
Inhibition of the Receptor for Advanced Glycation End Products Enhances the Cytotoxic Effect of Gemcitabine in Murine Pancreatic Tumors

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture Conditions
2.2. RAGE Quantitative Enzyme-Linked Immunosorbent Assay (ELISA)
2.3. Cell Viability Assays
2.4. Expression and Purification of IgG 2A11
2.5. Generation of the Orthotopic Mouse Model of Pancreatic Cancer
2.6. Mouse Treatment
2.7. Immunohistochemistry
2.8. Western Blots
2.9. Statistical Analysis
3. Results
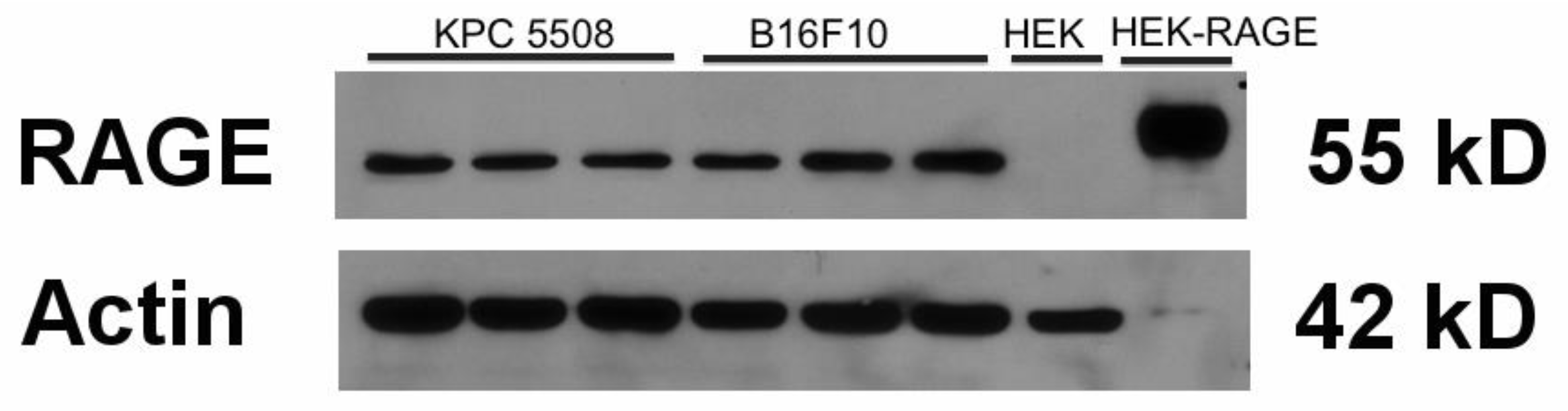
3.1. KPC #5508 Cells Express RAGE
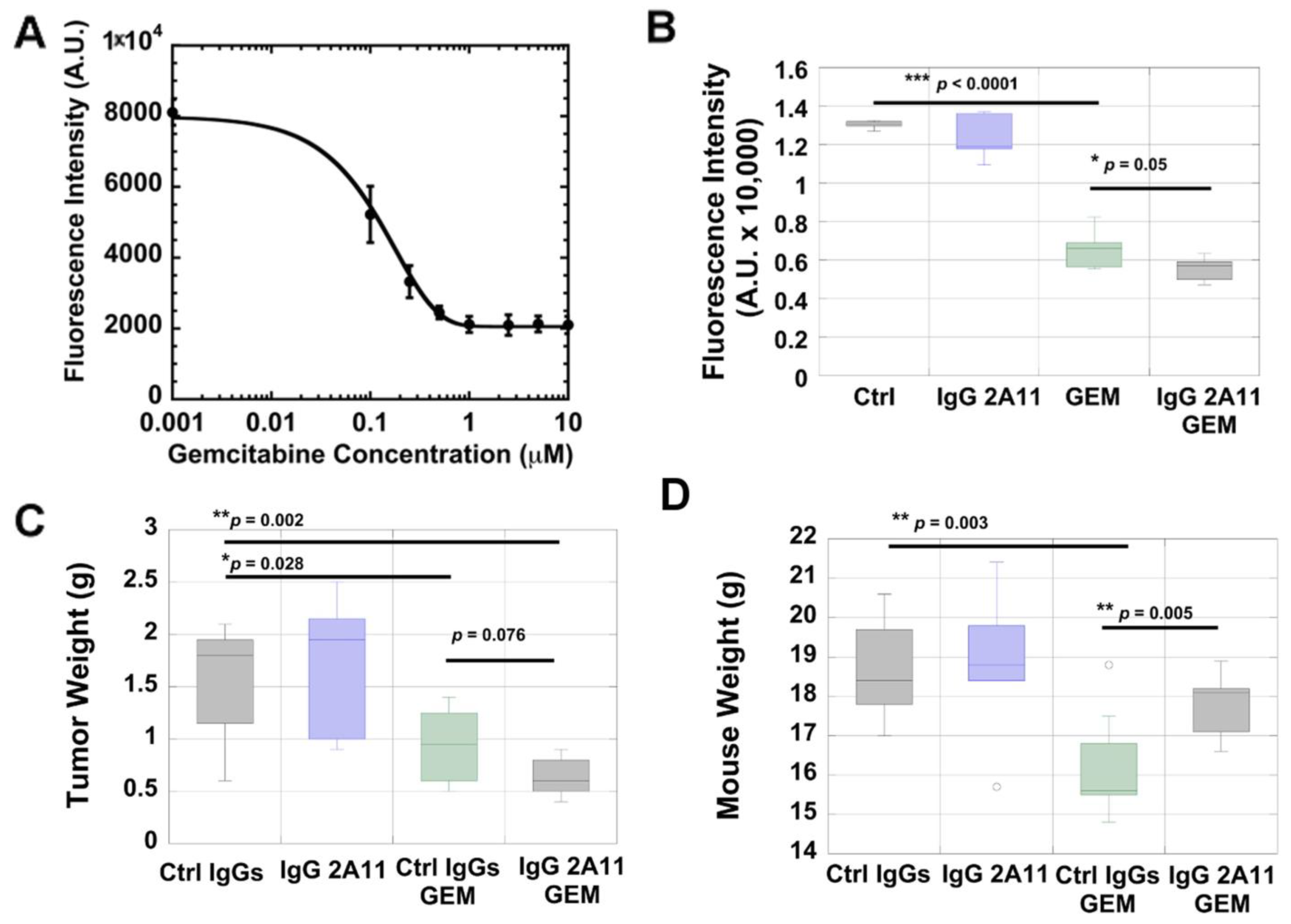
3.2. KPC #5508 Cells Are Sensitive to Gemcitabine In Vitro
3.3. RAGE Inhibition Enhances the Cytotoxic Effect of Gemcitabine In Vitro
3.4. RAGE Inhibition Enhances the Cytotoxic Effect of Gemcitabine in a Preclinical Mouse Model of Pancreatic Cancer
3.5. IgG 2A11 Combined with Gemcitabine Results in Reduced Body Weight Loss Compared to Treatment with Gemcitabine and Control IgGs
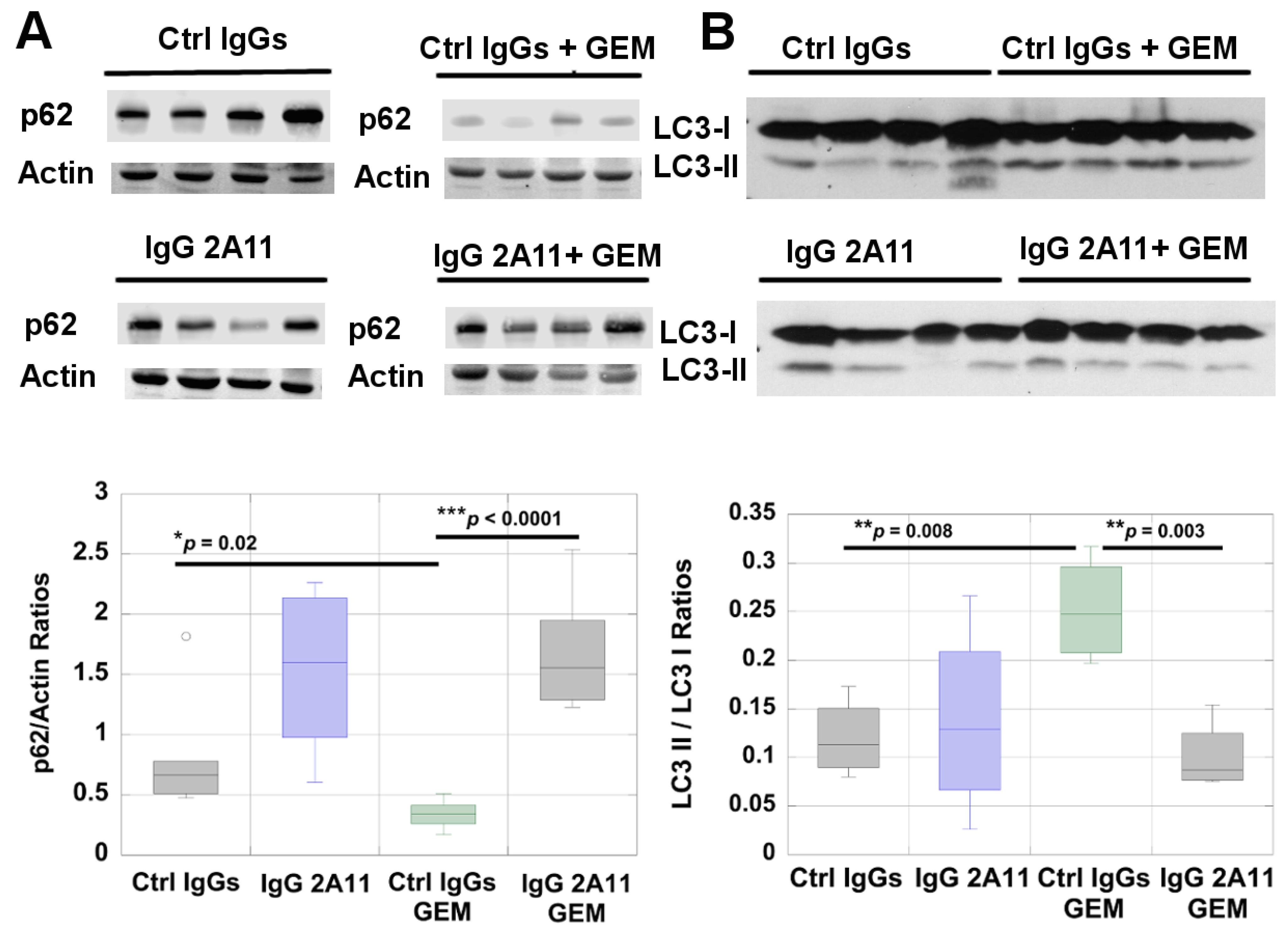
3.6. RAGE Inhibition Reduces Autophagy in Gemcitabine-Treated Tumors
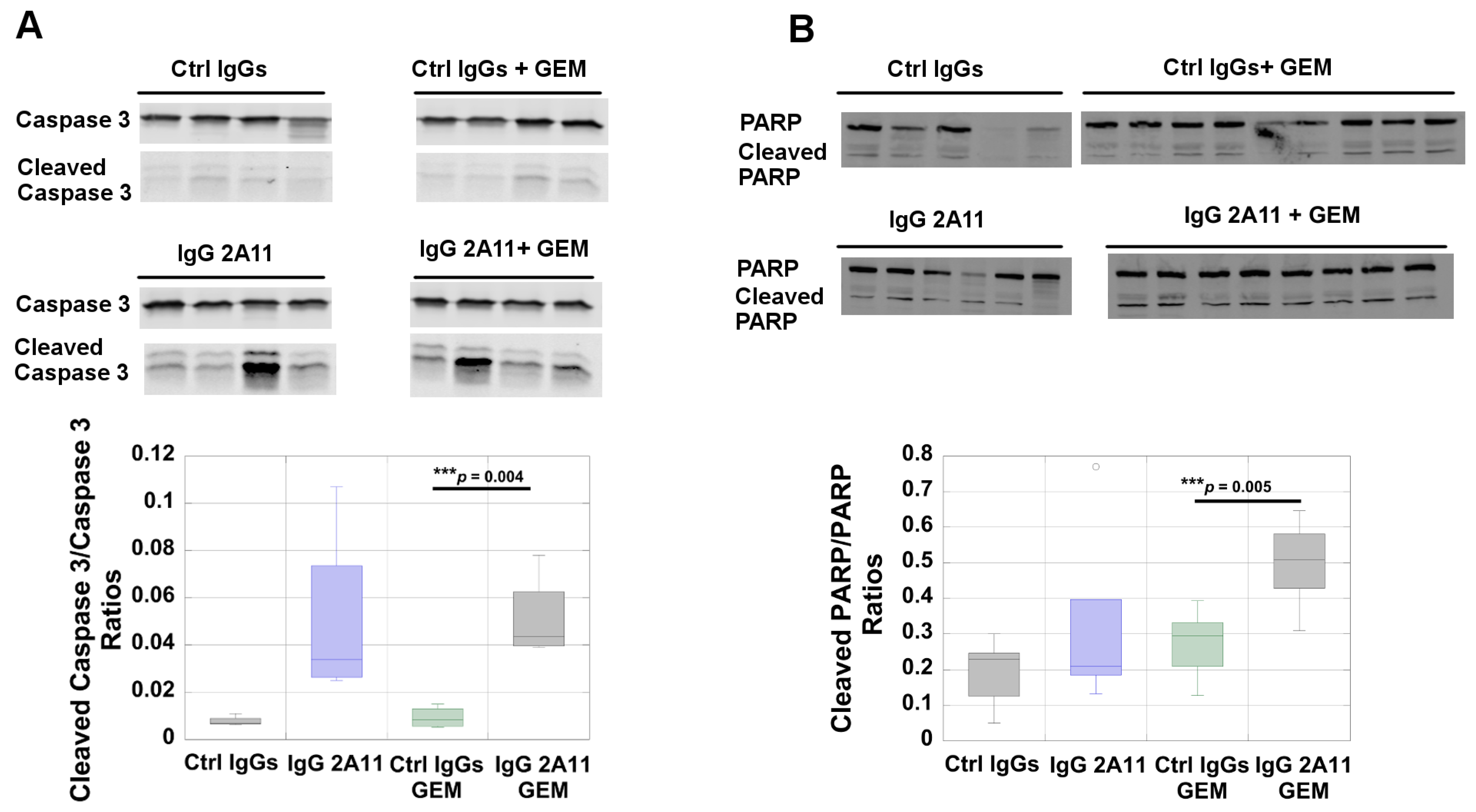
3.7. RAGE Inhibition Increases Poly(ADP-Ribose)Polymerase (PARP) Activation in Tumors Treated with Gemcitabine
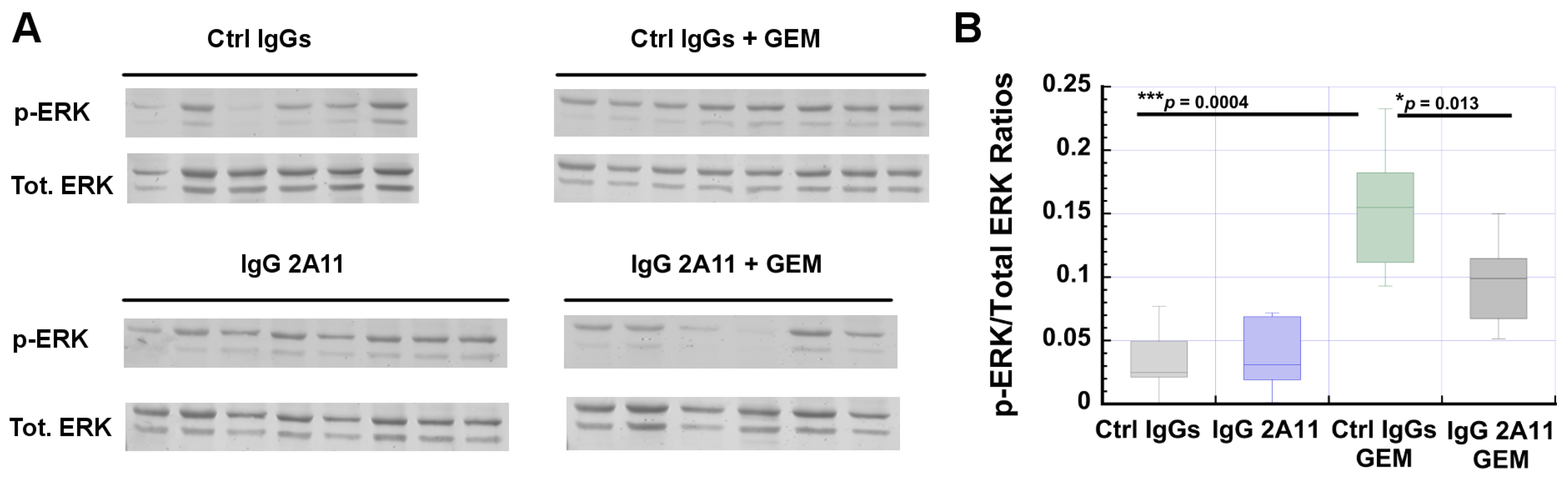
3.8. RAGE Inhibition Reverses Gemcitabine-Dependent Increases in ERK1/2 Activation
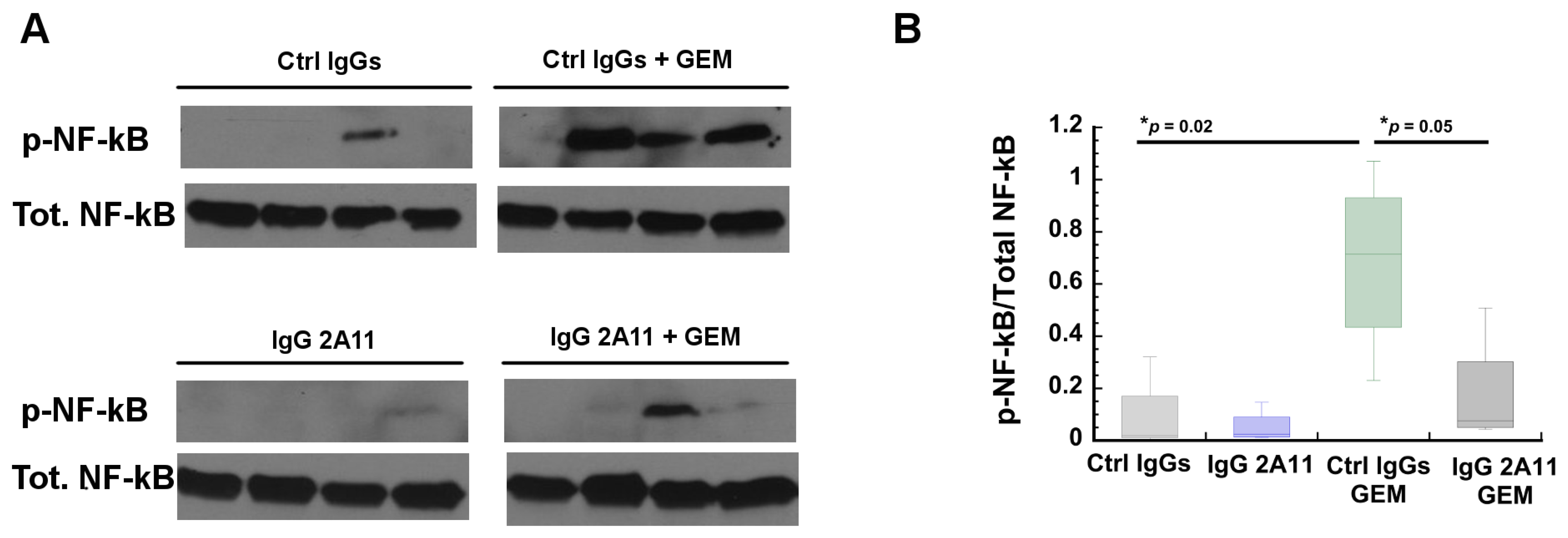
3.9. RAGE Inhibition Reverses Gemcitabine Mediated Increases in NF-κB Activation
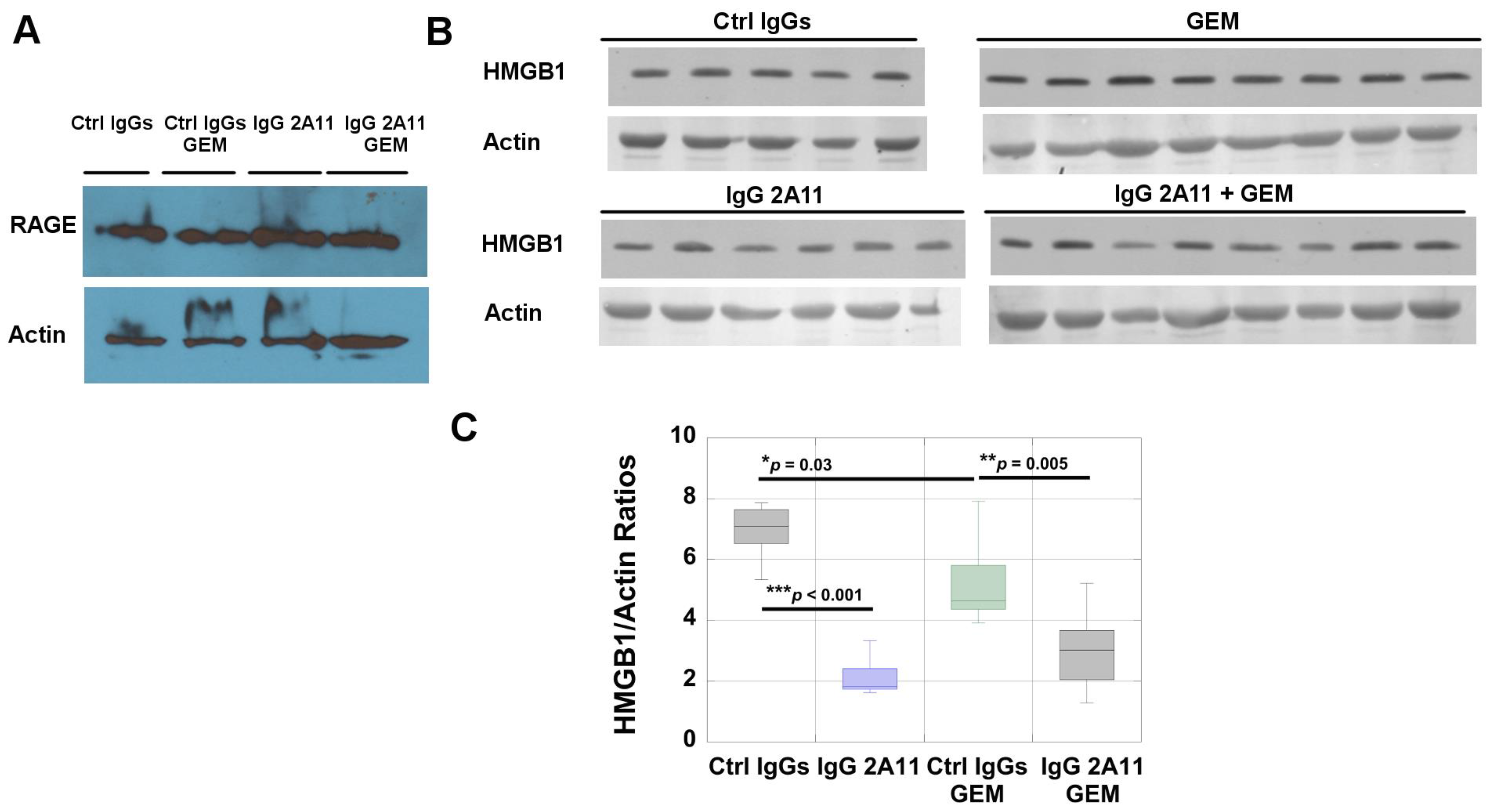
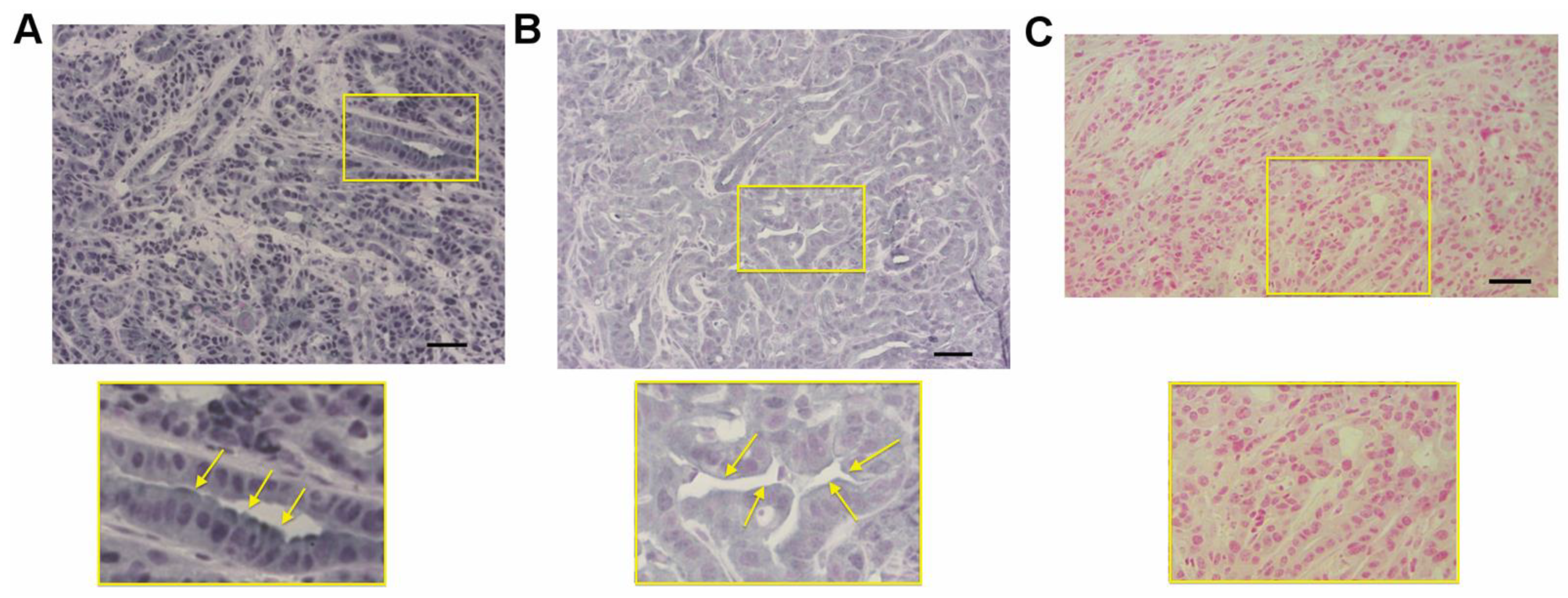
3.10. RAGE and HMGB1 Are Expressed in Murine Tumor Tissues
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmichael, J.; Fink, U.; Russell, R.C.; Spittle, M.F.; Harris, A.L.; Spiessi, G.; Blatter, J. Phase II study of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 1996, 73, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. 2017, 114, 139–152. [Google Scholar] [CrossRef]
- Chand, S.; O’Hayer, K.; Blanco, F.F.; Winter, J.M.; Brody, J.R. The Landscape of Pancreatic Cancer Therapeutic Resistance Mechanisms. Int. J. Biol. Sci. 2016, 12, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Whatcott, C.J.; Posner, R.G.; von Hoff, D.D.; Han, H. Desmoplasia and Chemoresistance in Pancreatic Cancer; Transworld Research Network: Trivandium, India, 2012. [Google Scholar]
- Kang, R.; Tang, D.; E Schapiro, N.; Livesey, K.M.; Farkas, A.M.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Tang, D.; Schapiro, N.E.; Lotze, M.T.; Zeh, H.J. Abstract B96: The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Tumor Biol. 2012, 33, 567–577. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Stern, D.M. Receptor for age (RAGE) is a gene within the major histocompatibility class III region: Implications for host response mechanisms in homeostasis and chronic disease. Front. Biosci. 2001, 6, 1151–1160. [Google Scholar]
- Brett, J.; Schmidt, A.M.; Du Yan, S.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the Distribution of a Newly Characterized Receptor for Advanced Glycation End Products in Tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar]
- Sorci, G.; Riuzzi, F.; Giambanco, I.; Donato, R. RAGE in tissue homeostasis, repair and regeneration. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Riuzzi, F.; Sorci, G.; Sagheddu, R.; Chiappalupi, S.; Salvadori, L.; Donato, R. RAGE in the pathophysiology of skeletal muscle. J. Cachex Sarcopenia Muscle 2018, 9, 1213–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Tempering the wrath of RAGE: An emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann. Med. 2009, 41, 408–422. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Munch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE ligands in cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Leclerc, E.; Vetter, S.W. The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 2706–2711. [Google Scholar] [CrossRef] [Green Version]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta BBA Mol. Cell Res. 2009, 1793, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.D.; Zhu, H.; Fu, J.; Yan, S.F.; Roher, A.; Tourtellotte, W.W.; Rajavashisth, T.; Chen, X.; Godman, G.C.; Stern, D.; et al. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: A proinflammatory pathway in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 5296–5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Rai, V.; Hudson, B.I.; Song, F.; Schmidt, A.M.; Barile, G.R. RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol. 2012, 274, 72–82. [Google Scholar] [CrossRef]
- Sirois, C.M.; Jin, T.; Miller, A.L.; Bertheloot, D.; Nakamura, H.; Horvath, G.L.; Mian, A.; Jiang, J.; Schrum, J.; Bossaller, L.; et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [Google Scholar] [CrossRef] [Green Version]
- Mizumoto, S.; Takahashi, J.; Sugahara, K. Receptor for Advanced Glycation End Products (RAGE) Functions as Receptor for Specific Sulfated Glycosaminoglycans, and Anti-RAGE Antibody or Sulfated Glycosaminoglycans Delivered in Vivo Inhibit Pulmonary Metastasis of Tumor Cells. J. Biol. Chem. 2012, 287, 18985–18994. [Google Scholar] [CrossRef] [Green Version]
- Foell, D.; Wittkowski, H.; Vogl, T.; Roth, J. S100 proteins expressed in phagocytes: A novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 2006, 81, 28–37. [Google Scholar] [CrossRef]
- Holzinger, D.; Foell, D.; Kessel, C. The role of S100 proteins in the pathogenesis and monitoring of autoinflammatory diseases. Mol. Cell. Pediatr. 2018, 5, 1–5. [Google Scholar] [CrossRef]
- Kessel, C.; Holzinger, D.; Foell, D. Phagocyte-derived S100 proteins in autoinflammation: Putative role in pathogenesis and usefulness as biomarkers. Clin. Immunol. 2013, 147, 229–241. [Google Scholar] [CrossRef]
- Schmidt, A.-M.; Hofmann, M.; Taguchi, A.; Du Yan, S.; Stern, D.M. RAGE: A Multiligand Receptor Contributing to the Cellular Response in Diabetic Vasculopathy and Inflammation. Semin. Thromb. Hemost. 2000, 26, 485–494. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, A.; Blood, D.C.; Del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE–amphoterin signalling suppresses tumour growth and metastases. Nat. Cell Biol. 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Kislinger, T.; Fu, C.; Huber, B.; Qu, W.; Taguchi, A.; Yan, S.; Hofmann, M.; FangYan, S.; Pischetsrieder, M.; Stern, D.; et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef] [Green Version]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-S.; Guh, J.-Y.; Chen, H.-C.; Hung, W.-C.; Lai, Y.-H.; Chuang, L.-Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Yeh, C.H.; Sturgis, L.; Haidacher, J.; Zhang, X.N.; Sherwood, S.J.; Bjercke, R.J.; Juhasz, O.; Crow, M.T.; Tilton, R.G.; Denner, L.; et al. Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-kappaB transcriptional activation and cytokine secretion. Diabetes 2001, 50, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta BBA Mol. Cell Res. 2000, 1498, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Yonekura, H.; Yamagishi, S.; Fujimori, H.; Yamamoto, Y.; Yamamoto, H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J. Biol. Chem. 2000, 275, 25781–25790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh, I.H.J. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 2012, 8, 989–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.M.; Du Yan, S.; Stern, D.M.; Yan, S.F. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Fürstenberger, G.; Muüller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, R. RAGE: A Single Receptor for Several Ligands and Different Cellular Responses: The Case of Certain S100 Proteins. Curr. Mol. Med. 2007, 7, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Qu, X.; Schmidt, A.M. Sp1-binding Elements in the Promoter of RAGE Are Essential for Amphoterin-mediated Gene Expression in Cultured Neuroblastoma Cells. J. Biol. Chem. 1998, 273, 30870–30878. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Schmidt, A.M. Characterization and Functional Analysis of the Promoter of RAGE, the Receptor for Advanced Glycation End Products. J. Biol. Chem. 1997, 272, 16498–16506. [Google Scholar] [CrossRef] [Green Version]
- DiNorcia, J.; Lee, M.K.; Moroziewicz, D.N.; Winner, M.; Suman, P.; Bao, F.; Remotti, H.E.; Zou, Y.S.; Yan, S.F.; Qiu, W.; et al. RAGE gene deletion inhibits the development and progression of ductal neoplasia and prolongs survival in a murine model of pancreatic cancer. J. Gastrointest. Surg. 2011, 16, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [Green Version]
- Prica, F.; Radon, T.; Cheng, Y.; Crnogorac-Jurcevic, T. The life and works of S100P—From conception to cancer. Am. J. Cancer Res. 2016, 6, 562–576. [Google Scholar]
- Arumugam, T.; Simeone, D.M.; Van Golen, K.; Logsdon, C.D. S100P Promotes Pancreatic Cancer Growth, Survival, and Invasion. Clin. Cancer Res. 2005, 11, 5356–5364. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.; Logsdon, C.D. S100P: A novel therapeutic target for cancer. Amino Acids 2011, 41, 893–899. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-Derived RAGE Antagonistic Peptide Reduces Tumor Growth and Metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.; Ramachandran, V.; Logsdon, C.D. Effect of Cromolyn on S100P Interactions With RAGE and Pancreatic Cancer Growth and Invasion in Mouse Models. J. Natl. Cancer Inst. 2006, 98, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Sun, D.; Peng, Z.; Pal, A.; Maxwell, D.S.; Bornmann, W.G.; Logsdon, C.D. Designing and Developing S100P Inhibitor 5-Methyl Cromolyn for Pancreatic Cancer Therapy. Mol. Cancer Ther. 2013, 12, 654–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arumugam, T.; Simeone, D.M.; Schmidt, A.M.; Logsdon, C.D. S100P Stimulates Cell Proliferation and Survival via Receptor for Activated Glycation End Products (RAGE). J. Biol. Chem. 2004, 279, 5059–5065. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.-G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Zeh, H.J., III; Lotze, M.T. High-mobility group box 1 and cancer. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2010, 1799, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.D.; Ito, N.; Lotze, M.T.; DeMarco, R.A.; Popovic, P.; Shand, S.H.; Watkins, S.; Winikoff, S.; Brown, C.K.; Bartlett, D.L.; et al. High mobility group box I (HMGB1) release from tumor cells after treatment: Implications for development of targeted chemoimmunotherapy. J. Immunother. 2007, 30, 596–606. [Google Scholar] [CrossRef]
- Liu, L.; Yang, M.; Kang, R.; Wang, Z.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; Lotze, M.T.; et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2010, 25, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhang, Z.; Zhang, Y.; Chen, X.; Guo, S.; Lei, Y.; Xu, Y.; Ji, C.; Bi, Z.; Wang, K. HMGB1-mediated autophagy modulates sensitivity of colorectal cancer cells to oxaliplatin via MEK/ERK signaling pathway. Cancer Biol. Ther. 2015, 16, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meghnani, V.; Wagh, A.; Indurthi, V.S.; Koladia, M.; Vetter, S.W.; Law, B.; Leclerc, E. The receptor for advanced glycation end products influences the expression of its S100 protein ligands in melanoma tumors. Int. J. Biochem. Cell Biol. 2014, 57, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.; Fritz, G.; Weibel, M.; Heizmann, C.W.; Galichet, A. S100B and S100A6 Differentially Modulate Cell Survival by Interacting with Distinct RAGE (Receptor for Advanced Glycation End Products) Immunoglobulin Domains. J. Biol. Chem. 2007, 282, 31317–31331. [Google Scholar] [CrossRef] [Green Version]
- Abramoff, M.D.; Magalhaes, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Saha, A.; Lee, Y.-C.; Zhang, Z.; Chandra, G.; Su, S.-B.; Mukherjee, A.B. Lack of an Endogenous Anti-inflammatory Protein in Mice Enhances Colonization of B16F10 Melanoma Cells in the Lungs. J. Biol. Chem. 2010, 285, 10822–10831. [Google Scholar] [CrossRef] [Green Version]
- Azizan, N.; Suter, M.A.; Liu, Y.; Logsdon, C.D. RAGE maintains high levels of NFkappaB and oncogenic Kras activity in pancreatic cancer. Biochem. Biophys. Res. Commun. 2017, 493, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Chapter 12 Monitoring Autophagic Degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Binenbaum, Y.; Na’Ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updates 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Woo, M.; Hakem, R.; Soengas, M.S.; Duncan, G.S.; Shahinian, A.; Kägi, D.; Hakem, A.; McCurrach, M.; Khoo, W.; Kaufman, S.A.; et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998, 12, 806–819. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Mechanisms of Caspase Activation and Inhibition during Apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; E Davidson, N.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar] [PubMed]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef]
- Yoon, H.; Min, J.K.; Lee, J.W.; Kim, D.G.; Hong, H.J. Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of AKT and extracellular signal-regulated kinase (ERK)1/2. Biochem. Biophys. Res. Commun. 2011, 405, 333–337. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, E.K.; Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Kang, H.S. Role of extracellular signal-regulated kinase (ERK)1/2 in multicellular resistance to docetaxel in MCF-7 cells. Int. J. Oncol. 2010, 37, 655–661. [Google Scholar]
- Zheng, C.; Jiao, X.; Jiang, Y.; Sun, S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J. Int. Med. Res. 2013, 41, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Mirmohammadsadegh, A.; Mota, R.; Gustrau, A.; Hassan, M.; Nambiar, S.; Marini, A.; Bojar, H.; Tannapfel, A.; Hengge, U.R. ERK1/2 is highly phosphorylated in melanoma metastases and protects melanoma cells from cisplatin-mediated apoptosis. J. Investig. Dermatol. 2007, 127, 2207–2215. [Google Scholar] [CrossRef] [Green Version]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends. Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Prabhu, L.; Mundade, R.; Korc, M.; Loehrer, P.J.; Lu, T. Critical role of NF-kappaB in pancreatic cancer. Oncotarget 2014, 5, 10969–10975. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, B.L.; Steel, H.C.; Theron, A.J.; Heyman, L.; Smit, T.; Ramdas, Y.; Anderson, R. High Mobility Group Box 1 in Human Cancer. Cells 2020, 9, 1664–1694. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells 2016, 5, 12–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, M.P.; Rachagani, S.; Souchek, J.J.; Mallya, K.; Johansson, S.L.; Batra, S.K. Novel Pancreatic Cancer Cell Lines Derived from Genetically Engineered Mouse Models of Spontaneous Pancreatic Adenocarcinoma: Applications in Diagnosis and Therapy. PLoS ONE 2013, 8, e80580. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Henderson, M.; Muth, S.; Murphy, A.; Zheng, L. Preclinical mouse models for immunotherapeutic and non-immunotherapeutic drug development for pancreatic ductal adenocarcinoma. Ann. Pancreat. Cancer 2020, 3, 7–22. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2008, 16, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Pramanik, K.C.; Makena, M.R.; Bhowmick, K.; Pandey, M.K. Advancement of NF-kappaB Signaling Pathway: A Novel Target in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3890–3904. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Mo, Z.C.; Yin, K.; Zhao, G.J.; Lv, Y.C.; Ouyang, X.P.; Jiang, Z.S.; Fu, Y.; Tang, C.K. Epigallocatechin-3-gallate prevents TNF-alpha-induced NF-kappaB activation thereby upregulating ABCA1 via the Nrf2/Keap1 pathway in macrophage foam cells. Int. J. Mol. Med. 2012, 29, 946–956. [Google Scholar]
- Chow, J.Y.; Ban, M.; Wu, H.L.; Nguyen, F.; Huang, M.; Chung, H.; Dong, H.; Carethers, J.M. TGF-beta downregulates PTEN via activation of NF-kappaB in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver. Physiol. 2010, 298, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Chiao, P.; Zhang, X.; Zhang, X.; Lazar, M.A.; Seto, E.; Young, H.A.; Ye, J. Coactivators and corepressors of NF-kappaB in IkappaB alpha gene promoter. J. Biol. Chem. 2005, 280, 21091–21098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, W.-S.; Kim, I.-W.; Hu, R.; Kong, A.-N.T. Modulation of AP-1 by natural chemopreventive compounds in human colon HT-29 cancer cell line. Pharm. Res. 2004, 21, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharm. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J. Cachex Sarcopenia Muscle 2020, 11, 929–946. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Provider/Catalog Number | Application | Dilution |
|---|---|---|---|
| P62 (SQSTM1) | Santa Cruz Biotechnologies/28359 | WB | 1/1000 |
| LC3A/B | Cell Signaling Technology/12741 | WB | 1/500 |
| Cleaved caspase 3 | Cell Signaling Technology/9662 | WB | 1/1000 |
| Caspase 3 | Cell Signaling Technology/9661 | WB | 1/1000 |
| PARP | Cell Signaling Technology/9542 | WB | 1/1000 |
| HMGB1 | Cell Signaling Technology/3935 | IHC | 1/1000 |
| HMGB1 | Thermo Fisher Scientific/PA5-27378 | WB | 1/100 |
| Phospho-ERK1/2 | Cell Signaling Technology/9101 | WB | 1/1000 |
| ERK1/2 | Cell Signaling Technology/4695 | WB | 1/1000 |
| Phospho-NF-KB p65 | Cell signaling Technology/3033 | WB | 1/1000 |
| Total NF-kB p65 | Cell Signaling Technology/8242 | WB | 1/1000 |
| RAGE | Thermo Fisher Scientific/PA1-075 | WB | 1/200 |
| RAGE | R&D Systems/AF1145 | IHC | 1/1000 |
| β-actin | Cell Signaling Technology/4967 | WB | 1/1000 |
| HRP-donkey anti-rabbit | Jackson ImmunoResearch/711-035-152 | WB | 1/25,000 |
| IRdye800 goat anti-rabbit | LI-COR/P/N 927-50000 | WB | 1/50,000 |
| IRdye680 goat anti-rabbit | LI-COR/P/N 926-68071 | WB | 1/50,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swami, P.; O’Connell, K.A.; Thiyagarajan, S.; Crawford, A.; Patil, P.; Radhakrishnan, P.; Shin, S.; Caffrey, T.C.; Grunkemeyer, J.; Neville, T.; et al. Inhibition of the Receptor for Advanced Glycation End Products Enhances the Cytotoxic Effect of Gemcitabine in Murine Pancreatic Tumors. Biomolecules 2021, 11, 526. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11040526
Swami P, O’Connell KA, Thiyagarajan S, Crawford A, Patil P, Radhakrishnan P, Shin S, Caffrey TC, Grunkemeyer J, Neville T, et al. Inhibition of the Receptor for Advanced Glycation End Products Enhances the Cytotoxic Effect of Gemcitabine in Murine Pancreatic Tumors. Biomolecules. 2021; 11(4):526. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11040526
Chicago/Turabian StyleSwami, Priyanka, Kelly A. O’Connell, Swetha Thiyagarajan, Ayrianne Crawford, Prathamesh Patil, Prakash Radhakrishnan, Simon Shin, Thomas C. Caffrey, James Grunkemeyer, Tammi Neville, and et al. 2021. "Inhibition of the Receptor for Advanced Glycation End Products Enhances the Cytotoxic Effect of Gemcitabine in Murine Pancreatic Tumors" Biomolecules 11, no. 4: 526. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11040526