
Potential of Induced Pluripotent Stem Cells for Use in Gene Therapy: History, Molecular Bases, and Medical Perspectives

 , , , ,
, , , ,
Abstract
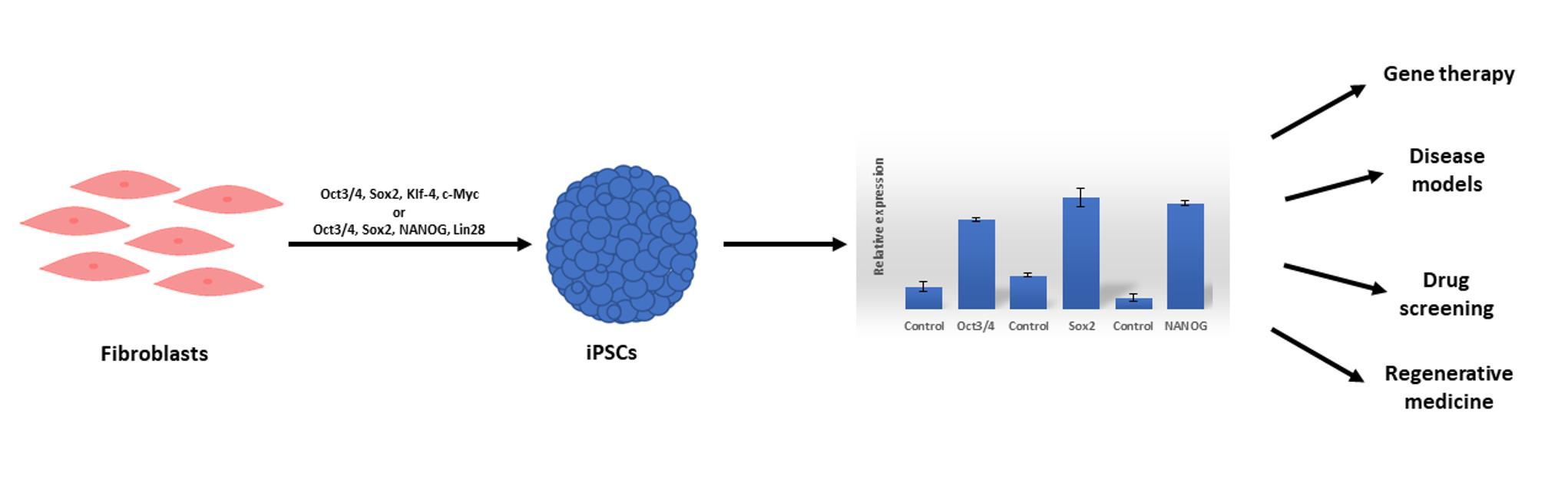
:
1. Introduction
2. What Are Stem Cells?
2.1. Where Do the Stem Cells Originate From?
2.2. Classification of Stem Cells Based on Their Stemness
3. Molecular Mechanisms of Somatic Cells Reprogramming Into iPSCs
3.1. Molecular Bases of Cell Reprogramming with Yamanaka and Thomson’s Factors
3.2. Vectors in iPSCs Reprogramming
4. Perspectives for iPSCs Use in Disease Therapies
4.1. Sandhoff Ddisease
4.2. SCID—Severe Combined Immunodeficiency
4.3. Severe Congenital Neutropenia
4.4. Hemophilia A
4.5. Osteogenesis Imperfecta
4.6. Myotonic Dystrophy
4.7. Retinitis Pigmentosa
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ramalho-Santos, M.; Willenbring, H. On the Origin of the Term “Stem Cell”. Cell Stem Cell 2007, 1, 35–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.J.; Shah, N.M.; Anderson, D.J. Regulatory Mechanisms in Stem Cell Biology. Cell 1997, 88, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Dulak, J.; Szade, K.; Szade, A.; Nowak, W.; Józkowicz, A. Adult stem cells: Hopes and hypes of regenerative medicine. Acta Biochim. Pol. 2015, 62, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, E.; Dazzi, F. Bone Marrow Transplantation 1957-2019. Front Immunol. 2019, 10, 1246. [Google Scholar] [CrossRef] [Green Version]
- Passweg, J.R.; Baldomero, H.; Bader, P.; Bonini, C.; Cesaro, S.; Dreger, P.; Duarte, R.F.; Dufour, C.; Kuball, J.; Farge-Bancel, D.; et al. Hematopoietic stem cell transplantation in Europe 2014: More than 40 000 transplants annually. Bone Marrow Transplant. 2016, 51, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.A. Human embryonic stem cell research: Ethical and legal issues. Nat. Rev. Genet. 2001, 2, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Attwood, S.W.; Edel, M.J. iPS-Cell Technology and the Problem of Genetic Instability-Can It Ever Be Safe for Clinical Use? J. Clin. Med. 2019, 28, 288. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nat. Cell Biol. 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, M.; Koryciak-Komarska, H.; Czekaj, P.; Sebesta, R.; Czekaj, T.M.; Urbanek, K.; Likus, W.; Malinowska-Kolodziej, I.; Plewka, D.; Nowaczyk-Dura, G.; et al. The comparison of multipotential for differentiation of progenitor mesenchymal-like stem cells obtained from livers of young and old rats. Folia Histochem. Cytobiol. 2007, 45, 245–254. [Google Scholar] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Seaberg, R.M.; van der Kooy, D. Stem and progenitor cells: The premature desertion of rigorous definitions. Trends Neurosci. 2003, 26, 125–131. [Google Scholar] [CrossRef]
- Klimanskaya, I.; Chung, Y.; Meisner, L.; Johnson, J.; West, M.D.; Lanza, R. Human embryonic stem cells derived without feeder cells. Lancet 2005, 365, 1636–1641. [Google Scholar] [CrossRef]
- Grigsby, C.L.; Leong, K.W. Balancing protection and release of DNA: Tools to address a bottleneck of non-viral gene delivery. J. R. Soc. Interface 2010, 7, S67–S82. [Google Scholar] [CrossRef]
- Kim, J.B.; Zaehres, H.; Wu, G.; Gentile, L.; Ko, K.; Sebastiano, V.; Araúzo-Bravo, M.J.; Ruau, D.; Han, D.W.; Zenke, M.; et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nat. Cell Biol. 2008, 454, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.-H.; Choi, J.; Pei, C.-Z. Cellular Functions of OCT-3/4 Regulated by Ubiquitination in Proliferating Cells. Cancers 2020, 12, 663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, A.J.M.; Stoop, H.; Biermann, K.; van Gurp, R.J.H.L.M.; Swartzman, E.; Cribbes, S.; Ferlinz, A.; Shannon, M.; Oosterhuis, W.; Looijenga, L.H.J. Expression and interdependencies of pluripotency factors LIN28, OCT3/4, NANOG, and Sox2 in human tes-ticular germ cells and tumours of the testis. Int. J. Androl. 2011, 34, e160–e174. [Google Scholar] [CrossRef]
- Shi, G.; Jin, Y. Role of Oct4 in maintaining and regaining stem cell pluripotency. Stem Cell Res. Ther. 2010, 1, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovic, S.; Pucéat, M. Oct-3/4: Not Just a Gatekeeper of Pluripotency for Embryonic Stem Cell, a Cell Fate Instructor through a Gene Dosage Effect. Cell Cycle 2007, 6, 8–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterneckert, J.; Höing, S.; Schöler, H.R. Concise Review: Oct4 and More: The Reprogramming Expressway. Stem Cells 2011, 30, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zeineddine, D.; Hammoud, A.A.; Mortada, M.; Boeuf, H. The Oct4 protein: More than a magic stemness marker. Am. J. Stem Cells 2014, 3, 74–82. [Google Scholar]
- Zeineddine, D.; Papadimou, E.; Chebli, K.; Gineste, M.; Liu, J.; Grey, C.; Thurig, S.; Behfar, A.; Wallace, V.A.; Skerjanc, I.S.; et al. Oct-3/4 Dose Dependently Regulates Specification of Embryonic Stem Cells toward a Cardiac Lineage and Early Heart Development. Dev. Cell 2006, 11, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Ciu, W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J. Stem Cells 2014, 6, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, J.; Yashiro, M.; Sakurai, K.; Kubo, N.; Tanaka, H.; Muguruma, K.; Sawada, T.; Ohira, M.; Hirakawa, K. Role of the Stemness Factors Sox2, Oct3/4, and Nanog in Gastric Carcinoma. J. Surg. Res. 2012, 174, 130–135. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Hirano, K.; Nagata, S.; Tada, T. Sox2 expression effects on direct reprogramming efficiency as determined by alternative somatic cell fate. Stem Cell Res. 2011, 6, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Carey, B.W.; Markoulaki, S.; Hanna, J.H.; Faddah, D.A.; Buganim, Y.; Kim, J.; Ganz, K.; Steine, E.J.; Cassady, J.P.; Creyghton, M.P.; et al. Reprogramming Factor Stoichiometry Influences the Epigenetic State and Biological Properties of Induced Pluripotent Stem Cells. Cell Stem Cell 2011, 9, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzino, A. Concise Review: The Sox2-Oct4 Connection: Critical Players in a Much Larger Interdependent Network Integrated at Multiple Levels. Stem Cells 2013, 31, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Kehler, J.; Tolkunova, E.; Koschorz, B.; Pesce, M.; Gentile, L.; Boiani, M.; Lomelí, H.; Nagy, A.; McLaughlin, K.J.; Schöler, H.R.; et al. Oct4 is required for primordial germ cell survival. EMBO Rep. 2004, 5, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Li, W.; Zhou, H.; Wie, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Sing, S. Reprogramming of Human Primary Somatic Cells buy OCT4 and Chemical Compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Daley, G.Q. Lin28: Primal regulator of growth and metabolism in stem cells. Cell Stem Cell 2013, 12, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef] [Green Version]
- Vogt, E.J.; Meglicki, M.; Hartung, K.I.; Borsuk, E.; Behr, R. Importance of the pluripotency factor LIN28 in the mammalian nucleolus during early embryonic development. Development 2012, 139, 4514–4523. [Google Scholar] [CrossRef] [Green Version]
- Hanna, H.; Saha, K.; Pando, B.; van Zon, J.; Lengner, C.J.; Creyghton, M.P.; van Oudenaarden, A.; Jaenisch, R. Direct cell re-programming is a stochastic process amenable to acceleration. Nature 2009, 462, 595–601. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Golipour, A.; David, L.; Liu, Y.; Jayakumaran, G.; Hirsch, C.L.; Trcka, D.; Wrana, J.L. A Late Transition in Somatic Cell Re-programming Requires Regulators Distinct From the Pluripotency Network. Cell Stem Cell 2012, 11, 769–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Su, Y.; Huang, C.; Yin, Y.; Zhu, J.; Knupp, A.; Chu, A.; Tang, Y. FOXH1 Is Regulated by NANOG and LIN28 for Early-stage Reprogramming. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Zare, M.; Soleimani, M.; Mohammadian, M.; Akbarzadeh, A.; Havasi, P.; Zarghami, N. Efficient biotechnological approach for lentiviral transduction of induced pluripotent stem cells. Artif. Cells Nanomed. Biotechnol. 2014, 44, 743–748. [Google Scholar] [CrossRef]
- Bayart, E.; Cohen-Haguenauer, O. Technological Overview of iPS Induction from Human Adult Somatic Cells. Curr. Gene Ther. 2013, 13, 73–92. [Google Scholar] [CrossRef]
- Chang, C.-W.; Lai, Y.-S.; Pawlik, K.M.; Liu, K.; Sun, C.-W.; Li, C.; Schoeb, T.R.; Townes, T.M. Polycistronic Lentiviral Vector for “Hit and Run” Reprogramming of Adult Skin Fibroblasts to Induced Pluripotent Stem Cells. Stem Cells 2009, 27, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Sebe, A.; Ivics, Z. Reprogramming of Human Fibroblasts to Induced Pluripotent Stem Cells with Sleeping Beauty Transpos-on-Based Stable Gene Delivery. Transposons and Retrotransposons. Methods Mol. Biol. 2016, 1400, 419–427. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, S.L.; Sivanandam, V.G.; Kochanek, S. Homologous and heterologous recombination between adenovirus vector DNA and chromosomal DNA. J. Gene Med. 2008, 10, 1176–1189. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.-J.; Lee, C.-S.; Kwon, Y.-W.; Paek, J.S.; Lee, S.-H.; Hur, J.; Lee, E.J.; Roh, T.-Y.; Chu, I.-S.; Leem, S.-H.; et al. Induction of pluripotent stem cells from adult somatic cells by protein-based reprogramming without genetic manipulation. Blood 2010, 116, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.J.; Hong, Y.J.; Do, J.T. Cellular Reprogramming Using Protein and Cell-Penetrating Peptides. Int. J. Mol. Sci. 2017, 18, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, I.M.; Weitzman, M.D. GENE THERAPY: Twenty-First Century Medicine. Annu. Rev. Biochem. 2005, 74, 711–738. [Google Scholar] [CrossRef] [Green Version]
- Far, H.H.; Patria, Y.N.; Motazedian, A.; Elefanty, A.G.; Stanley, E.G.; Lamande, S.R.; Bateman, J.F. Generation of a heterozygous COL1A1 (c.3969_3970insT) osteogenesis imperfecta mutation human iPSC line, MCRIi001-A-1, using CRISPR/Cas9 editing. Stem Cell Res. 2019, 37, 101449. [Google Scholar] [CrossRef]
- Matsui, H.; Fujimoto, N.; Sasakawa, N.; Ohinata, Y.; Shima, M.; Yamanaka, S.; Sugimoto, M.; Hotta, A. Delivery of Full-Length Factor VIII Using a piggyBac Transposon Vector to Correct a Mouse Model of Hemophilia A. PLoS ONE 2014, 9, e104957. [Google Scholar] [CrossRef] [Green Version]
- Olgasi, C.; Talmon, M.; Merlin, S.; Cucci, A.; Richaud-Patin, Y.; Ranaldo, G.; Colangelo, D.; Di Scipio, F.; Berta, G.N.; Borsotti, C.; et al. Patient-Specific iPSC-Derived Endothelial Cells Provide Long-Term Phenotypic Correction of Hemophilia A. Stem Cell Rep. 2018, 11, 1391–1406. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.; Gao, K.; Cortez-Toledo, E.; Agu, E.; Hyllen, A.A.; Conroy, K.; Pan, G.; Nolta, J.A.; Wang, A.; Zhou, P. Endothelial cells derived from patients’ induced pluripotent stem cells for sustained factor VIII delivery and the treatment of hemophilia A. Stem Cells Transl. Med. 2020, 9, 686–696. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, W.-H.; Hsu, C.-W.; Nguyen, H.V.; Tsai, Y.-T.; Chan, L.; Nagasaki, T.; Maumenee, I.H.; Yannuzzi, L.A.; Hoang, Q.V.; et al. Gene Therapy in Patient-specific Stem Cell Lines and a Preclinical Model of Retinitis Pigmentosa With Membrane Frizzled-related Protein Defects. Mol. Ther. 2014, 22, 1688–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tsai, Y.-T.; Hsu, C.-W.; Erol, D.; Yang, J.; Wu, W.-H.; Davis, R.J.; Egli, D.; Tsang, S.H. Long-term Safety and Efficacy of Human-Induced Pluripotent Stem Cell (iPS) Grafts in a Preclinical Model of Retinitis Pigmentosa. Mol. Med. 2012, 18, 1312–1319. [Google Scholar] [CrossRef]
- Morishima, T.; Watanabe, K.-I.; Niwa, A.; Hirai, H.; Saida, S.; Tanaka, T.; Kato, I.; Umeda, K.; Hiramatsu, H.; Saito, M.K.; et al. Genetic correction of HAX1 in induced pluripotent stem cells from a patient with severe congenital neutropenia improves defective granulopoiesis. Haematologica 2013, 99, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Hiramoto, T.; Ebihara, Y.; Mizoguchi, Y.; Nakamura, K.; Yamaguchi, K.; Ueno, K.; Nariai, N.; Mochizuki, S.; Yamamoto, S.; Na-gasaki, M.; et al. Wnt3a stimulates the maturation of impaired neu-trophils developed from severe congenital neutropenia patient-derived pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 3023–3028. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.H.; Kuehle, J.; Reimann, C.; Mlambo, T.; Alzubi, J.; Maeder, M.L.; Riedel, H.; Fisch, P.; Cantz, T.; Rudolph, C.; et al. Rescue of DNA-PK Signaling and T-Cell Differentiation by Targeted Genome Editing in a prkdc Deficient iPSC Disease Model. PLoS Genet. 2015, 11, e1005239. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Narayanan, K.; Lu, H.; Choo, Y.; Du, C.; Wiradharma, N.; Yang, Y.-Y.; Wan, A.C. Delivery of reprogramming factors into fibroblasts for generation of non-genetic induced pluripotent stem cells using a cationic bolaamphiphile as a non-viral vector. Biomaterials 2013, 34, 5336–5343. [Google Scholar] [CrossRef] [PubMed]
- Menon, T.; Firth, A.L.; Scripture-Adams, D.D.; Galic, Z.; Qualls, S.J.; Gilmore, W.B.; Ke, E.; Singer, O.; Anderson, L.S.; Bornzin, A.R.; et al. Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell 2015, 16, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef] [Green Version]
- Xia, G.; Gao, Y.; Jin, S.; Subramony, S.; Terada, N.; Ranum, L.P.; Swanson, M.S.; Ashizawa, T. Genome Modification Leads to Phenotype Reversal in Human Myotonic Dystrophy Type 1 Induced Pluripotent Stem Cell-Derived Neural Stem Cells. Stem Cells 2015, 33, 1829–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allende, M.L.; Cook, E.K.; Larman, B.C.; Nugent, A.; Brady, J.M.; Golebiowski, D.; Sena-Esteves, M.; Tifft, C.J.; Proia, R.L. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 2018, 59, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Bley, A.E.; Giannikopoulos, O.A.; Hayden, D.; Kubilus, K.; Tifft, C.J.; Eichler, F.S. Natural history of infantile G(M2) gangli-osidosis. Pediatrics 2011, 128, e1233–e1241. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Song, H.; Ming, G.-L. Brain organoids: Advances, applications and challenges. Development 2019, 146, 166074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodbine, L.; Gennery, A.R.; Jeggo, P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair 2014, 16, 84–96. [Google Scholar] [CrossRef]
- Kohn, D.B. Eliminating SCID row: New approaches to SCID. Hematology 2014, 2014, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Song, E.Y.; Kwon, Y.-W.; Oh, S.; Park, H.; Kim, N.-H.; Roh, E.Y. Usefulness of the Hematopoietic Stem Cell Donor Pool as a Source of HLA-Homozygous Induced Pluripotent Stem Cells for Haplobanking: Combined Analysis of the Cord Blood Inventory and Bone Marrow Donor Registry. Biol. Blood Marrow Transplant. 2020, 26, e202–e208. [Google Scholar] [CrossRef]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Primers 2017, 8, 17032. [Google Scholar] [CrossRef]
- Peyvandi, F.; Garagiola, I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia 2019, 25, 738–746. [Google Scholar] [CrossRef]
- Pipe, S.W. Gene therapy for hemophilia. Pediatr. Blood Cancer 2018, 65, e26865. [Google Scholar] [CrossRef]
- Peyvandi, F.; Garagiola, I.; Young, G. The past and future of haemophilia: Diagnosis, treatments, and its complications. Lancet 2016, 388, 187–197. [Google Scholar] [CrossRef]
- Deshpande, M.C.; Garnett, M.C.; Vamvakaki, M.; Bailey, L.; Armes, S.P.; Stolnik, S. Influence of polymer architecture on the structure of complexes formed by PEG–tertiary amine methacrylate copolymers and phosphorothioate oligonucleotide. J. Control. Release 2002, 81, 185–199. [Google Scholar] [CrossRef]
- He, E.; Yue, C.Y.; Simeon, F.; Zhou, L.H.; Too, H.P.; Tam, K.C.; Tam, M.K. Polyplex formation between four-arm poly(ethylene oxide)-b-poly(2-(diethylamino)ethyl methacrylate) and plasmid DNA in gene delivery. J. Biomed. Mater. Res. Part A 2009, 91, 708–718. [Google Scholar] [CrossRef]
- Georgiou, T.K.; Vamvakaki, M.; Patrickios, C.S.; Yamasaki, E.N.; Phylactou, L.A. Nanoscopic Cationic Methacrylate Star Homopolymers: Synthesis by Group Transfer Polymerization, Characterization and Evaluation as Transfection Reagents. Biomacromolecules 2004, 5, 2221–2229. [Google Scholar] [CrossRef]
- Tarnowski, M.; Sieroń, A.L. [Osteogenesis imperfecta--etiology, characteristics, current and future treatment]. Wiad. Lek. 2008, 61, 166–172. [Google Scholar]
- Tarnowski, M.; Szydło, A.; Anioł, J.; Koryciak-Komarska, H.; Lesiak, M.; Gutmajster, E.; Sieron, A.L.; Kusz, D. Optimization of Genetic Engineering and Homologous Recombination of Collagen Type I Genes in Rat Bone Marrow Mesenchymal Stem Cells (MSC). Cell. Reprogramming 2010, 12, 275–282. [Google Scholar] [CrossRef]
- Bird, T.D. Myotonic Dystrophy Type GeneReviews Seattle (WA); [Updated 2019]; University of Washington: Seattle, WA, USA, 1999. [Google Scholar] [PubMed]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An Efficient Nonviral Method to Generate Integration-Free Human-Induced Pluripotent Stem Cells from Cord Blood and Peripheral Blood Cells. Stem Cells 2012, 31, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.-H.; Agarwal, S.; Park, I.-H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integra-tion-free humaniPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of Induced Pluripotent Stem Cells from Human Terminally Differentiated Circulating T Cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Nishishita, N.; Takenaka, C.; Fusaki, N.; Kawamata, S. Generation of human induced pluripotent stem cells from cord blood cells. J. Stem Cells 2011, 6, 101–1088. [Google Scholar] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Stem Cell Origin | Stem Cell Type | Potential to Differentiation |
|---|---|---|
| Embryonic germ stem cells, e.g., zygotes | Totipotent | 3 germ layers and their derivatives and extraembryonic tissue |
| Embryonic stem cells | Pluripotent | 3 germ layers and their derivatives |
| iPSCs | Trophectoderm | |
| Neural stem cells | Multipotent | Glial and neuronal cells |
| Stem cells in bone marrow, hematopoietic stem cells | Multipotent | All blood cell types |
| Mesenchymal stem cells | Multipotent | cells of the mesenchymal lineage (adipocytes, osteocytes, and chondrocytes) |
| Adult stem cells, e.g., epithelial stem cells, skin stem cells | Multipotent | e.g., stem cells within the bulge, intestinal epithelium |
| Unipotent | e.g., the interfollicular epidermis, sebaceous glands, intestinal epithelium | |
| Adult stem cells, e.g., myeloid | Oligopotent | Granulocytes, monocytes, platelets |
| Adult stem cells, e.g., lymphoid | Lymphocytes, natural killer cells | |
| Spermatogenic stem cells | Unipotent | Sperm cells |
| Disease (OMIM Number) | Affected Genes/Proteins | Clinical Features/ Symptoms | iPSCs Application in In Vitro Models | iPSCs Have Potential Applications in Autologous Transplants |
|---|---|---|---|---|
| Osteogenesis imperfecta (#166200) | COL1A1/COL1A2 | lower bone mass and fragility; Short and inclined arms; Shortened legs; Blue-gray sclera of the eye | [49] | - |
| Haemophilia A (#306700) | Procoagulation factor VIII | Spontaneous haemorrhages | [50] | [51,52] |
| Retinitis pigmentosa (#500004) | Frizzled-Related Protein (MFRP) | The disruption and loss pf cells in the retina | [53] | [54] |
| Severe congenital neutropenia (#618752) | HAX1 | recurring infections | [55,56] | - |
| SCID (#602450) | Genes responsible for the proper function of immune cells (mostly T- lymphocytes, but also B-lymphocytes) | T-cell impairment; somatic cells’ sensitivity to radiation; | [57] | [58,59] |
| Myotonic Dystrophy (#602668) | DMPK1 | Myopathy; myotonia; cardiac conduction defects; | [60] | [61] |
| Sandhoff disease (#268800) | β-hexosaminidase A and B deficiency | Neurodegeneration; | [62] | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fus-Kujawa, A.; Mendrek, B.; Trybus, A.; Bajdak-Rusinek, K.; Stepien, K.L.; Sieron, A.L. Potential of Induced Pluripotent Stem Cells for Use in Gene Therapy: History, Molecular Bases, and Medical Perspectives. Biomolecules 2021, 11, 699. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050699
Fus-Kujawa A, Mendrek B, Trybus A, Bajdak-Rusinek K, Stepien KL, Sieron AL. Potential of Induced Pluripotent Stem Cells for Use in Gene Therapy: History, Molecular Bases, and Medical Perspectives. Biomolecules. 2021; 11(5):699. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050699
Chicago/Turabian StyleFus-Kujawa, Agnieszka, Barbara Mendrek, Anna Trybus, Karolina Bajdak-Rusinek, Karolina L. Stepien, and Aleksander L. Sieron. 2021. "Potential of Induced Pluripotent Stem Cells for Use in Gene Therapy: History, Molecular Bases, and Medical Perspectives" Biomolecules 11, no. 5: 699. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050699