–360. [Google Scholar]
- Casu, B.; Naggi, A.; Torri, G. Heparin-derived heparan sulfate mimics to modulate heparan sulfate-protein interaction in inflammation and cancer. Matrix Biol. 2010, 29, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Rudd, T.R.; Preston, M.D.; Yates, E.A. The nature of the conserved basic amino acid sequences found among 437 heparin binding proteins determined by network analysis. Mol. Biosyst. 2017, 13, 852–865. [Google Scholar] [CrossRef] [Green Version]
- Duchesne, L.; Tissot, B.; Rudd, T.R.; Dell, A.; Fernig, D.G. N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding. J. Biol. Chem. 2006, 281, 27178–27189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Multhaupt, H.A.; Couchman, J.R. Heparan sulfate biosynthesis: Methods for investigation of the heparanosome. J. Histochem. Cytochem. 2012, 60, 908–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitovic, D.; Chatzinikolaou, G.; Tsiaoussis, J.; Tsatsakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Insights into targeting colon cancer cell fate at the level of proteoglycans/glycosaminoglycans. Curr. Med. Chem. 2012, 19, 4247–4258. [Google Scholar] [CrossRef]
- Lindahl, U.; Kjellen, L. Pathophysiology of heparan sulphate: Many diseases, few drugs. J. Intern. Med. 2013, 273, 555–571. [Google Scholar] [CrossRef] [Green Version]
- Rabenstein, D.L. Heparin and heparan sulfate: Structure and function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of Heparin and Related Drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Lever, R.; Page, C.P. Mast cell glycosaminoglycans. Glycoconj. J. 2017, 34, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolset, S.O.; Tveit, H. Serglycin--structure and biology. Cell Mol. Life Sci. 2008, 65, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Jacobsson, K.G.; Lindahl, U. Degradation of heparin proteoglycan in cultured mouse mastocytoma cells. Biochem. J. 1987, 246, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Olivera, A.; Beaven, M.A.; Metcalfe, D.D. Mast cells signal their importance in health and disease. J. Allergy Clin. Immunol. 2018, 142, 381–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oie, C.I.; Olsen, R.; Smedsrod, B.; Hansen, J.B. Liver sinusoidal endothelial cells are the principal site for elimination of unfractionated heparin from the circulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G520–G528. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.B.; Liu, J. Multi-faceted substrate specificity of heparanase. Matrix Biol. 2013, 32, 223–227. [Google Scholar] [CrossRef]
- Lauder, R.M. Chondroitin sulphate: A complex molecule with potential impacts on a wide range of biological systems. Complement. Ther. Med. 2009, 17, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Lauder, R.M.; Huckerby, T.N.; Nieduszynski, I.A. A fingerprinting method for chondroitin/dermatan sulfate and hyaluronan oligosaccharides. Glycobiology 2000, 10, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Malavaki, C.; Mizumoto, S.; Karamanos, N.; Sugahara, K. Recent advances in the structural study of functional chondroitin sulfate and dermatan sulfate in health and disease. Connect. Tissue Res. 2008, 49, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Mikami, T. Chondroitin/dermatan sulfate in the central nervous system. Curr. Opin. Struct. Biol. 2007, 17, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Mikami, T.; Yamada, S.; Faissner, A.; Muramatsu, T.; Sugahara, K. Heparin-binding growth factor, pleiotrophin, mediates neuritogenic activity of embryonic pig brain-derived chondroitin sulfate/dermatan sulfate hybrid chains. J. Biol. Chem. 2005, 280, 9180–9191. [Google Scholar] [CrossRef] [Green Version]
- Hopwood, J.J.; Robinson, H.C. The molecular-weight distribution of glycosaminoglycans. Biochem. J. 1973, 135, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Krusius, T.; Finne, J.; Margolis, R.K.; Margolis, R.U. Identification of an O-glycosidic mannose-linked sialylatedtetrasaccharide and keratan sulfate oligosaccharides in the chondroitin sulfate proteoglycan of brain. J. Biol. Chem. 1986, 261, 8237–8242. [Google Scholar] [CrossRef]
- Funderburgh, J.L. Keratan sulfate biosynthesis. IUBMB Life 2002, 54, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Muramatsu, T.; Murase, A.; Yuasa, S.; Uchimura, K.; Kadomatsu, K. N-Acetylglucosamine 6-O-sulfotransferase-1 is required for brain keratan sulfate biosynthesis and glial scar formation after brain injury. Glycobiology 2006, 16, 702–710. [Google Scholar] [CrossRef]
- Uchimura, K. Keratan sulfate: Biosynthesis, structures, and biological functions. Methods Mol. Biol. 2015, 1229, 389–400. [Google Scholar]
- Leiphrakpam, P.D.; Patil, P.P.; Remmers, N.; Swanson, B.; Grandgenett, P.M.; Qiu, F.; Yu, F.; Radhakrishnan, P. Role of keratan sulfate expression in human pancreatic cancer malignancy. Sci. Rep. 2019, 9, 9665. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Hayatsu, N.; Kaneko, M.K.; Ogasawara, S.; Hamano, T.; Takahashi, S.; Nishikawa, R.; Matsutani, M.; Mishima, K.; Narimatsu, H. Increased expression of highly sulfated keratan sulfate synthesized in malignant astrocytic tumors. Biochem. Biophys. Res. Commun. 2008, 369, 1041–1046. [Google Scholar] [CrossRef]
- Itano, N.; Sawai, T.; Yoshida, M.; Lenas, P.; Yamada, Y.; Imagawa, M.; Shinomura, T.; Hamaguchi, M.; Yoshida, Y.; Ohnuki, Y.; et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J. Biol. Chem. 1999, 274, 25085–25092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef]
- Vigetti, D.; Viola, M.; Karousou, E.; De Luca, G.; Passi, A. Metabolic control of hyaluronan synthases. Matrix Biol. 2014, 35, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourguignon, L.Y.; Zhu, H.; Shao, L.; Chen, Y.W. CD44 interaction with tiam1 promotes Rac1 signaling and hyaluronic acid-mediated breast tumor cell migration. J. Biol. Chem. 2000, 275, 1829–1838. [Google Scholar] [CrossRef] [Green Version]
- Kouvidi, K.; Berdiaki, A.; Tzardi, M.; Karousou, E.; Passi, A.; Nikitovic, D.; Tzanakakis, G.N. Receptor for hyaluronic acid- mediated motility (RHAMM) regulates HT1080 fibrosarcoma cell proliferation via a beta-catenin/c-myc signaling axis. Biochim. Biophys. Acta 2016, 1860, 814–824. [Google Scholar] [CrossRef]
- Kavasi, R.M.; Berdiaki, A.; Spyridaki, I.; Papoutsidakis, A.; Corsini, E.; Tsatsakis, A.; Tzanakakis, G.N.; Nikitovic, D. Contact allergen (PPD and DNCB)-induced keratinocyte sensitization is partly mediated through a low molecular weight hyaluronan (LMWHA)/TLR4/NF-kappaB signaling axis. Toxicol. Appl. Pharmacol. 2019, 377, 114632. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Takahashi, M. CD44-dependent intracellular and extracellular catabolism of hyaluronic acid by hyaluronidase-1 and -2. J. Biol. Chem. 2007, 282, 5597–5607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, J.; Li, F. Hyaluronidase and Chondroitinase. Adv. Exp. Med. Biol. 2017, 925, 75–87. [Google Scholar] [PubMed]
- Yamada, S. Role of hyaluronidases in the catabolism of chondroitin sulfate. Adv. Exp. Med. Biol. 2015, 842, 185–197. [Google Scholar]
- Stern, R.; Jedrzejas, M.J. Hyaluronidases: Their genomics, structures, and mechanisms of action. Chem. Rev. 2006, 106, 818–839. [Google Scholar] [CrossRef] [Green Version]
- Puissant, E.; Gilis, F.; Dogne, S.; Flamion, B.; Jadot, M.; Boonen, M. Subcellular trafficking and activity of Hyal-1 and its processed forms in murine macrophages. Traffic 2014, 15, 500–515. [Google Scholar] [CrossRef]
- Tan, J.X.; Wang, X.Y.; Su, X.L.; Li, H.Y.; Shi, Y.; Wang, L.; Ren, G.S. Upregulation of HYAL1 expression in breast cancer promoted tumor cell proliferation, migration, invasion and angiogenesis. PLoS ONE 2011, 6, e22836. [Google Scholar] [CrossRef] [Green Version]
- McAtee, C.O.; Barycki, J.J.; Simpson, M.A. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv. Cancer Res. 2014, 123, 1–34. [Google Scholar]
- Andre, B.; Duterme, C.; Van Moer, K.; Mertens-Strijthagen, J.; Jadot, M.; Flamion, B. Hyal2 is a glycosylphosphatidylinositol-anchored, lipid raft-associated hyaluronidase. Biochem. Biophys. Res. Commun. 2011, 411, 175–179. [Google Scholar] [CrossRef]
- Karthikeyan, R.; Koushik, O. Nano drug delivery systems to overcome cancer drug resistance-a review. J. Nanomed. Nanotechnol. 2016, 7, 2. [Google Scholar]
- Jiao, Y.; Pang, X.; Zhai, G. Advances in Hyaluronic Acid-Based Drug Delivery Systems. Curr. Drug Targets 2016, 17, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Song, X.; Wen, Y.; Zhu, J.L.; Li, J. Injectable Thermoresponsive Hydrogel Formed by Alginate-g-Poly(N-isopropylacrylamide) That Releases Doxorubicin-Encapsulated Micelles as a Smart Drug Delivery System. ACS Appl. Mater. Interfaces 2017, 9, 35673–35682. [Google Scholar] [CrossRef] [PubMed]
- Sedyakina, N.; Kuskov, A.; Velonia, K.; Feldman, N.; Lutsenko, S.; Avramenko, G. Modulation of Entrapment Efficiency and In Vitro Release Properties of BSA-Loaded Chitosan Microparticles Cross-Linked with Citric Acid as a Potential Protein-Drug Delivery System. Materials 2020, 13, 1989. [Google Scholar] [CrossRef] [PubMed]
- Cong, Z.; Shi, Y.; Wang, Y.; Niu, J.; Chen, N.; Xue, H. A novel controlled drug delivery system based on alginate hydrogel/chitosan micelle composites. Int. J. Biol. Macromol. 2018, 107 Pt A, 855–864. [Google Scholar] [CrossRef]
- Kuskov, A.N.; Kulikov, P.P.; Goryachaya, A.V.; Shtilman, M.I.; Tzatzarakis, M.N.; Tsatsakis, A.M.; Velonia, K. Self-assembled amphiphilic poly-N-vinylpyrrolidone nanoparticles as carriers for hydrophobic drugs: Stability aspects. J. Appl. Polym. Sci. 2018, 135, 45637. [Google Scholar] [CrossRef]
- Song, Z.; Lin, Y.; Zhang, X.; Feng, C.; Lu, Y.; Gao, Y.; Dong, C. Cyclic RGD peptide-modified liposomal drug delivery system for targeted oral apatinib administration: Enhanced cellular uptake and improved therapeutic effects. Int. J. Nanomed. 2017, 12, 1941–1958. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Wu, D. Biodegradable dendrimers for drug delivery. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 90, 713–727. [Google Scholar] [CrossRef]
- Mehmood, A.; Ghafar, H.; Yaqoob, S.; Gohar, F.; Ahmad, B. Mesoporous Silica Nanoparticles: A Review. J. Dev. Drugs 2017, 6, 2. [Google Scholar] [CrossRef]
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Tsatsakis, A.; Stratidakis, A.K.; Goryachaya, A.V.; Tzatzarakis, M.N.; Stivaktakis, P.D.; Docea, A.O.; Berdiaki, A.; Nikitovic, D.; Velonia, K.; Shtilman, M.I.; et al. In vitro blood compatibility and in vitro cytotoxicity of amphiphilic poly-N-vinylpyrrolidone nanoparticles. Food Chem. Toxicol. 2019, 127, 42–52. [Google Scholar] [CrossRef]
- Kuskov, A.N.; Kulikov, P.P.; Shtilman, M.I.; Rakitskii, V.N.; Tsatsakis, A.M. Amphiphilic poly-N-vynilpyrrolidone nanoparticles: Cytotoxicity and acute toxicity study. Food Chem. Toxicol. 2016, 96, 273–279. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Lazarovits, J.; Chen, Y.Y.; Song, F.; Ngo, W.; Tavares, A.J.; Zhang, Y.N.; Audet, J.; Tang, B.; Lin, Q.; Tleugabulova, M.C.; et al. Synthesis of Patient-Specific Nanomaterials. Nano Lett. 2019, 19, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Escareno, N.; Topete, A.; Taboada, P.; Daneri-Navarro, A. Rational Surface Engineering of Colloidal Drug Delivery Systems for Biological Applications. Curr. Top. Med. Chem. 2018, 18, 1224–1241. [Google Scholar] [CrossRef] [PubMed]
- Wyss, P.P.; Lamichhane, S.P.; Abed, A.; Vonwil, D.; Kretz, O.; Huber, T.B.; Sarem, M.; Shastri, V.P. Renal clearance of polymeric nanoparticles by mimicry of glycan surface of viruses. Biomaterials 2020, 230, 119643. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yao, W.; Wang, X.; Xie, C.; Zhang, J.; Jiang, X. Bioreducible heparin-based nanogel drug delivery system. Biomaterials 2015, 39, 260–268. [Google Scholar] [CrossRef]
- Rippe, M.; Stefanello, T.F.; Kaplum, V.; Britta, E.A.; Garcia, F.P.; Poirot, R.; Companhoni, M.V.P.; Nakamura, C.V.; Szarpak-Jankowska, A.; Auzely-Velty, R. Heparosan as a potential alternative to hyaluronic acid for the design of biopolymer-based nanovectors for anticancer therapy. Biomater. Sci. 2019, 7, 2850–2860. [Google Scholar] [CrossRef]
- Li, X.M.; Wu, Z.Z.; Zhang, B.; Pan, Y.; Meng, R.; Chen, H.Q. Fabrication of chitosan hydrochloride and carboxymethyl starch complex nanogels as potential delivery vehicles for curcumin. Food Chem. 2019, 293, 197–203. [Google Scholar] [CrossRef]
- Wang, J.; Ma, W.; Guo, Q.; Li, Y.; Hu, Z.; Zhu, Z.; Wang, X.; Zhao, Y.; Chai, X.; Tu, P. The effect of dual-functional hyaluronic acid-vitamin E succinate micelles on targeting delivery of doxorubicin. Int. J. Nanomed. 2016, 11, 5851–5870. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Xu, Y.; An, S.; Zhang, M.; Hu, Y.; Wang, L.; Peng, Q. Near-infrared induced phase-shifted ICG/Fe3O4 loaded PLGA nanoparticles for photothermal tumor ablation. Sci. Rep. 2017, 7, 5490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhan, X.; Xiong, J.; Peng, S.; Huang, W.; Joshi, R.; Cai, Y.; Liu, Y.; Li, R.; Yuan, K.; et al. Temperature-dependent cell death patterns induced by functionalized gold nanoparticle photothermal therapy in melanoma cells. Sci. Rep. 2018, 8, 8720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Choi, H.; Choi, E.S.; Park, M.H.; Ryu, J.H. Hyaluronic Acid-Coated Nanomedicine for Targeted Cancer Therapy. Pharmaceutics 2019, 11, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Sun, J.; Zhang, W.; Zhao, Y.; Zhang, S. Drug delivery systems based on CD44-targeted glycosaminoglycans for cancer therapy. Carbohydr. Polym. 2021, 251, 117103. [Google Scholar] [CrossRef]
- Bishnoi, M.; Jain, A.; Hurkat, P.; Jain, S.K. Chondroitin sulphate: A focus on osteoarthritis. Glycoconj. J. 2016, 33, 693–705. [Google Scholar] [CrossRef]
- Hulsopple, C. Musculoskeletal Therapies: Musculoskeletal Injection Therapy. FP Essent. 2018, 470, 21–26. [Google Scholar] [PubMed]
- Keen, M.A. Hyaluronic Acid in Dermatology. Skinmed 2017, 15, 441–448. [Google Scholar] [PubMed]
- Choi, K.Y.; Saravanakumar, G.; Park, J.H.; Park, K. Hyaluronic acid-based nanocarriers for intracellular targeting: Interfacial interactions with proteins in cancer. Colloids Surf. B Biointerfaces 2012, 99, 82–94. [Google Scholar] [CrossRef] [Green Version]
- Berdiaki, A.; Perisynaki, E.; Stratidakis, A.; Kulikov, P.P.; Kuskov, A.N.; Stivaktakis, P.; Henrich-Noack, P.; Luss, A.L.; Shtilman, M.M.; Tzanakakis, G.N.; et al. Assessment of Amphiphilic Poly-N-vinylpyrrolidone Nanoparticles’ Biocompatibility with Endothelial Cells in Vitro and Delivery of an Anti-Inflammatory Drug. Mol. Pharm. 2020, 17, 4212–4225. [Google Scholar] [CrossRef] [PubMed]
- Hajebi, S.; Rabiee, N.; Bagherzadeh, M.; Ahmadi, S.; Rabiee, M.; Roghani-Mamaqani, H.; Tahriri, M.; Tayebi, L.; Hamblin, M.R. Stimulus-responsive polymeric nanogels as smart drug delivery systems. Acta Biomater. 2019, 92, 1–18. [Google Scholar] [CrossRef]
- Linhardt, R.J.; Claude, S. Hudson Award address in carbohydrate chemistry. Heparin: Structure and activity. J. Med. Chem. 2003, 46, 2551–2564. [Google Scholar] [CrossRef]
- Zhao, F.; Ma, M.L.; Xu, B. Molecular hydrogels of therapeutic agents. Chem. Soc. Rev. 2009, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Nurunnabi, M.; Khatun, Z.; Moon, W.C.; Lee, G.; Lee, Y.K. Heparin based nanoparticles for cancer targeting and noninvasive imaging. Quant. Imaging Med. Surg. 2012, 2, 219–226. [Google Scholar]
- Min, K.A.; Yu, F.; Yang, V.C.; Zhang, X.; Rosania, G.R. Transcellular Transport of Heparin-coated Magnetic Iron Oxide Nanoparticles (Hep-MION) Under the Influence of an Applied Magnetic Field. Pharmaceutics 2010, 2, 119–135. [Google Scholar] [CrossRef]
- Cho, K.J.; Moon, H.T.; Park, G.E.; Jeon, O.C.; Byun, Y.; Lee, Y.K. Preparation of sodium deoxycholate (DOC) conjugated heparin derivatives for inhibition of angiogenesis and cancer cell growth. Bioconjug. Chem. 2008, 19, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- She, W.; Li, N.; Luo, K.; Guo, C.; Wang, G.; Geng, Y.; Gu, Z. Dendronized heparin-doxorubicin conjugate based nanoparticle as pH-responsive drug delivery system for cancer therapy. Biomaterials 2013, 34, 2252–2264. [Google Scholar] [CrossRef]
- Park, K.; Kim, K.; Kwon, I.C.; Kim, S.K.; Lee, S.; Lee, D.Y.; Byun, Y. Preparation and characterization of self-assembled nanoparticles of heparin-deoxycholic acid conjugates. Langmuir 2004, 20, 11726–11731. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, G.Y.; Kim, Y.S.; Yu, M.; Park, R.W.; Kim, I.S.; Kim, S.Y.; Byun, Y. Heparin-deoxycholic acid chemical conjugate as an anticancer drug carrier and its antitumor activity. J. Control. Release 2006, 114, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Baier, G.; Winzen, S.; Messerschmidt, C.; Frank, D.; Fichter, M.; Gehring, S.; Mailander, V.; Landfester, K. Heparin-based nanocapsules as potential drug delivery systems. Macromol. Biosci. 2015, 15, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P. Glucuronyl C5-epimerase an enzyme converting glucuronic acid to iduronic acid in heparan sulfate/heparin biosynthesis. Prog. Mol. Biol. Transl. Sci. 2010, 93, 59–78. [Google Scholar] [PubMed]
- Yang, X.; Du, H.; Liu, J.; Zhai, G. Advanced nanocarriers based on heparin and its derivatives for cancer management. Biomacromolecules 2015, 16, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Ghiselli, G. Heparin Binding Proteins as Therapeutic Target: An Historical Account and Current Trends. Medicines 2019, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, R.J.; Esko, J.D.; Tor, Y. Targeting heparin and heparan sulfate protein interactions. Org. Biomol. Chem. 2017, 15, 5656–5668. [Google Scholar] [CrossRef]
- Ghofrani, M.; Shirmard, L.R.; Dehghankelishadi, P.; Amini, M.; Dorkoosh, F.A. Development of Octreotide-Loaded Chitosan and Heparin Nanoparticles: Evaluation of Surface Modification Effect on Physicochemical Properties and Macrophage Uptake. J. Pharm. Sci. 2019, 108, 3036–3045. [Google Scholar] [CrossRef]
- Duckworth, C.A.; Guimond, S.E.; Sindrewicz, P.; Hughes, A.J.; French, N.S.; Lian, L.Y.; Yates, E.A.; Pritchard, D.M.; Rhodes, J.M.; Turnbull, J.E.; et al. Chemically modified, non-anticoagulant heparin derivatives are potent galectin-3 binding inhibitors and inhibit circulating galectin-3-promoted metastasis. Oncotarget 2015, 6, 23671–23687. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.; Liu, Y.; Zhang, H.; Zhang, Z.; Gao, H.; He, Q. Antitumor and Antimetastasis Activities of Heparin-based Micelle Served As Both Carrier and Drug. ACS Appl. Mater. Interfaces 2016, 8, 9577–9589. [Google Scholar] [CrossRef]
- Wang, J.; Yang, Y.; Zhang, Y.; Huang, M.; Zhou, Z.; Luo, W.; Tang, J.; Wang, J.; Xiao, Q.; Chen, H.; et al. Dual-Targeting Heparin-Based Nanoparticles that Re-Assemble in Blood for Glioma Therapy through Both Anti-Proliferation and Anti-Angiogenesis. Adv. Funct. Mater. 2016, 26, 7873–7885. [Google Scholar] [CrossRef]
- Rodriguez-Torres, M.D.P.; Diaz-Torres, L.A.; Millan-Chiu, B.E.; Garcia-Contreras, R.; Hernandez-Padron, G.; Acosta-Torres, L.S. Antifungal and Cytotoxic Evaluation of Photochemically Synthesized Heparin-Coated Gold and Silver Nanoparticles. Molecules 2020, 25, 2849. [Google Scholar] [CrossRef] [PubMed]
- Groult, H.; Poupard, N.; Herranz, F.; Conforto, E.; Bridiau, N.; Sannier, F.; Bordenave, S.; Piot, J.M.; Ruiz-Cabello, J.; Fruitier-Arnaudin, I.; et al. Family of Bioactive Heparin-Coated Iron Oxide Nanoparticles with Positive Contrast in Magnetic Resonance Imaging for Specific Biomedical Applications. Biomacromolecules 2017, 18, 3156–3167. [Google Scholar] [CrossRef]
- Fazilati, M. Anti-neoplastic applications of heparin coated magnetic nanoparticles against human ovarian cancer. J. Inorg. Organomet. Polym. Mater. 2014, 24, 551–559. [Google Scholar] [CrossRef]
- Zhang, J.; Shin, M.C.; David, A.E.; Zhou, J.; Lee, K.; He, H.; Yang, V.C. Long-circulating heparin-functionalized magnetic nanoparticles for potential application as a protein drug delivery platform. Mol. Pharm. 2013, 10, 3892–3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shin, M.C.; Yang, V.C. Magnetic targeting of novel heparinized iron oxide nanoparticles evaluated in a 9L-glioma mouse model. Pharm. Res. 2014, 31, 579–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Hu, B.; Yuan, X.; Cai, L.; Gao, H.; Yang, Q. Nanogel: A Versatile Nano-Delivery System for Biomedical Applications. Pharmaceutics 2020, 12, 290. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Peng, J.; Han, M.; Omar, M.; Hu, D.; Ke, X.; Lu, N. A low-molecular-weight heparin-coated doxorubicin-liposome for the prevention of melanoma metastasis. J. Drug Target. 2015, 23, 335–346. [Google Scholar] [CrossRef]
- Joung, Y.K.; Jang, J.Y.; Choi, J.H.; Han, D.K.; Park, K.D. Heparin-conjugated pluronic nanogels as multi-drug nanocarriers for combination chemotherapy. Mol. Pharm. 2013, 10, 685–693. [Google Scholar] [CrossRef]
- Kandil, R.; Merkel, O.M. Recent Progress of Polymeric Nanogels for Gene Delivery. Curr. Opin. Colloid Interface Sci. 2019, 39, 11–23. [Google Scholar] [CrossRef]
- Liu, Y.; Lang, T.; Zheng, Z.; Cheng, H.; Huang, X.; Wang, G.; Yin, Q.; Li, Y. In Vivo Environment-Adaptive Nanocomplex with Tumor Cell-Specific Cytotoxicity Enhances T Cells Infiltration and Improves Cancer Therapy. Small 2019, 15, e1902822. [Google Scholar] [CrossRef]
- Tran, T.H.; Bae, B.C.; Lee, Y.K.; Na, K.; Huh, K.M. Heparin-folate-retinoic acid bioconjugates for targeted delivery of hydrophobic photosensitizers. Carbohydr. Polym. 2013, 92, 1615–1624. [Google Scholar] [CrossRef]
- Park, I.K.; Tran, T.H.; Oh, I.H.; Kim, Y.J.; Cho, K.J.; Huh, K.M.; Lee, Y.K. Ternary biomolecular nanoparticles for targeting of cancer cells and anti-angiogenesis. Eur. J. Pharm. Sci. 2010, 41, 148–155. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Linhardt, R.J. Lessons learned from the contamination of heparin. Nat. Prod. Rep. 2009, 26, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Mousa, S.A.; Petersen, L.J. Anti-cancer properties of low-molecular-weight heparin: Preclinical evidence. Thromb. Haemost. 2009, 102, 258–267. [Google Scholar]
- Khan, A.R.; Yang, X.; Du, X.; Yang, H.; Liu, Y.; Khan, A.Q.; Zhai, G. Chondroitin sulfate derived theranostic and therapeutic nanocarriers for tumor-targeted drug delivery. Carbohydr. Polym. 2020, 233, 115837. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pei, X.; Du, Y.; Li, Y. Quaternized chitosan/rectorite intercalative materials for a gene delivery system. Nanotechnology 2008, 19, 375102. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhang, Z.; Jing, P. Black rice anthocyanins embedded in self-assembled chitosan/chondroitin sulfate nanoparticles enhance apoptosis in HCT-116 cells. Food Chem. 2019, 301, 125280. [Google Scholar] [CrossRef] [PubMed]
- Jardim, K.V.; Joanitti, G.A.; Azevedo, R.B.; Parize, A.L. Physico-chemical characterization and cytotoxicity evaluation of curcumin loaded in chitosan/chondroitin sulfate nanoparticles. Mater. Sci. Eng. 2015, 56, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Zu, M.; Ma, L.; Zhang, X.; Xie, D.; Kang, Y.; Xiao, B. Chondroitin sulfate-functionalized polymeric nanoparticles for colon cancer-targeted chemotherapy. Colloids Surf. B Biointerfaces 2019, 177, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, N.; Yan, L.; Gao, F.; Ji, D.; Zhang, S.; Zhang, L.; Li, Y.; Xiao, Y. Preparation, characterisation and in vitro and in vivo evaluation of CD44-targeted chondroitin sulphate-conjugated doxorubicin PLGA nanoparticles. Carbohydr. Polym. 2019, 213, 17–26. [Google Scholar] [CrossRef]
- Nikitovic, D.; Juranek, I.; Wilks, M.F.; Tzardi, M.; Tsatsakis, A.; Tzanakakis, G.N. Anthracycline-dependent cardiotoxicity and extracellular matrix remodeling. Chest 2014, 146, 1123–1130. [Google Scholar] [CrossRef]
- Germanakis, I.; Kalmanti, M.; Parthenakis, F.; Nikitovic, D.; Stiakaki, E.; Patrianakos, A.; Vardas, P.E. Correlation of plasma N-terminal pro-brain natriuretic peptide levels with left ventricle mass in children treated with anthracyclines. Int. J. Cardiol. 2006, 108, 212–215. [Google Scholar] [CrossRef]
- Wang, X.F.; Ren, J.; He, H.Q.; Liang, L.; Xie, X.; Li, Z.X.; Zhao, J.G.; Yu, J.M. Self-assembled nanoparticles of reduction-sensitive poly (lactic-co-glycolic acid)-conjugated chondroitin sulfate A for doxorubicin delivery: Preparation, characterization and evaluation. Pharm. Dev. Technol. 2019, 24, 794–802. [Google Scholar] [CrossRef]
- Liang, S.; Duan, Y.; Zhang, J.; Xing, Z.; Chen, X.; Yang, Y.; Li, Q. Chemically conjugating poly(amidoamine) with chondroitin sulfate to promote CD44-mediated endocytosis for miR-34a delivery. J. Control. Release 2015, 213, e95–e96. [Google Scholar] [CrossRef]
- Miller, T.; Goude, M.C.; McDevitt, T.C.; Temenoff, J.S. Molecular engineering of glycosaminoglycan chemistry for biomolecule delivery. Acta Biomater. 2014, 10, 1705–1719. [Google Scholar] [CrossRef] [Green Version]
- Iwaki, J.; Minamisawa, T.; Tateno, H.; Kominami, J.; Suzuki, K.; Nishi, N.; Nakamura, T.; Hirabayashi, J. Desulfatedgalactosaminoglycans are potential ligands for galectins: Evidence from frontal affinity chromatography. Biochem. Biophys. Res. Commun. 2008, 373, 206–212. [Google Scholar] [CrossRef]
- Yip, G.W.; Smollich, M.; Gotte, M. Therapeutic value of glycosaminoglycans in cancer. Mol. Cancer Ther. 2006, 5, 2139–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belting, M. Glycosaminoglycans in cancer treatment. Thromb. Res. 2014, 133 (Suppl. 2), S95–S101. [Google Scholar] [CrossRef]
- Caterson, B.; Melrose, J. Keratan sulfate, a complex glycosaminoglycan with unique functional capability. Glycobiology 2018, 28, 182–206. [Google Scholar] [CrossRef]
- Lee, J.Y.; Spicer, A.P. Hyaluronan: A multifunctional, megaDalton, stealth molecule. Curr. Opin. Cell Biol. 2000, 12, 581–586. [Google Scholar] [CrossRef]
- Zhong, L.; Liu, Y.; Xu, L.; Li, Q.; Zhao, D.; Li, Z.; Zhang, H.; Kan, Q.; Sun, J.; He, Z. Exploring the relationship of hyaluronic acid molecular weight and active targeting efficiency for designing hyaluronic acid-modified nanoparticles. Asian J. Pharm. Sci. 2019, 14, 521–530. [Google Scholar] [CrossRef]
- Mizrahy, S.; Goldsmith, M.; Leviatan-Ben-Arye, S.; Kisin-Finfer, E.; Redy, O.; Srinivasan, S.; Shabat, D.; Godin, B.; Peer, D. Tumor targeting profiling of hyaluronan-coated lipid based-nanoparticles. Nanoscale 2014, 6, 3742–3752. [Google Scholar] [CrossRef] [PubMed]
- Landesman-Milo, D.; Goldsmith, M.; Leviatan Ben-Arye, S.; Witenberg, B.; Brown, E.; Leibovitch, S.; Azriel, S.; Tabak, S.; Morad, V.; Peer, D. Hyaluronan grafted lipid-based nanoparticles as RNAi carriers for cancer cells. Cancer Lett. 2013, 334, 221–227. [Google Scholar] [CrossRef]
- Yang, C.; Li, C.; Zhang, P.; Wu, W.; Jiang, X. Radox responsive hyaluronic acid nanogels for treating RHAMM (CD168) over-expressive cancer, both primary and metastatic tumors. Theranostics 2017, 7, 1719–1734. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.G.; Kovar, J.L.; Loughman, E.; Elowsky, C.; Oakley, G.G.; Simpson, M.A. Spontaneous metastasis of prostate cancer is promoted by excess hyaluronan synthesis and processing. Am. J. Pathol. 2009, 174, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, M.A. Concurrent expression of hyaluronan biosynthetic and processing enzymes promotes growth and vascularization of prostate tumors in mice. Am. J. Pathol. 2006, 169, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Azpurua, J.; Hine, C.; Vaidya, A.; Myakishev-Rempel, M.; Ablaeva, J.; Mao, Z.; Nevo, E.; Gorbunova, V.; Seluanov, A. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 2013, 499, 346–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, L.; Polyak, A.; Mathe, D.; Kiraly, R.; Thuroczy, J.; Terez, M.; Janoki, G.; Ting, Y.; Bucci, L.R.; Schauss, A.G. Absorption, uptake and tissue affinity of high-molecular-weight hyaluronan after oral administration in rats and dogs. J. Agric. Food Chem. 2008, 56, 10582–10593. [Google Scholar] [CrossRef] [PubMed]
- Laurent, T.C.; Fraser, J.R. Hyaluronan. FASEB J. 1992, 6, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.V.; Rho, J.G.; Um, W.; Ek, P.K.; Nguyen, V.Q.; Oh, B.H.; Kim, W.; Park, J.H. Hyaluronic Acid Nanoparticles as Nanomedicine for Treatment of Inflammatory Diseases. Pharmaceutics 2020, 12, 931. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, K.; Park, T.G. Hyaluronic acid-paclitaxel conjugate micelles: Synthesis, characterization, and antitumor activity. Bioconjug. Chem. 2008, 19, 1319–1325. [Google Scholar] [CrossRef]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Jin, Z.; Han, J.; Cho, S.; Nguyen, V.D.; Ko, S.Y.; Park, J.O.; Park, S. Preparation of HIFU-triggered tumor-targeted hyaluronic acid micelles for controlled drug release and enhanced cellular uptake. Colloids Surf. B Biointerfaces 2016, 143, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Goltsche, K.; Cheng, L.; Xie, F.; Meng, F.; Deng, C.; Zhong, Z.; Haag, R. Hyaluronic acid-shelled acid-activatable paclitaxel prodrug micelles effectively target and treat CD44-overexpressing human breast tumor xenografts in vivo. Biomaterials 2016, 84, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, P.; Banerjee, S.; Padhye, S.; Sarkar, F.H.; Iyer, A.K. Hyaluronic Acid Engineered Nanomicelles Loaded with 3,4-Difluorobenzylidene Curcumin for Targeted Killing of CD44+ Stem-Like Pancreatic Cancer Cells. Biomacromolecules 2015, 16, 3042–3053. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- ComertOnder, F.; SagbasSuner, S.; Sahiner, N.; Ay, M.; Ozpolat, B. Delivery of Small Molecule EF2 Kinase Inhibitor for Breast and Pancreatic Cancer Cells Using Hyaluronic Acid Based Nanogels. Pharm. Res. 2020, 37, 63. [Google Scholar] [CrossRef] [PubMed]
- Ossipov, D.A. Nanostructured hyaluronic acid-based materials for active delivery to cancer. Expert Opin. Drug Deliv. 2010, 7, 681–703. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Moon, M.J.; PoililSurendran, S.; Jeong, Y.Y. Biomedical Applications of Hyaluronic Acid-Based Nanomaterials in Hyperthermic Cancer Therapy. Pharmaceutics 2019, 11, 306. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Y.; Wang, L.; Wang, X.; Tu, P. Comparison of hyaluronic acid-based micelles and polyethylene glycol-based micelles on reversal of multidrug resistance and enhanced anticancer efficacy in vitro and in vivo. Drug Deliv. 2018, 25, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Warren, D.S.; Sutherland, S.P.H.; Kao, J.Y.; Weal, G.R.; Mackay, S.M. The Preparation and Simple Analysis of a Clay Nanoparticle Composite Hydrogel. J. Chem. Educ. 2017, 94, 1772–1779. [Google Scholar] [CrossRef]
- Lee, H.; Mok, H.; Lee, S.; Oh, Y.K.; Park, T.G. Target-specific intracellular delivery of siRNA using degradable hyaluronic acid nanogels. J. Control. Release 2007, 119, 245–252. [Google Scholar] [CrossRef]
- Yanqi, Y.; Jicheng, Y.; Zhen, G. Versatile Protein Nanogels Prepared by In Situ Polymerization. Macromol. Chem. Phys. 2015, 217, 333–343. [Google Scholar]
- Chen, Y.Y.; Wu, H.C.; Sun, J.S.; Dong, G.C.; Wang, T.W. Injectable and thermoresponsive self-assembled nanocomposite hydrogel for long-term anticancer drug delivery. Langmuir 2013, 29, 3721–3729. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, X.; Yao, X.; Zhang, Y.; Wu, W.; Jiang, X. Hyaluronic acid nanogels with enzyme-sensitive cross-linking group for drug delivery. J. Control. Release 2015, 205, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Jiang, Y.; Zhang, J.; Klok, H.A.; Zhong, Z. pH-Sensitive Coiled-Coil Peptide-Cross-Linked Hyaluronic Acid Nanogels: Synthesis and Targeted Intracellular Protein Delivery to CD44 Positive Cancer Cells. Biomacromolecules 2018, 19, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Yeagle, P.L. Cholesterol and the cell membrane. Biochim. Biophys. Acta 1985, 822, 267–287. [Google Scholar] [CrossRef]
- Park, W.; Kim, K.S.; Bae, B.C.; Kim, Y.H.; Na, K. Cancer cell specific targeting of nanogels from acetylated hyaluronic acid with low molecular weight. Eur. J. Pharm. Sci. 2010, 40, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Simon-Yarza, T.; Mielcarek, A.; Couvreur, P.; Serre, C. Nanoparticles of Metal-Organic Frameworks: On the Road to In Vivo Efficacy in Biomedicine. Adv. Mater. 2018, 30, e1707365. [Google Scholar] [CrossRef] [PubMed]
- Shu, F.; DaojunLv, A.; Song, X.-L.; Huang, B.; Wang, C.; Yu, Y.; Zhao, S.-C. Fabrication of a hyaluronic acid conjugated metal organic framework for targeted drug delivery and magnetic resonance imaging. RSC Adv. 2018, 8, 6581–6589. [Google Scholar] [CrossRef] [Green Version]
- Hainfeld, J.F.; Dilmanian, F.; Slatkin, D.N.; Smilowitz, H.M. Radiotherapy enhancement with gold nanoparticles. J. Pharm. Pharmacol. 2008, 60, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manju, S.; Sreenivasan, K. Gold nanoparticles generated and stabilized by water soluble curcumin-polymer conjugate: Blood compatibility evaluation and targeted drug delivery onto cancer cells. J. Colloid Interface Sci. 2012, 368, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.S.; Raja, M.D.; Sundar, D.S.; GoverAntoniraj, M.; Ruckmani, K. Hyaluronic acid co-functionalized gold nanoparticle complex for the targeted delivery of metformin in the treatment of liver cancer (HepG2 cells). Carbohydr. Polym. 2015, 128, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Vyas, D.; Lopez-Hisijos, N.; Gandhi, S.; El-Dakdouki, M.; Basson, M.D.; Walsh, M.F.; Huang, X.; Vyas, A.K.; Chaturvedi, L.S. Doxorubicin-Hyaluronan Conjugated Super-Paramagnetic Iron Oxide Nanoparticles (DOX-HA-SPION) Enhanced Cytoplasmic Uptake of Doxorubicin and Modulated Apoptosis, IL-6 Release and NF-kappaB Activity in Human MDA-MB-231 Breast Cancer Cells. J. Nanosci. Nanotechnol. 2015, 15, 6413–6422. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Zhou, Y.; Liu, Z.; Li, J.; Zhang, D.; Chen, J.; Cai, Z. Cisplatin Loaded Hyaluronic Acid Modified TiO2 Nanoparticles for Neoadjuvant Chemotherapy of Ovarian Cancer. J. Nanomater. 2015, 2015, e390358. [Google Scholar] [CrossRef] [Green Version]
- Liberman, A.; Mendez, N.; Trogler, W.C.; Kummel, A.C. Synthesis and surface functionalization of silica nanoparticles for nanomedicine. Surf. Sci. Rep. 2014, 69, 132–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nairi, V.; Magnolia, S.; Piludu, M.; Nieddu, M.; Caria, C.A.; Sogos, V.; Vallet-Regi, M.; Monduzzi, M.; Salis, A. Mesoporous silica nanoparticles functionalized with hyaluronic acid. Effect of the biopolymer chain length on cell internalization. Colloids Surf. B Biointerfaces 2018, 168, 50–59. [Google Scholar] [CrossRef]
- Trial of FOLF(HA)Iri With Cetuximab in mCRC (Chime). Available online: https://clinicaltrials.gov/ct2/show/NCT02216487 (accessed on 9 December 2020).
- Alamgeer, M.; Neil Watkins, D.; Banakh, I.; Kumar, B.; Gough, D.J.; Markman, B.; Ganju, V. A phase IIa study of HA-irinotecan, formulation of hyaluronic acid and irinotecan targeting CD44 in extensive-stage small cell lung cancer. Investig. New Drugs 2018, 36, 288–298. [Google Scholar] [CrossRef]
- Cai, S.; Zhang, T.; Forrest, W.C.; Yang, Q.; Groer, C.; Mohr, E.; Aires, D.J.; Axiak-Bechtel, S.M.; Flesner, B.K.; Henry, C.J.; et al. Phase I-II clinical trial of hyaluronan-cisplatin nanoconjugate in dogs with naturally occurring malignant tumors. Am. J. Vet. Res. 2016, 77, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.D.; Pantoliano, M.W.; Springer, B.A. Energetic characterization of the basic fibroblast growth factor-heparin interaction: Identification of the heparin binding domain. Biochemistry 1994, 33, 3831–3840. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Bohlke, K.; Khorana, A.A.; Kuderer, N.M.; Lee, A.Y.; Arcelus, J.I.; Balaban, E.P.; Clarke, J.M.; Flowers, C.R.; Francis, C.W.; et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American society of clinical oncology clinical practice guideline update 2014. J. Clin. Oncol. 2015, 33, 654–656. [Google Scholar] [CrossRef]
- Laubli, H.; Varki, A.; Borsig, L. Antimetastatic Properties of Low Molecular Weight Heparin. J. Clin. Oncol. 2016, 34, 2560–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulaney, S.B.; Huang, X. Strategies in synthesis of heparin/heparan sulfate oligosaccharides: 2000-present. Adv. Carbohydr. Chem. Biochem. 2012, 67, 95–136. [Google Scholar] [PubMed] [Green Version]
- Mohamed, S.; Ferro, V. Synthetic Approaches to L-Iduronic Acid and L-Idose: Key Building Blocks for the Preparation of Glycosaminoglycan Oligosaccharides. Adv. Carbohydr. Chem. Biochem. 2015, 72, 21–61. [Google Scholar]
- MacDonald, A.; Priess, M.; Curran, J.; Guess, J.; Farutin, V.; Oosterom, I.; Chu, C.L.; Cochran, E.; Zhang, L.; Getchell, K.; et al. Multitargeting Heparan Sulfate Mimetic, Targets Tumor and Stromal Compartments in Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Gacche, R.N.; Meshram, R.J. Targeting tumor micro-environment for design and development of novel anti-angiogenic agents arresting tumor growth. Prog. Biophys Mol. Biol. 2013, 113, 333–354. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Presta, M. Fibroblast growth factors/fibroblast growth factor receptors as targets for the development of anti-angiogenesis strategies. Curr. Pharm. Des. 2007, 13, 2025–2044. [Google Scholar] [CrossRef]
- Kessler, T.; Fehrmann, F.; Bieker, R.; Berdel, W.E.; Mesters, R.M. Vascular endothelial growth factor and its receptor as drug targets in hematological malignancies. Curr. Drug Targets 2007, 8, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Ferro, V.; Fewings, K.; Palermo, M.C.; Li, C. Large-scale preparation of the oligosaccharide phosphate fraction of Pichia holstii NRRL Y-2448 phosphomannan for use in the manufacture of PI-88. Carbohydr. Res. 2001, 332, 183–189. [Google Scholar] [CrossRef]
- Ferro, V.; Li, C.; Fewings, K.; Palermo, M.C.; Linhardt, R.J.; Toida, T. Determination of the composition of the oligosaccharide phosphate fraction of Pichia (Hansenula) holstii NRRL Y-2448 phosphomannan by capillary electrophoresis and HPLC. Carbohydr. Res. 2002, 337, 139–146. [Google Scholar] [CrossRef]
- Ono, K.; Ishihara, M.; Ishikawa, K.; Ozeki, Y.; Deguchi, H.; Sato, M.; Hashimoto, H.; Saito, Y.; Yura, H.; Kurita, A.; et al. Periodate-treated, non-anticoagulant heparin-carrying polystyrene (NAC-HCPS) affects angiogenesis and inhibits subcutaneous induced tumour growth and metastasis to the lung. Br. J. Cancer 2002, 86, 1803–1812. [Google Scholar] [CrossRef] [Green Version]
- Cassinelli, G.; Favini, E.; Dal Bo, L.; Tortoreto, M.; De Maglie, M.; Dagrada, G.; Pilotti, S.; Zunino, F.; Zaffaroni, N.; Lanzi, C. Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget 2016, 7, 47848–47863. [Google Scholar] [CrossRef]
- Chalkiadaki, G.; Nikitovic, D.; Berdiaki, A.; Katonis, P.; Karamanos, N.K.; Tzanakakis, G.N. Heparin plays a key regulatory role via a p53/FAK-dependent signaling in melanoma cell adhesion and migration. IUBMB Life 2011, 63, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Li, J.-P. Heparanase–Discovery and Targets. In Heparanase: From Basic Research to Clinical Applications; Vlodavsky, I., Sanderson, R.D., Ilan, N., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 61–69. [Google Scholar]
- Mohan, C.D.; Hari, S.; Preetham, H.D.; Rangappa, S.; Barash, U.; Ilan, N.; Nayak, S.C.; Gupta, V.K.; Basappa; Vlodavsky, I.; et al. Targeting Heparanase in Cancer: Inhibition by Synthetic, Chemically Modified, and Natural Compounds. iScience 2019, 15, 360–390. [Google Scholar] [CrossRef] [Green Version]
- Hammond, E.; Khurana, A.; Shridhar, V.; Dredge, K. The Role of Heparanase and Sulfatases in the Modification of Heparan Sulfate Proteoglycans within the Tumor Microenvironment and Opportunities for Novel Cancer Therapeutics. Front. Oncol. 2014, 4, 195. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Gross-Cohen, M.; Weissmann, M.; Ilan, N.; Sanderson, R.D. Opposing Functions of Heparanase-1 and Heparanase-2 in Cancer Progression. Trends Biochem. Sci. 2018, 43, 18–31. [Google Scholar] [CrossRef]
- Gutter-Kapon, L.; Alishekevitz, D.; Shaked, Y.; Li, J.P.; Aronheim, A.; Ilan, N.; Vlodavsky, I. Heparanase is required for activation and function of macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, E7808–E7817. [Google Scholar] [CrossRef] [Green Version]
- Edovitsky, E.; Elkin, M.; Zcharia, E.; Peretz, T.; Vlodavsky, I. Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. J. Natl. Cancer Inst. 2004, 96, 1219–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updates 2016, 29, 54–75. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, G.; Nian, J.; Yu, M.; Chen, S.; Zhang, Y.; Yang, G.; Yang, L.; Cheng, P.; Yan, C.; et al. Elevated heparanase expression is associated with poor prognosis in breast cancer: A study based on systematic review and TCGA data. Oncotarget 2017, 8, 43521–43535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purushothaman, A.; Sanderson, R.D. Heparanase: A Dynamic Promoter of Myeloma Progression. Adv. Exp. Med. Biol. 2020, 1221, 331–349. [Google Scholar] [PubMed]
- Gohji, K.; Okamoto, M.; Kitazawa, S.; Toyoshima, M.; Dong, J.; Katsuoka, Y.; Nakajima, M. Heparanase protein and gene expression in bladder cancer. J. Urol. 2001, 166, 1286–1290. [Google Scholar] [CrossRef]
- Purushothaman, A.; Uyama, T.; Kobayashi, F.; Yamada, S.; Sugahara, K.; Rapraeger, A.C.; Sanderson, R.D. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood 2010, 115, 2449–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumi, Y.; Miyake, M.; Shimada, K.; Fujii, T.; Hori, S.; Morizawa, Y.; Nakai, Y.; Anai, S.; Tanaka, N.; Konishi, N.; et al. Inhibition of Heparanase Expression Results in Suppression of Invasion, Migration and Adhesion Abilities of Bladder Cancer Cells. Int. J. Mol. Sci. 2020, 21, 3789. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.; Gotte, M. Involvement of Syndecan-1 and Heparanase in Cancer and Inflammation. Adv. Exp. Med. Biol. 2020, 1221, 97–135. [Google Scholar]
- Ramani, V.C.; Vlodavsky, I.; Ng, M.; Zhang, Y.; Barbieri, P.; Noseda, A.; Sanderson, R.D. Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biol. 2016, 55, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.A.; Purushothaman, A.; Ramani, V.C.; Vlodavsky, I.; Sanderson, R.D. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J. Biol. Chem. 2013, 288, 10093–10099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandari, S.K.; Purushothaman, A.; Ramani, V.C.; Brinkley, G.J.; Chandrashekar, D.S.; Varambally, S.; Mobley, J.A.; Zhang, Y.; Brown, E.E.; Vlodavsky, I.; et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol. 2018, 65, 104–118. [Google Scholar] [CrossRef]
- Ritchie, J.P.; Ramani, V.C.; Ren, Y.; Naggi, A.; Torri, G.; Casu, B.; Penco, S.; Pisano, C.; Carminati, P.; Tortoreto, M.; et al. SST 0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin. Cancer Res. 2011, 17, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Pala, D.; Rivara, S.; Mor, M.; Milazzo, F.M.; Roscilli, G.; Pavoni, E.; Giannini, G. Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology 2016, 26, 640–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, M.; Chatterjee, M.; Grasso, M.; Specchia, G.; Magen, H.; Einsele, H.; Celeghini, I.; Barbieri, P.; Paoletti, D.; Pace, S.; et al. Phase I study of the heparanase inhibitor roneparstat: An innovative approach for ultiple myeloma therapy. Haematologica 2018, 103, e469–e472. [Google Scholar] [CrossRef]
- Ferro, V.; Dredge, K.; Liu, L.; Hammond, E.; Bytheway, I.; Li, C.; Johnstone, K.; Karoli, T.; Davis, K.; Copeman, E.; et al. PI-88 and novel heparan sulfate mimetics inhibit angiogenesis. Semin. Thromb. Hemost. 2007, 33, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Dredge, K.; Hammond, E.; Davis, K.; Li, C.P.; Liu, L.; Johnstone, K.; Handley, P.; Wimmer, N.; Gonda, T.J.; Gautam, A.; et al. The PG500 series: Novel heparan sulfate mimetics as potent angiogenesis and heparanase inhibitors for cancer therapy. Investig. New Drugs 2010, 28, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.D.; Robinson, W.A.; Millward, M.J.; Powell, A.; Price, T.J.; Thomson, D.B.; Walpole, E.T.; Haydon, A.M.; Creese, B.R.; Roberts, K.L.; et al. A phase II study of the heparanase inhibitor PI-88 in patients with advanced melanoma. Investig. New Drugs 2008, 26, 89–94. [Google Scholar] [CrossRef]
- Chhabra, M.; Ferro, V. PI-88 and Related Heparan Sulfate Mimetics. Adv. Exp. Med. Biol. 2020, 1221, 473–491. [Google Scholar] [PubMed]
- Ferro, V.; Liu, L.; Johnstone, K.D.; Wimmer, N.; Karoli, T.; Handley, P.; Rowley, J.; Dredge, K.; Li, C.P.; Hammond, E.; et al. Discovery of PG545: A highly potent and simultaneous inhibitor of angiogenesis, tumor growth, and metastasis. J. Med. Chem. 2012, 55, 3804–3813. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, M.; Bhattacharya, U.; Feld, S.; Hammond, E.; Ilan, N.; Vlodavsky, I. The heparanase inhibitor PG545 is a potent anti-lymphoma drug: Mode of action. Matrix Biol. 2019, 77, 58–72. [Google Scholar] [CrossRef]
- Barash, U.; Cohen-Kaplan, V.; Arvatz, G.; Gingis-Velitski, S.; Levy-Adam, F.; Nativ, O.; Shemesh, R.; Ayalon-Sofer, M.; Ilan, N.; Vlodavsky, I. A novel human heparanase splice variant, T5, endowed with protumorigenic characteristics. FASEB J. 2010, 24, 1239–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neagu, M.; Constantin, C.; Caruntu, C.; Dumitru, C.; Surcel, M.; Zurac, S. Inflammation: A key process in skin tumorigenesis. Oncol. Lett. 2019, 17, 4068–4084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neagu, M.; Constantin, C.; Longo, C. Chemokines in the melanoma metastasis biomarkers portrait. J. Immunoass. Immunochem. 2015, 36, 559–566. [Google Scholar] [CrossRef]
- Khaldoyanidi, S.K.; Goncharova, V.; Mueller, B.; Schraufstatter, I.U. Hyaluronan in the healthy and malignant hematopoietic microenvironment. Adv. Cancer Res. 2014, 123, 149–189. [Google Scholar]
- Neagu, M.; Constantin, C.; Popescu, I.D.; Zipeto, D.; Tzanakakis, G.; Nikitovic, D.; Fenga, C.; Stratakis, C.A.; Spandidos, D.A.; Tsatsakis, A.M. Inflammation and Metabolism in Cancer Cell-Mitochondria Key Player. Front. Oncol. 2019, 9, 348. [Google Scholar] [CrossRef] [Green Version]
- Rios de la Rosa, J.M.; Tirella, A.; Gennari, A.; Stratford, I.J.; Tirelli, N. The CD44-Mediated Uptake of Hyaluronic Acid-Based Carriers in Macrophages. Adv. Heal. Mater. 2017, 6, 4. [Google Scholar] [CrossRef]
- Bulman, A.; Neagu, M.; Constantin, C. Immunomics in Skin Cancer-Improvement in Diagnosis, Prognosis and Therapy Monitoring. Curr. Proteom. 2013, 10, 202–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.C.; Chen, S.H.; Chiang, W.H.; Huang, C.W.; Lo, C.L.; Chern, C.S.; Chiu, H.C. Tumor Microenvironment-Responsive Nanoparticle Delivery of Chemotherapy for Enhanced Selective Cellular Uptake and Transportation within Tumor. Biomacromolecules 2016, 17, 3883–3892. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Zhou, X.; Liu, M.; Hou, X.; Cheng, Z.; Chen, D. Development of dual-targeted nano-dandelion based on an oligomeric hyaluronic acid polymer targeting tumor-associated macrophages for combination therapy of non-small cell lung cancer. Drug Deliv. 2019, 26, 1265–1279. [Google Scholar] [CrossRef] [Green Version]
- Parayath, N.N.; Gandham, S.K.; Leslie, F.; Amiji, M.M. Improved anti-tumor efficacy of paclitaxel in combination with MicroRNA-125b-based tumor-associated macrophage repolarization in epithelial ovarian cancer. Cancer Lett. 2019, 461, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, X.; Ren, Y.; Cao, F.; Hou, L.; Zhang, Z. An in situ microenvironmental nano-regulator to inhibit the proliferation and metastasis of 4T1 tumor. Theranostics 2019, 9, 3580–3594. [Google Scholar] [CrossRef] [PubMed]
- Alaniz, L.; Rizzo, M.; Garcia, M.G.; Piccioni, F.; Aquino, J.B.; Malvicini, M.; Atorrasagasti, C.; Bayo, J.; Echeverria, I.; Sarobe, P.; et al. Low molecular weight hyaluronan preconditioning of tumor-pulsed dendritic cells increases their migratory ability and induces immunity against murine colorectal carcinoma. Cancer Immunol. Immunother. 2011, 60, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Liu, T.; Shi, C.; Zhang, X.; Chen, X. Bioconjugated Manganese Dioxide Nanoparticles Enhance Chemotherapy Response by Priming Tumor-Associated Macrophages toward M1-like Phenotype and Attenuating Tumor Hypoxia. ACS Nano 2016, 10, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Yoon, H.Y.; Shin, J.M.; Saravanakumar, G.; Noh, K.H.; Song, K.H.; Jeon, J.H.; Kim, D.W.; Lee, K.M.; Kim, K.; et al. A polymeric conjugate foreignizing tumor cells for targeted immunotherapy in vivo. J. Control. Release 2015, 199, 98–105. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Huang, L.; Hou, X.; Zhong, C.; Bachir, Z.A.; Lan, M.; Chen, R.; Gao, F. Efficient ovalbumin delivery using a novel multifunctional micellar platform for targeted melanoma immunotherapy. Int. J. Pharm. 2019, 560, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.M.; Oh, S.J.; Kwon, S.; Deepagan, V.G.; Lee, M.; Song, S.H.; Lee, H.J.; Kim, S.; Song, K.H.; Kim, T.W.; et al. A PEGylated hyaluronic acid conjugate for targeted cancer immunotherapy. J. Control. Release 2017, 267, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Yan, M.; Liu, Y.; Liu, L.; Ma, G. Photothermally Controlled MHC Class I Restricted CD8(+) T-Cell Responses Elicited by Hyaluronic Acid Decorated Gold Nanoparticles as a Vaccine for Cancer Immunotherapy. Adv. Healthc. Mater. 2018, 7, e1701439. [Google Scholar] [CrossRef]
- Watanabe, K.; Tsuchiya, Y.; Kawaguchi, Y.; Sawada, S.; Ayame, H.; Akiyoshi, K.; Tsubata, T. The use of cationic nanogels to deliver proteins to myeloma cells and primary T lymphocytes that poorly express heparan sulfate. Biomaterials 2011, 32, 5900–5905. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, S.R.; Tampa, M.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Caruntu, A.; Lupu, M.; Matei, C.; Constantin, C.; Neagu, M. Tumour Microenvironment in Skin Carcinogenesis. Adv. Exp. Med. Biol. 2020, 1226, 123–142. [Google Scholar] [PubMed]
- Rossi, G.R.; Trindade, E.S.; Souza-Fonseca-Guimaraes, F. Tumor Microenvironment-Associated Extracellular Matrix Components Regulate NK Cell Function. Front. Immunol. 2020, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Melstrom, L.G.; Salazar, M.D.; Diamond, D.J. The pancreatic cancer microenvironment: A true double agent. J. Surg. Oncol. 2017, 116, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Ancuceanu, R.; Neagu, M. Immune based therapy for melanoma. Indian J. Med. Res. 2016, 143, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Li, X.; Thompson, C.B.; Huang, Z.; Araiza, F.; Osgood, R.; Wei, G.; Feldmann, M.; Frost, G.I.; Shepard, H.M. Effective targeting of the tumor microenvironment for cancer therapy. Anticancer Res. 2012, 32, 1203–1212. [Google Scholar]
- Borad, M.J.; Ramanathan, R.K.; Bessudo, A.; LoRusso, P.; Shepard, H.M.; Maneval, D.C.; Jiang, P.; Zhu, J.; Frost, G.I.; Infante, J.R. Targeting hyaluronan (HA) in tumor stroma: A phase I study to evaluate the safety, pharmacokinetics (PK), andpharmacodynamics (PD) of pegylated hyaluronidase (PEGPH20) in patients with solid tumors. J. Clin. Oncol. 2012, 30, 2579. [Google Scholar] [CrossRef]
- Wong, K.M.; Horton, K.J.; Coveler, A.L.; Hingorani, S.R.; Harris, W.P. Targeting the Tumor Stroma: The Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20). Curr. Oncol. Rep. 2017, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Bollyky, P.L.; Wu, R.P.; Falk, B.A.; Lord, J.D.; Long, S.A.; Preisinger, A.; Teng, B.; Holt, G.E.; Standifer, N.E.; Braun, K.R.; et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc. Natl. Acad. Sci. USA 2011, 108, 7938–7943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clift, R.; Souratha, J.; Garrovillo, S.A.; Zimmerman, S.; Blouw, B. Remodeling the Tumor Microenvironment Sensitizes Breast Tumors to Anti-Programmed Death-Ligand 1 Immunotherapy. Cancer Res. 2019, 79, 4149–4159. [Google Scholar] [CrossRef] [Green Version]
- Hermano, E.; Lerner, I.; Elkin, M. Heparanase enzyme in chronic inflammatory bowel disease and colon cancer. Cell. Mol. Life Sci. 2012, 69, 2501–2513. [Google Scholar] [CrossRef]
- Mayfosh, A.J.; Baschuk, N.; Hulett, M.D. Leukocyte Heparanase: A Double-Edged Sword in Tumor Progression. Front. Oncol. 2019, 9, 331. [Google Scholar] [CrossRef]
- Weissmann, M.; Arvatz, G.; Horowitz, N.; Feld, S.; Naroditsky, I.; Zhang, Y.; Ng, M.; Hammond, E.; Nevo, E.; Vlodavsky, I.; et al. Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 704–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef]
- Singha, N.C.; Nekoroski, T.; Zhao, C.; Symons, R.; Jiang, P.; Frost, G.I.; Huang, Z.; Shepard, H.M. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol. Cancer Ther. 2015, 14, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Very, N.; Lefebvre, T.; El Yazidi-Belkoura, I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget 2018, 9, 1380–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.A.; Mason, R.W.; Kobayashi, H.; Yamaguchi, S.; Tomatsu, S. Advances in glycosaminoglycan detection. Mol. Genet. Metab. 2020, 130, 101–109. [Google Scholar] [CrossRef]
- Pshezhetsky, A.V. Crosstalk between 2 organelles: Lysosomal storage of heparan sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type C. Rare Dis. 2015, 3, e1049793. [Google Scholar] [CrossRef] [Green Version]
- Ponsiglione, A.M.; Russo, M.; Torino, E. Glycosaminoglycans and Contrast Agents: The Role of Hyaluronic Acid as MRI Contrast Enhancer. Biomolecules 2020, 10, 1612. [Google Scholar] [CrossRef]
- Guzzo, T.; Barile, F.; Marras, C.; Bellini, D.; Mandaliti, W.; Nepravishta, R.; Paci, M.; Topai, A. Stability Evaluation and Degradation Studies of DAC((R)) Hyaluronic-Polylactide Based Hydrogel by DOSY NMR Spectroscopy. Biomolecules 2020, 10, 1478. [Google Scholar] [CrossRef]
- Whitmore, E.K.; Vesenka, G.; Sihler, H.; Guvench, O. Efficient Construction of Atomic-Resolution Models of Non-Sulfated Chondroitin Glycosaminoglycan Using Molecular Dynamics Data. Biomolecules 2020, 10, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadi, J.; Dobra, K.; Hjerpe, A. Multiplex Soluble Biomarker Analysis from Pleural Effusion. Biomolecules 2020, 10, 1113. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Kouvidi, K.; Kavasi, R.M.; Berdiaki, A.; Tzanakakis, G.N. Hyaluronan/Hyaladherins-a Promising Axis for Targeted Drug Delivery in Cancer. Curr. Drug Deliv. 2016, 13, 500–511. [Google Scholar] [CrossRef] [PubMed]


| HA-Based NP Types | Composition | Drug/Conjugate | Human Cancer Type | Reference |
|---|---|---|---|---|
| Micelles | HA-b-dendritic oligoglycerol | paclitaxel | breast | [174] |
| HA-copoly(styrene maleic acid) | 3,4-difluorobenzylidene curcumin | pancreatic | [175] | |
| Nanogels | Coiled-coil-peptide-cross-linked-HA | GY(EIAALEK)3GC (E3) and GY(KIAALKE)3GC (K3) | breast | [186] |
| Acetylated HA with low molecular weight 1,2,3-with degrees of acetylation 0.8, 2.1, 2.6 acetyl groups per unit (2 glucose rings) | Doxorubicin | cervical | [188] | |
| Inorganic | HA super-paramagnetic iron oxide | Doxorubicin | breast | [194] |
| HA-titanium dioxide | Cisplatin | ovarian | [195] |
| Therapy Target | Drug | Cancer Type | Stage | Reference |
|---|---|---|---|---|
| Antagonists of angiogenic growth factors | necuparanib | Pancreatic cancer | 3D model, animal tumor model, Phase I/II clinical trial in combination with standard therapy | [206] |
| PI-88 (muparfosfat) | General tumor angiogenesis | In vitro, animal models | [210,211] | |
| NAC-HCPS | Lung tumor | Animal model | [212] | |
| Hep SST0001 (roneparstat) | Sarcoma | Animal models Section 2 | [213] | |
| Heparanase Inhibitors Section 3 | SST0001 (roneparstat) | Multiple myeloma Section 4 | Animal model, Clinical trial Section 5 | [232,233] |
| PI-88 (muparfosfat) | Hepatocellular Carcinoma. melanoma | Clinical trial | [235,236] | |
| PI-88 analogs (PC545-pixatimod) | Human lymphoma | Animal model, Clinical trial | [237,238] |
| Target | Therapy | Cancer Type | Stage | Reference |
|---|---|---|---|---|
| Hyaluronan | PEGylated recombinant hyaluronidase Section 6 | Solid tumors | phase I study | [263] |
| Non-small lung cancer | Animal model | [262] | ||
| Refractory locally advanced or metastatic gastric adenocarcinoma and Non-small cell lung carcinoma | A phase 1b trial of PEGPH20 with pembrolizumab (NCT02563548) | [264] | ||
| Heparanase | Heparanase inhibitors | Colon carcinoma | Animal model | [267,271] |
| Human lymphoma | In vitro cellular model | [239] | ||
| Heparanase neutralizing antibody | Human follicular and diffused non-Hodgkin’s B-lymphomas | Animal model | [269] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berdiaki, A.; Neagu, M.; Giatagana, E.-M.; Kuskov, A.; Tsatsakis, A.M.; Tzanakakis, G.N.; Nikitovic, D. Glycosaminoglycans: Carriers and Targets for Tailored Anti-Cancer Therapy. Biomolecules 2021, 11, 395. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030395
Berdiaki A, Neagu M, Giatagana E-M, Kuskov A, Tsatsakis AM, Tzanakakis GN, Nikitovic D. Glycosaminoglycans: Carriers and Targets for Tailored Anti-Cancer Therapy. Biomolecules. 2021; 11(3):395. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030395
Chicago/Turabian StyleBerdiaki, Aikaterini, Monica Neagu, Eirini-Maria Giatagana, Andrey Kuskov, Aristidis M. Tsatsakis, George N. Tzanakakis, and Dragana Nikitovic. 2021. "Glycosaminoglycans: Carriers and Targets for Tailored Anti-Cancer Therapy" Biomolecules 11, no. 3: 395. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030395