
Immune Cell Modulation of the Extracellular Matrix Contributes to the Pathogenesis of Pancreatic Cancer


Abstract
:1. Introduction
2. The Pancreatic Adenocarcinoma Tumor Microenvironment
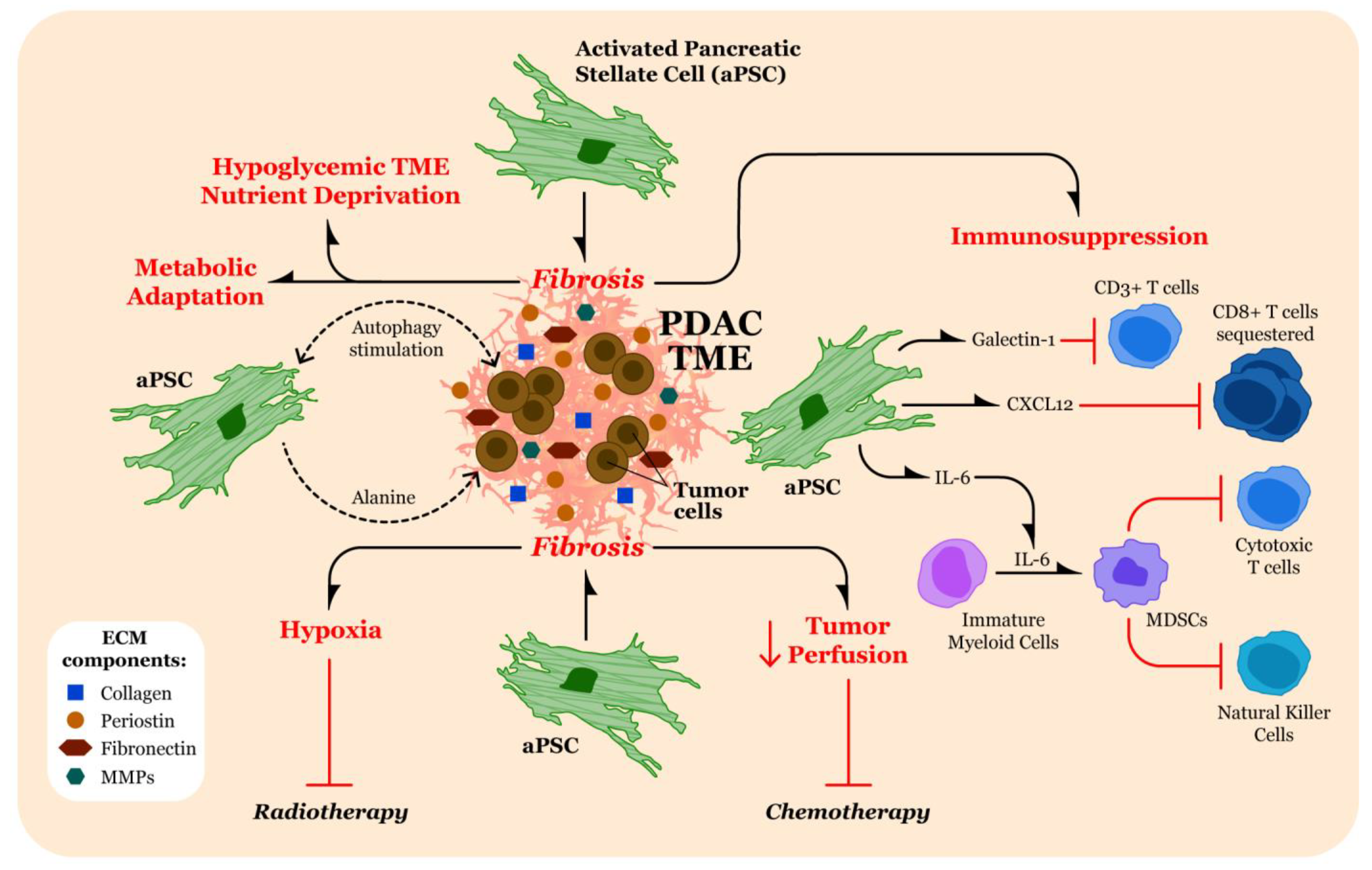
3. Contributions of Fibrosis in the TME to Pathogenesis of Pancreatic Cancer
3.1. Therapeutic Resistance through Limiting Penetration of Cytotoxic Agents
3.2. Promotion of Hypoxia in the TME
3.3. Altering Tumor Cell Metabolism
3.4. Immunomodulation
4. Strategies for Targeting Fibrosis in PDAC
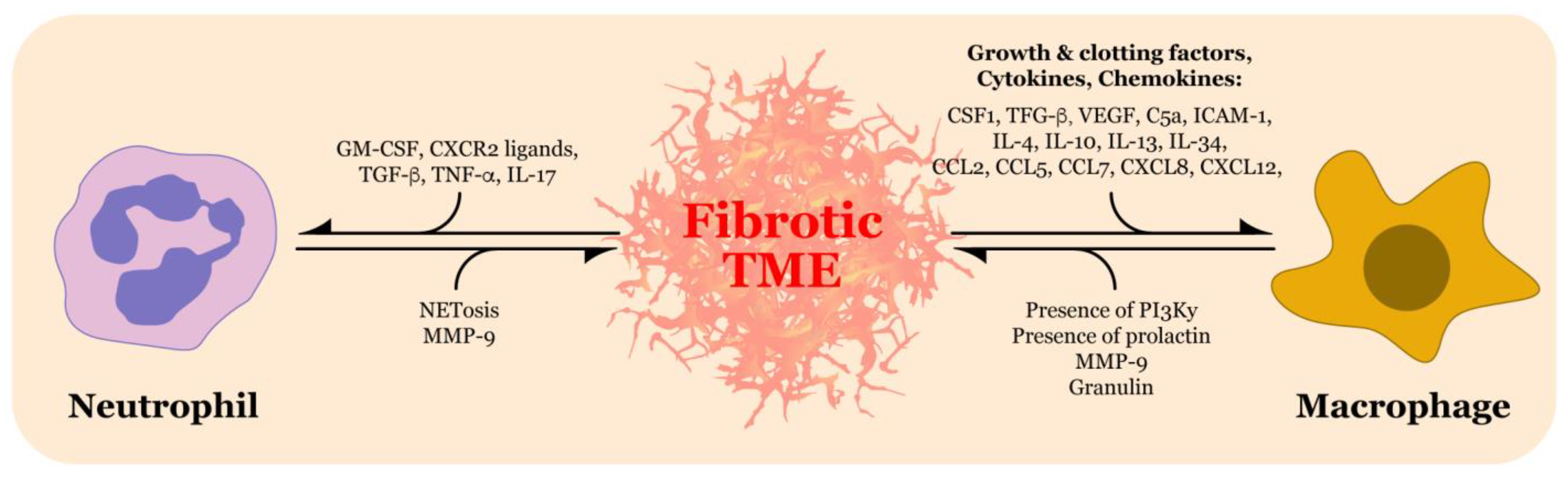
5. Immune Characterization of the TME and Impact on the ECM
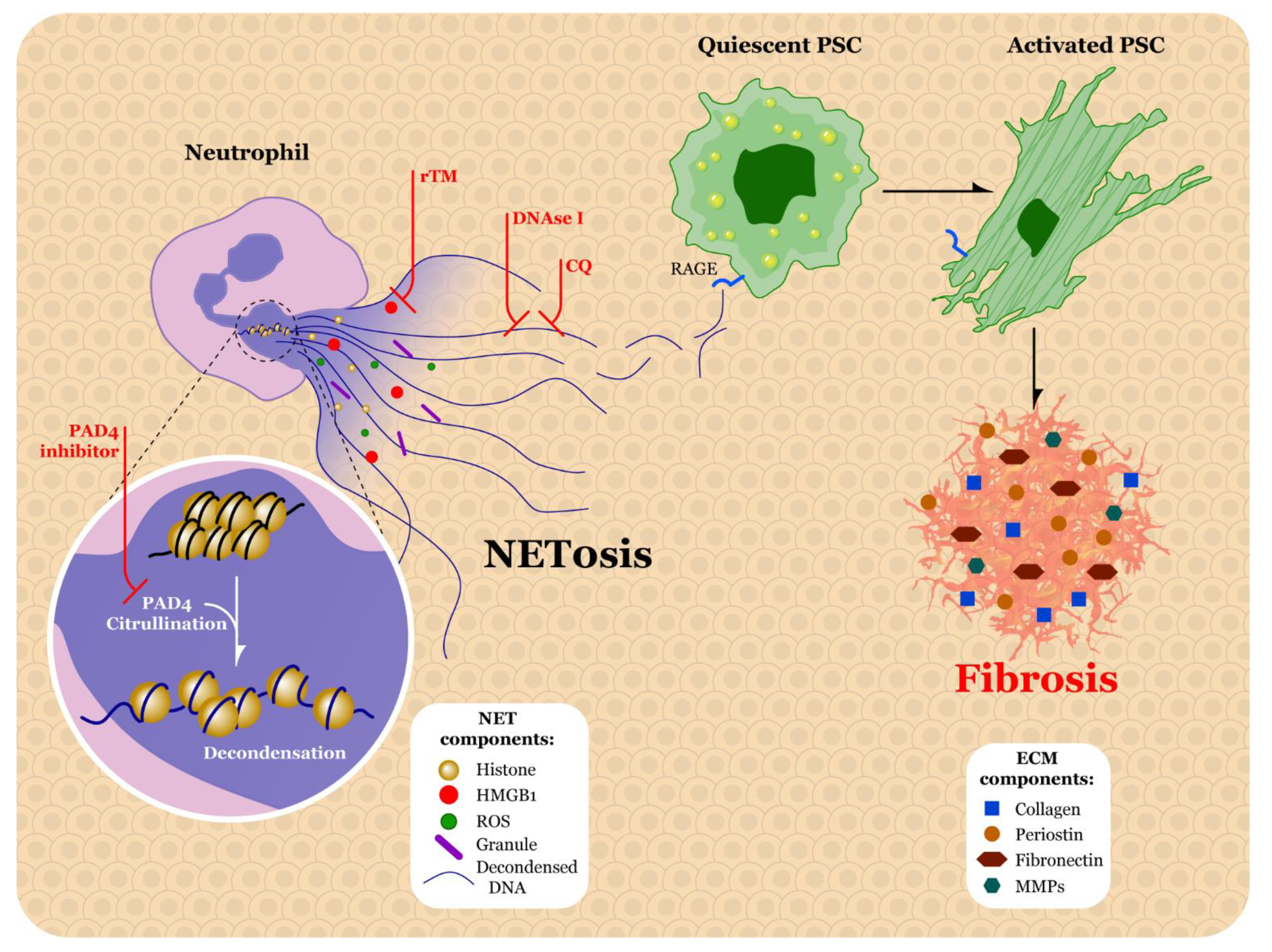
5.1. Neutrophils
5.2. Factors Promoting Neutrophil Extracellular Traps in PDAC
5.3. Therapeutic Strategies for Targeting Neutrophils and NETs
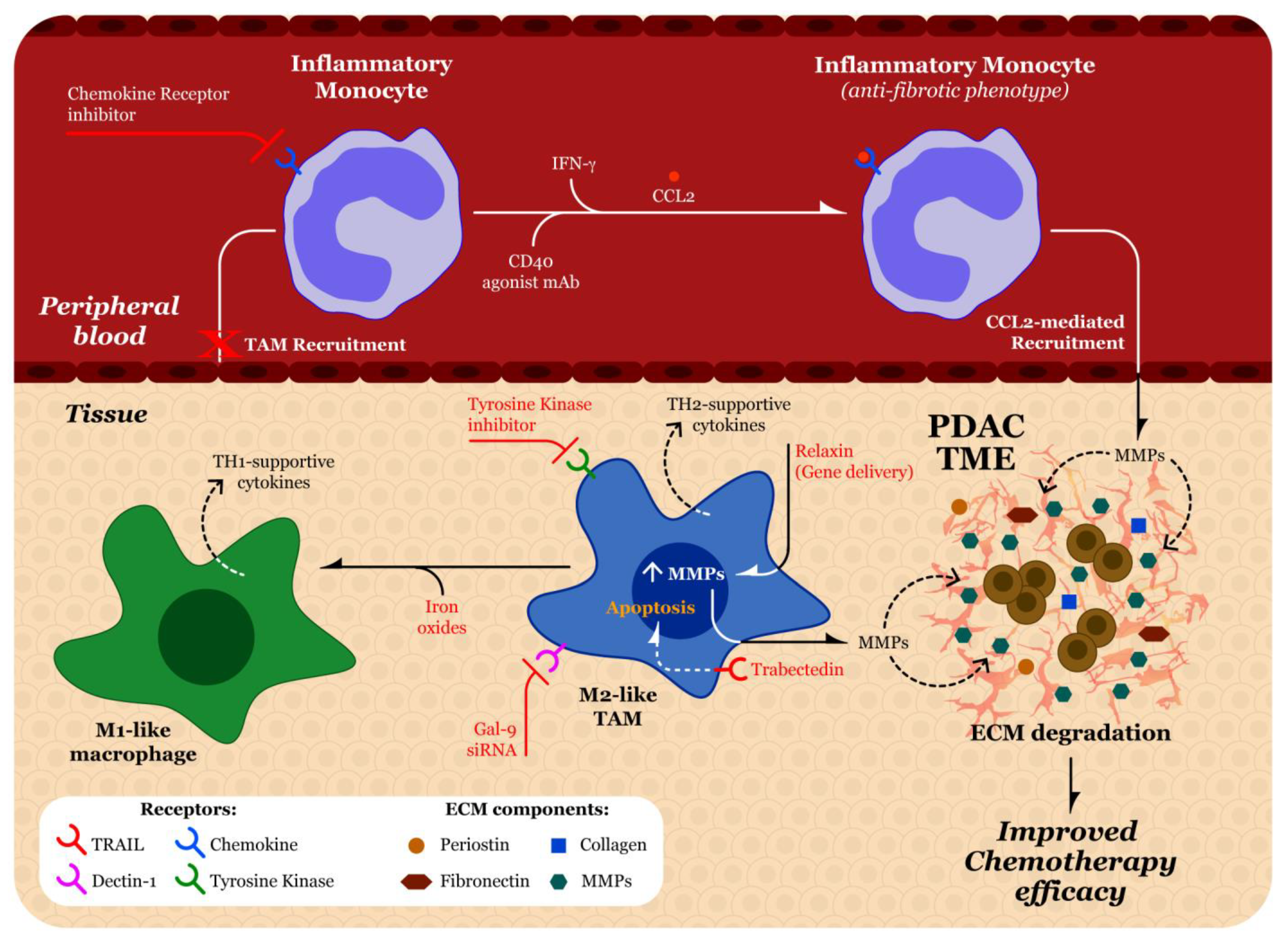
5.4. Tumor Associated Macrophages
5.5. Effect of TAMs on ECM
5.6. Recruitment of Macrophages
5.7. Strategies to Target Macrophages
5.8. Beyond Neutrophils and Macrophages
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Tavakkoli, A.; Singal, A.G.; Waljee, A.K.; Elmunzer, B.J.; Pruitt, S.L.; McKey, T.; Rubenstein, J.H.; Scheiman, J.M.; Murphy, C.C. Racial disparities and trends in pancreatic cancer incidence and mortality in the united states. Clin. Gastroenterol. Hepatol. 2020, 18, 171–178.e110. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Melstrom, L.G.; Salazar, M.D.; Diamond, D.J. The pancreatic cancer microenvironment: A true double agent. J. Surg. Oncol. 2017, 116, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The extracellular matrix and pancreatic cancer: A complex relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [Green Version]
- DeClerck, Y.A. Desmoplasia: A response or a niche? Cancer Discov. 2012, 2, 772–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef] [Green Version]
- Suklabaidya, S.; Dash, P.; Das, B.; Suresh, V.; Sasmal, P.K.; Senapati, S. Experimental models of pancreatic cancer desmoplasia. Lab. Investig. 2018, 98, 27–40. [Google Scholar] [CrossRef]
- von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.; Cui, M.; Yang, G.; Wang, H.; Feng, M.; You, L.; Zhao, Y. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol. Cancer 2018, 17, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soundararajan, M.; Kannan, S. Fibroblasts and mesenchymal stem cells: Two sides of the same coin? J. Cell Physiol. 2018, 233, 9099–9109. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. Caf subpopulations: A new reservoir of stromal targets in pancreatic cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. Tgfbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bae, J.S. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [Green Version]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the tumor stroma for treatment of pancreatic cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, B.; Liao, Q.; Niu, Z.; Zhou, L.; Zhao, Y. Cancer-associated fibroblasts in pancreatic adenocarcinoma. Future Oncol. 2015, 11, 2603–2610. [Google Scholar] [CrossRef]
- Allam, A.; Thomsen, A.R.; Gothwal, M.; Saha, D.; Maurer, J.; Brunner, T.B. Pancreatic stellate cells in pancreatic cancer: In focus. Pancreatology 2017, 17, 514–522. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, B.; Li, W.; Zhou, J.; Gao, F.; Wang, X.; Cai, M.; Sun, Z. Pancreatic stellate cells: A rising translational physiology star as a potential stem cell type for beta cell neogenesis. Front. Physiol. 2019, 10, 218. [Google Scholar] [CrossRef]
- Jin, G.; Hong, W.; Guo, Y.; Bai, Y.; Chen, B. Molecular mechanism of pancreatic stellate cells activation in chronic pancreatitis and pancreatic cancer. J. Cancer 2020, 11, 1505–1515. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Kim, J.W.; Park, H.S.; Lee, E.Y.; Yoon, K.H. Pancreatic stellate cells in the islets as a novel target to preserve the pancreatic beta-cell mass and function. J. Diabetes Investig. 2020, 11, 268–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bynigeri, R.R.; Jakkampudi, A.; Jangala, R.; Subramanyam, C.; Sasikala, M.; Rao, G.V.; Reddy, D.N.; Talukdar, R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J. Gastroenterol. 2017, 23, 382–405. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Garcea, G.; Neal, C.P.; Pattenden, C.J.; Steward, W.P.; Berry, D.P. Molecular prognostic markers in pancreatic cancer: A systematic review. Eur J. Cancer 2005, 41, 2213–2236. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Ohlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ecm during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlund, D.; Lundin, C.; Ardnor, B.; Oman, M.; Naredi, P.; Sund, M. Type iv collagen is a tumour stroma-derived biomarker for pancreas cancer. Br. J. Cancer 2009, 101, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drifka, C.R.; Loeffler, A.G.; Mathewson, K.; Keikhosravi, A.; Eickhoff, J.C.; Liu, Y.; Weber, S.M.; Kao, W.J.; Eliceiri, K.W. Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection. Oncotarget 2016, 7, 76197–76213. [Google Scholar] [CrossRef] [Green Version]
- Biancur, D.E.; Kimmelman, A.C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 67–75. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Shah, V.M.; Sheppard, B.C.; Sears, R.C.; Alani, A.W. Hypoxia: Friend or foe for drug delivery in pancreatic cancer. Cancer Lett. 2020, 492, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef]
- Sorensen, B.S.; Horsman, M.R. Tumor hypoxia: Impact on radiation therapy and molecular pathways. Front. Oncol. 2020, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef] [Green Version]
- Riess, J.G. Understanding the fundamentals of perfluorocarbons and perfluorocarbon emulsions relevant to in vivo oxygen delivery. Artif. Cells Blood Substit. Immobil. Biotechnol. 2005, 33, 47–63. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a kras-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Sharma, N.S.; Gupta, V.K.; Garrido, V.T.; Hadad, R.; Durden, B.C.; Kesh, K.; Giri, B.; Ferrantella, A.; Dudeja, V.; Saluja, A.; et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-pd1 therapy. J. Clin. Investig. 2020, 130, 451–465. [Google Scholar] [CrossRef] [Green Version]
- New, M.; Tooze, S. The role of autophagy in pancreatic cancer-recent advances. Biology 2019, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Lo Re, A.E.; Fernandez-Barrena, M.G.; Almada, L.L.; Mills, L.D.; Elsawa, S.F.; Lund, G.; Ropolo, A.; Molejon, M.I.; Vaccaro, M.I.; Fernandez-Zapico, M.E. Novel akt1-gli3-vmp1 pathway mediates kras oncogene-induced autophagy in cancer cells. J. Biol. Chem. 2012, 287, 25325–25334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgulu, K.; Diakopoulos, K.N.; Kaya-Aksoy, E.; Ciecielski, K.J.; Ai, J.; Lesina, M.; Algul, H. The role of autophagy in pancreatic cancer: From bench to the dark bedside. Cells 2020, 9, 1063. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Mace, T.A.; Bloomston, M.; Lesinski, G.B. Pancreatic cancer-associated stellate cells: A viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunology 2013, 2, e24891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.H.; Hong, T.M.; Cheng, H.W.; Pan, S.H.; Liang, Y.R.; Hong, H.C.; Chiang, W.F.; Wong, T.Y.; Shieh, D.B.; Shiau, A.L.; et al. Galectin-1-mediated tumor invasion and metastasis, up-regulated matrix metalloproteinase expression, and reorganized actin cytoskeletons. Mol. Cancer Res. 2009, 7, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Yuan, Z.; Xue, X.; Lu, Z.; Zhang, Y.; Wang, H.; Chen, M.; An, Y.; Wei, J.; Zhu, Y.; et al. High expression of galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int. J. Cancer 2012, 130, 2337–2348. [Google Scholar] [CrossRef]
- Looi, C.K.; Chung, F.F.; Leong, C.O.; Wong, S.F.; Rosli, R.; Mai, C.W. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 162. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester cd8+ t cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and pancreatic cancer: Focus on metabolism, cytokines, and immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [Green Version]
- Tantau, A.; Leucuta, D.C.; Tantau, M.; Botan, E.; Zaharie, R.; Mandrutiu, A.; Tomuleasa, I.C. Inflammation, tumoral markers and interleukin-17, -10, and -6 profiles in pancreatic adenocarcinoma and chronic pancreatitis. Dig. Dis. Sci. 2020. [Google Scholar]
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, 102655. [Google Scholar] [CrossRef] [PubMed]
- Saka, D.; Gokalp, M.; Piyade, B.; Cevik, N.C.; Arik Sever, E.; Unutmaz, D.; Ceyhan, G.O.; Demir, I.E.; Asimgil, H. Mechanisms of t-cell exhaustion in pancreatic cancer. Cancers 2020, 12, 2274. [Google Scholar] [CrossRef]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef]
- Farajzadeh Valilou, S.; Keshavarz-Fathi, M.; Silvestris, N.; Argentiero, A.; Rezaei, N. The role of inflammatory cytokines and tumor associated macrophages (tams) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev. 2018, 39, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Zambirinis, C.P.; Seifert, L.; Akkad, N.; Mohan, N.; Werba, G.; Barilla, R.; Torres-Hernandez, A.; Hundeyin, M.; Mani, V.R.K.; et al. Gammadelta t cells support pancreatic oncogenesis by restraining alphabeta t cell activation. Cell 2016, 166, 1485–1499.e1415. [Google Scholar] [CrossRef] [Green Version]
- Seifert, A.M.; List, J.; Heiduk, M.; Decker, R.; von Renesse, J.; Meinecke, A.C.; Aust, D.E.; Welsch, T.; Weitz, J.; Seifert, L. Gamma-delta t cells stimulate il-6 production by pancreatic stellate cells in pancreatic ductal adenocarcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 3233–3240. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012, 2, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Ferrone, C.R. Neoadjuvant Therapy for Resectable Pancreatic Cancer: An Evolving Paradigm Shift. Front. Oncol. 2019, 9, 1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Amanam, I.; Chung, V. Current and future therapies for advanced pancreatic cancer. J. Surg. Oncol. 2017, 116, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Suker, M.; Beumer, B.R.; Sadot, E.; Marthey, L.; Faris, J.E.; Mellon, E.A.; El-Rayes, B.F.; Wang-Gillam, A.; Lacy, J.; Hosein, P.J.; et al. FOLFIRINOX for locally advanced pancreatic cancer: A systematic review and patient-level meta-analysis. Lancet Oncol. 2016, 17, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Byrne, J.D.; Jajja, M.R.N.; O’Neill, A.T.; Schorzman, A.N.; Keeler, A.W.; Luft, J.C.; Zamboni, W.C.; DeSimone, J.M.; Yeh, J.J. Impact of formulation on the iontophoretic delivery of the FOLFIRINOX regimen for the treatment of pancreatic cancer. Cancer Chemother. Pharmacol. 2018, 81, 991–998. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Boker, V.; Kleeff, J. Targeting and reprograming cancer-associated fibroblasts and the tumor microenvironment in pancreatic cancer. Cancers 2021, 13, 697. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim Biophys Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic cancer associated fibroblasts (caf): Under-explored target for pancreatic cancer treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Huang, H.; Brekken, R.A. Recent advances in understanding cancer-associated fibroblasts in pancreatic cancer. Am. J. Physiol. Cell Physiol. 2020, 319, C233–c243. [Google Scholar] [CrossRef]
- Blaine, S.A.; Ray, K.C.; Branch, K.M.; Robinson, P.S.; Whitehead, R.H.; Means, A.L. Epidermal growth factor receptor regulates pancreatic fibrosis. Am. J. Physiol. Gastrointest Liver Physiol. 2009, 297, G434–G441. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [Green Version]
- Pitarresi, J.R.; Liu, X.; Avendano, A.; Thies, K.A.; Sizemore, G.M.; Hammer, A.M.; Hildreth, B.E., 3rd; Wang, D.J.; Steck, S.A.; Donohue, S.; et al. Disruption of stromal hedgehog signaling initiates rnf5-mediated proteasomal degradation of pten and accelerates pancreatic tumor growth. Life Sci. Alliance 2018, 1, e201800190. [Google Scholar] [CrossRef] [Green Version]
- Reinehr, R.; Zoller, S.; Klonowski-Stumpe, H.; Kordes, C.; Haussinger, D. Effects of angiotensin ii on rat pancreatic stellate cells. Pancreas 2004, 28, 129–137. [Google Scholar] [CrossRef]
- Masamune, A.; Hamada, S.; Kikuta, K.; Takikawa, T.; Miura, S.; Nakano, E.; Shimosegawa, T. The angiotensin ii type i receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand. J. Gastroenterol. 2013, 48, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Loh, W.M.; Gopinath, S.C.B.; Bonam, S.R.; Fareez, I.M.; Mac Guad, R.; Sim, M.S.; Wu, Y.S. Selective phytochemicals targeting pancreatic stellate cells as new anti- fibrotic agents for chronic pancreatitis and pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 399–413. [Google Scholar] [CrossRef]
- Elechalawar, C.K.; Hossen, M.N.; Shankarappa, P.; Peer, C.J.; Figg, W.D.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Targeting pancreatic cancer cells and stellate cells using designer nanotherapeutics in vitro. Int. J. Nanomed. 2020, 15, 991–1003. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, E.F. The toxins of william b. Coley and the treatment of bone and soft-tissue sarcomas. Iowa. Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Caswell, C.C.; Oliver-Kozup, H.; Han, R.; Lukomska, E.; Lukomski, S. Scl1, the multifunctional adhesin of group a streptococcus, selectively binds cellular fibronectin and laminin, and mediates pathogen internalization by human cells. FEMS Microbiol. Lett. 2010, 303, 61–68. [Google Scholar] [CrossRef] [Green Version]
- McNitt, D.H.; Choi, S.J.; Keene, D.R.; Van De Water, L.; Squeglia, F.; Berisio, R.; Lukomski, S. Surface-exposed loops and an acidic patch in the scl1 protein of group a streptococcus enable scl1 binding to wound-associated fibronectin. J. Biol. Chem. 2018, 293, 7796–7810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNitt, D.H.; Choi, S.J.; Allen, J.L.; Hames, R.A.; Weed, S.A.; Van De Water, L.; Berisio, R.; Lukomski, S. Adaptation of the group a streptococcus adhesin scl1 to bind fibronectin type iii repeats within wound-associated extracellular matrix: Implications for cancer therapy. Mol. Microbiol. 2019, 112, 800–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNitt, D.H.; Van De Water, L.; Marasco, D.; Berisio, R.; Lukomski, S. Streptococcal collagen-like protein 1 binds wound fibronectin: Implications in pathogen targeting. Curr. Med. Chem. 2019, 26, 1933–1945. [Google Scholar] [CrossRef]
- Oliver-Kozup, H.; Martin, K.H.; Schwegler-Berry, D.; Green, B.J.; Betts, C.; Shinde, A.V.; Van De Water, L.; Lukomski, S. The group a streptococcal collagen-like protein-1, scl1, mediates biofilm formation by targeting the extra domain a-containing variant of cellular fibronectin expressed in wounded tissue. Mol. Microbiol. 2013, 87, 672–689. [Google Scholar] [CrossRef] [Green Version]
- Oliver-Kozup, H.A.; Elliott, M.; Bachert, B.A.; Martin, K.H.; Reid, S.D.; Schwegler-Berry, D.E.; Green, B.J.; Lukomski, S. The streptococcal collagen-like protein-1 (scl1) is a significant determinant for biofilm formation by group a streptococcus. BMC Microbiol. 2011, 11, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachert, B.A.; Choi, S.J.; LaSala, P.R.; Harper, T.I.; McNitt, D.H.; Boehm, D.T.; Caswell, C.C.; Ciborowski, P.; Keene, D.R.; Flores, A.R.; et al. Unique footprint in the scl1.3 locus affects adhesion and biofilm formation of the invasive m3-type group a streptococcus. Front. Cell Infect. Microbiol. 2016, 6, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, S.; Veracini, L.; Grall, D.; Butori, C.; Schaub, S.; Audebert, S.; Camoin, L.; Baudelet, E.; Radwanska, A.; Beghelli-de la Forest Divonne, S.; et al. Fibronectin-guided migration of carcinoma collectives. Nat. Commun. 2017, 8, 14105. [Google Scholar] [CrossRef]
- Astrof, S.; Crowley, D.; George, E.L.; Fukuda, T.; Sekiguchi, K.; Hanahan, D.; Hynes, R.O. Direct test of potential roles of eiiia and eiiib alternatively spliced segments of fibronectin in physiological and tumor angiogenesis. Mol. Cell Biol. 2004, 24, 8662–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumra, H.; Reinhardt, D.P. Fibronectin-targeted drug delivery in cancer. Adv. Drug Deliv. Rev. 2016, 97, 101–110. [Google Scholar] [CrossRef]
- Han, Z.; Zhang, S.; Fujiwara, K.; Zhang, J.; Li, Y.; Liu, J.; van Zijl, P.C.M.; Lu, Z.R.; Zheng, L.; Liu, G. Extradomain-b fibronectin-targeted dextran-based chemical exchange saturation transfer magnetic resonance imaging probe for detecting pancreatic cancer. Bioconjug. Chem. 2019, 30, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Qiao, P.; Ayat, N.R.; Vaidya, A.; Gao, S.; Sun, W.; Chou, S.; Han, Z.; Gilmore, H.; Winter, J.M.; Lu, Z.R. Magnetic resonance molecular imaging of extradomain b fibronectin improves imaging of pancreatic cancer tumor xenografts. Front. Oncol. 2020, 10, 586727. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.K.; Du, W.X.; Li, R.G.; Yang, J.; Li, J.; Li, F.; Tan, H.B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Diegelmann, R.F.; Evans, M.C. Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. 2004, 9, 283–289. [Google Scholar] [CrossRef]
- Tazzyman, S.; Lewis, C.E.; Murdoch, C. Neutrophils: Key mediators of tumour angiogenesis. Int. J. Exp. Pathol. 2009, 90, 222–231. [Google Scholar] [CrossRef]
- Malech, H.L.; Deleo, F.R.; Quinn, M.T. The role of neutrophils in the immune system: An overview. Methods Mol. Biol. 2014, 1124, 3–10. [Google Scholar]
- Nemeth, T.; Sperandio, M.; Mocsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated cxcr2(+) neutrophils and ccr2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.T.; Zhao, Y.L.; Peng, L.S.; Chen, N.; Chen, W.; Lv, Y.P.; Mao, F.Y.; Zhang, J.Y.; Cheng, P.; Teng, Y.S.; et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through gm-csf-pd-l1 pathway. Gut 2017, 66, 1900–1911. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The role of tumor-associated neutrophils in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; Ferrara, N. The complex role of neutrophils in tumor angiogenesis and metastasis. Cancer Immunol. Res. 2016, 4, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, O.E.; Borregaard, N. Neutrophil extracellular traps-the dark side of neutrophils. J. Clin. Investig. 2016, 126, 1612–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta 2013, 1829, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Takesue, S.; Ohuchida, K.; Shinkawa, T.; Otsubo, Y.; Matsumoto, S.; Sagara, A.; Yonenaga, A.; Ando, Y.; Kibe, S.; Nakayama, H.; et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancerassociated fibroblasts. Int. J. Oncol. 2020, 56, 596–605. [Google Scholar]
- Miller-Ocuin, J.L.; Liang, X.; Boone, B.A.; Doerfler, W.R.; Singhi, A.D.; Tang, D.; Kang, R.; Lotze, M.T.; Zeh, H.J., 3rd. DNA released from neutrophil extracellular traps (nets) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology 2019, 8, e1605822. [Google Scholar] [CrossRef]
- Boone, B.A.; Orlichenko, L.; Schapiro, N.E.; Loughran, P.; Gianfrate, G.C.; Ellis, J.T.; Singhi, A.D.; Kang, R.; Tang, D.; Lotze, M.T.; et al. The receptor for advanced glycation end products (rage) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015, 22, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Xu, H.X.; Zhang, S.R.; Li, H.; Wang, W.Q.; Gao, H.L.; Wu, C.T.; Xu, J.Z.; Qi, Z.H.; Li, S.; et al. Tumor-infiltrating nets predict postsurgical survival in patients with pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2019, 26, 635–643. [Google Scholar] [CrossRef]
- Jung, H.S.; Gu, J.; Kim, J.E.; Nam, Y.; Song, J.W.; Kim, H.K. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS ONE 2019, 14, e0216055. [Google Scholar] [CrossRef] [Green Version]
- Hisada, Y.; Grover, S.P.; Maqsood, A.; Houston, R.; Ay, C.; Noubouossie, D.F.; Cooley, B.C.; Wallen, H.; Key, N.S.; Thalin, C.; et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica 2020, 105, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Boone, B.A.; Murthy, P.; Miller-Ocuin, J.; Doerfler, W.R.; Ellis, J.T.; Liang, X.; Ross, M.A.; Wallace, C.T.; Sperry, J.L.; Lotze, M.T.; et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer 2018, 18, 678. [Google Scholar] [CrossRef] [Green Version]
- Abdol Razak, N.; Elaskalani, O.; Metharom, P. Pancreatic cancer-induced neutrophil extracellular traps: A potential contributor to cancer-associated thrombosis. Int. J. Mol. Sci. 2017, 18, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajioka, H.; Kagawa, S.; Ito, A.; Yoshimoto, M.; Sakamoto, S.; Kikuchi, S.; Kuroda, S.; Yoshida, R.; Umeda, Y.; Noma, K.; et al. Targeting neutrophil extracellular traps with thrombomodulin prevents pancreatic cancer metastasis. Cancer Lett. 2021, 497, 1–13. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via ccdc25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e20190354. [Google Scholar] [CrossRef]
- Teijeira, A.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. Cxcr1 and cxcr2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity 2020, 52, 856–871.e858. [Google Scholar] [CrossRef]
- Zambirinis, C.P.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. Tlr9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Torezani, G.S.; Braga, C.A.; Palhano, F.L.; Kelly, J.W.; Saraiva, E.M.; Foguel, D. Amyloid fibrils trigger the release of neutrophil extracellular traps (nets), causing fibril fragmentation by net-associated elastase. J. Biol Chem. 2012, 287, 37206–37218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, H.; Jones, J.O.; Janowitz, T.; Hoffmann, M.; Euler, M.; Martins, C.P.; Welsh, S.J.; Shields, J.D. Stromal-driven and amyloid beta-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat. Commun. 2021, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Werner, S. The cornified envelope: A first line of defense against reactive oxygen species. J. Investig. Dermatol. 2011, 131, 1409–1411. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yuan, R.; Ren, T.; Yang, B.; Miao, H.; Liu, L.; Cai, C.; Yang, Y.; Hu, Y.; Jiang, C.; et al. Role of sciellin in gallbladder cancer proliferation and formation of neutrophil extracellular traps. Cell Death Dis. 2021, 12, 30. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, K.; Geng, L.; Sun, J.; Xu, W.; Liu, D.; Gong, S.; Zhu, Y. Identification of candidate diagnostic and prognostic biomarkers for pancreatic carcinoma. EBioMedicine 2019, 40, 382–393. [Google Scholar] [CrossRef] [Green Version]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. Netosis, complement, and coagulation: A triangular relationship. Cell Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef]
- Shrestha, B.; Ito, T.; Kakuuchi, M.; Totoki, T.; Nagasato, T.; Yamamoto, M.; Maruyama, I. Recombinant thrombomodulin suppresses histone-induced neutrophil extracellular trap formation. Front. Immunol. 2019, 10, 2535. [Google Scholar] [CrossRef]
- Helms, J.; Clere-Jehl, R.; Bianchini, E.; Le Borgne, P.; Burban, M.; Zobairi, F.; Diehl, J.L.; Grunebaum, L.; Toti, F.; Meziani, F.; et al. Thrombomodulin favors leukocyte microvesicle fibrinolytic activity, reduces netosis and prevents septic shock-induced coagulopathy in rats. Ann. Intensive. Care 2017, 7, 118. [Google Scholar] [CrossRef]
- Liu, S.; Su, X.; Pan, P.; Zhang, L.; Hu, Y.; Tan, H.; Wu, D.; Liu, B.; Li, H.; Li, Y.; et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci. Rep. 2016, 6, 37252. [Google Scholar] [CrossRef] [Green Version]
- Lefrancais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3, e98178. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Coagulopathy and thromboembolic events in patients with sars-cov-2 infection: Pathogenesis and management strategies. Ann. Hematol. 2020, 99, 1953–1965. [Google Scholar] [CrossRef]
- Park, H.H.; Park, W.; Lee, Y.Y.; Kim, H.; Seo, H.S.; Choi, D.W.; Kwon, H.K.; Na, D.H.; Kim, T.H.; Choy, Y.B.; et al. Bioinspired dnase-i-coated melanin-like nanospheres for modulation of infection-associated netosis dysregulation. Adv. Sci. (Weinh.) 2020, 7, 2001940. [Google Scholar] [CrossRef]
- Xia, Y.; He, J.; Zhang, H.; Wang, H.; Tetz, G.; Maguire, C.A.; Wang, Y.; Onuma, A.; Genkin, D.; Tetz, V.; et al. Aav-mediated gene transfer of dnase i in the liver of mice with colorectal cancer reduces liver metastasis and restores local innate and adaptive immune response. Mol. Oncol. 2020, 14, 2920–2935. [Google Scholar] [CrossRef]
- Smith, C.K.; Vivekanandan-Giri, A.; Tang, C.; Knight, J.S.; Mathew, A.; Padilla, R.L.; Gillespie, B.W.; Carmona-Rivera, C.; Liu, X.; Subramanian, V.; et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: An additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis. Rheumatol. 2014, 66, 2532–2544. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Ikari, J.; Anazawa, R.; Tanaka, N.; Katsumata, Y.; Shimada, A.; Suzuki, E.; Tatsumi, K. Pad4 deficiency improves bleomycin-induced neutrophil extracellular traps and fibrosis in mouse lung. Am. J. Respir. Cell Mol. Biol. 2020, 63, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Franken, L.; Schiwon, M.; Kurts, C. Macrophages: Sentinels and regulators of the immune system. Cell Microbiol. 2016, 18, 475–487. [Google Scholar] [CrossRef]
- van Dalen, F.J.; van Stevendaal, M.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular repolarisation of tumour-associated macrophages. Molecules 2018, 24, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, M.; Rees, A.; Cronin, J.G.; Nair, M.; Jones, N.; Thornton, C.A. Macrophage plasticity in reproduction and environmental influences on their function. Front. Immunol. 2020, 11, 607328. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef]
- Malekghasemi, S.; Majidi, J.; Baghbanzadeh, A.; Abdolalizadeh, J.; Baradaran, B.; Aghebati-Maleki, L. Tumor-associated macrophages: Protumoral macrophages in inflammatory tumor microenvironment. Adv. Pharm. Bull. 2020, 10, 556–565. [Google Scholar] [CrossRef]
- Pandol, S.J.; Edderkaoui, M. What are the macrophages and stellate cells doing in pancreatic adenocarcinoma? Front. Physiol. 2015, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Washington, M.K.; Chaturvedi, R.; Drosos, Y.; Revetta, F.L.; Weaver, C.J.; Buzhardt, E.; Yull, F.E.; Blackwell, T.S.; Sosa-Pineda, B.; et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab. Investig. 2014, 94, 409–421. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 2017, 47, 323–338.e326. [Google Scholar] [CrossRef]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. Taming pancreatic cancer: Combat with a double edged sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef] [Green Version]
- Tekin, C.; Aberson, H.L.; Waasdorp, C.; Hooijer, G.K.J.; de Boer, O.J.; Dijk, F.; Bijlsma, M.F.; Spek, C.A. Macrophage-secreted mmp9 induces mesenchymal transition in pancreatic cancer cells via par1 activation. Cell Oncol. (Dordr.) 2020, 43, 1161–1174. [Google Scholar] [CrossRef]
- Tandon, M.; Coudriet, G.M.; Criscimanna, A.; Socorro, M.; Eliliwi, M.; Singhi, A.D.; Cruz-Monserrate, Z.; Bailey, P.; Lotze, M.T.; Zeh, H.; et al. Prolactin promotes fibrosis and pancreatic cancer progression. Cancer Res. 2019, 79, 5316–5327. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage pi3kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ucero, A.C.; Bakiri, L.; Roediger, B.; Suzuki, M.; Jimenez, M.; Mandal, P.; Braghetta, P.; Bonaldo, P.; Paz-Ares, L.; Fustero-Torre, C.; et al. Fra-2-expressing macrophages promote lung fibrosis in mice. J. Clin. Investig. 2019, 129, 3293–3309. [Google Scholar] [CrossRef]
- Venneri, M.A.; De Palma, M.; Ponzoni, M.; Pucci, F.; Scielzo, C.; Zonari, E.; Mazzieri, R.; Doglioni, C.; Naldini, L. Identification of proangiogenic tie2-expressing monocytes (tems) in human peripheral blood and cancer. Blood 2007, 109, 5276–5285. [Google Scholar] [CrossRef] [Green Version]
- Pucci, F.; Venneri, M.A.; Biziato, D.; Nonis, A.; Moi, D.; Sica, A.; Di Serio, C.; Naldini, L.; De Palma, M. A distinguishing gene signature shared by tumor-infiltrating tie2-expressing monocytes, blood "resident" monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 2009, 114, 901–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.E.; De Palma, M.; Naldini, L. Tie2-expressing monocytes and tumor angiogenesis: Regulation by hypoxia and angiopoietin-2. Cancer Res. 2007, 67, 8429–8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.J.; Hao, Y.X.; Yang, X.; Fu, X.L.; Shi, Y.; Yue, H.L.; Yin, P.; Dong, H.L.; Yu, P.W. Overexpression of tie2 is associated with poor prognosis in patients with gastric cancer. Oncol. Lett. 2018, 15, 8027–8033. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, G.; Potner, C.; Aust, G.; Schierle, K.; Dietel, C.; Benzing, C.; Krenzien, F.; Bartels, M.; Eichfeld, U.; Schmelzle, M.; et al. Tie2-expressing monocytes and m2-polarized macrophages impact survival and correlate with angiogenesis in adenocarcinoma of the pancreas. Oncotarget 2018, 9, 29715–29726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.B.; Tal, A.O.; Scholz, A.; De Palma, M.; Patel, S.; Urbich, C.; Biswas, S.K.; Murdoch, C.; Plate, K.H.; Reiss, Y.; et al. Angiopoietin-2 regulates gene expression in tie2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010, 70, 5270–5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habtezion, A.; Edderkaoui, M.; Pandol, S.J. Macrophages and pancreatic ductal adenocarcinoma. Cancer Lett. 2016, 381, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, J.; Jenmalm, M.C.; Matussek, A.; Geffers, R.; Berg, G.; Ernerudh, J. Macrophages at the fetal-maternal interface express markers of alternative activation and are induced by m-csf and il-10. J. Immunol. 2011, 187, 3671–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Calderon, B.; Carrero, J.A.; Ferris, S.T.; Sojka, D.K.; Moore, L.; Epelman, S.; Murphy, K.M.; Yokoyama, W.M.; Randolph, G.J.; Unanue, E.R. The pancreas anatomy conditions the origin and properties of resident macrophages. J. Exp. Med. 2015, 212, 1497–1512. [Google Scholar] [CrossRef] [Green Version]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Wang, N.; Wang, S.; Wang, X.; Zheng, Y.; Yang, B.; Zhang, J.; Pan, B.; Gao, J.; Wang, Z. Research trends in pharmacological modulation of tumor-associated macrophages. Clin. Transl. Med. 2021, 11, e288. [Google Scholar]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the ccl2/ccr2 axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with ccr2 inhibition in combination with folfirinox in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose- finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Five Prime Therapeutics Provides Update on Phase 2 Trial of Cabiralizumab Combined with Opdivo® in Pancreatic Cancer. 2020. Available online: https://www.businesswire.com/news/home/20200218005144/en/Five-Prime-Therapeutics-Provides-Update-on-Phase-2-Trial-of-Cabiralizumab-Combined-with-Opdivo%C2%AE-in-Pancreatic-Cancer (accessed on 16 June 2021).
- Razak, A.R.; Cleary, J.M.; Moreno, V.; Boyer, M.; Calvo Aller, E.; Edenfield, W.; Tie, J.; Harvey, R.D.; Rutten, A.; Shah, M.A.; et al. Safety and efficacy of amg 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J. Immunother Cancer 2020, 8, e001006. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact. Mater. 2021, 6, 1973–1987. [Google Scholar] [CrossRef]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro- inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, B.; Wu, L.; Xiao, H.; Ding, K.; Zheng, C.; Song, Q.; Sun, L.; Wang, L.; Zhang, Z. Amplified cancer immunotherapy of a surface-engineered antigenic microparticle vaccine by synergistically modulating tumor microenvironment. ACS Nano 2019, 13, 12553–12566. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Li, C.; Chu, Y.; et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials 2021, 268, 120546. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Brodowicz, T. Trabectedin in soft tissue sarcomas. Future Oncol. 2014, 10, s1–s5. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, K.; Igarashi, K.; Murakami, T.; Kiyuna, T.; Lwin, T.M.; Hwang, H.K.; Delong, J.C.; Clary, B.M.; Bouvet, M.; Unno, M.; et al. Mek inhibitors cobimetinib and trametinib, regressed a gemcitabine- resistant pancreatic-cancer patient-derived orthotopic xenograft (pdox). Oncotarget 2017, 8, 47490–47496. [Google Scholar] [CrossRef] [Green Version]
- Borgoni, S.; Iannello, A.; Cutrupi, S.; Allavena, P.; D’Incalci, M.; Novelli, F.; Cappello, P. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating t cells and restores their anti-tumor phenotype. Oncoimmunology 2018, 7, e1393596. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.; Patel, S.R. Chemotherapy for soft tissue sarcoma. Cancer 2016, 122, 2952–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belli, C.; Piemonti, L.; D’Incalci, M.; Zucchetti, M.; Porcu, L.; Cappio, S.; Doglioni, C.; Allavena, P.; Ceraulo, D.; Maggiora, P.; et al. Phase ii trial of salvage therapy with trabectedin in metastatic pancreatic adenocarcinoma. Cancer Chemother. Pharmacol. 2016, 77, 477–484. [Google Scholar] [CrossRef]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. Ifnγ and ccl2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef] [Green Version]
- Mardhian, D.F.; Storm, G.; Bansal, R.; Prakash, J. Nano-targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo. J. Control. Release 2018, 290, 1–10. [Google Scholar] [CrossRef]
- Hu, M.; Wang, Y.; Xu, L.; An, S.; Tang, Y.; Zhou, X.; Li, J.; Liu, R.; Huang, L. Relaxin gene delivery mitigates liver metastasis and synergizes with check point therapy. Nat. Commun. 2019, 10, 2993. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Y.; Hu, M.; Wang, M.; Liu, X.; Huang, L. Relaxin gene delivery modulates macrophages to resolve cancer fibrosis and synergizes with immune checkpoint blockade therapy. Sci. Adv. 2021, 7, eabb6596. [Google Scholar] [CrossRef] [PubMed]
- Mazzieri, R.; Pucci, F.; Moi, D.; Zonari, E.; Ranghetti, A.; Berti, A.; Politi, L.S.; Gentner, B.; Brown, J.L.; Naldini, L.; et al. Targeting the ang2/tie2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 2011, 19, 512–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, C.; Eichten, A.; Castanaro, C.; Pasnikowski, E.; Adler, A.; Lalani, A.S.; Papadopoulos, N.; Kyle, A.H.; Minchinton, A.I.; Yancopoulos, G.D.; et al. Angiopoietin-2 functions as a tie2 agonist in tumor models, where it limits the effects of vegf inhibition. Cancer Res. 2013, 73, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Lai, J.Y.; Do, J.; Liu, D.; Li, L.; Del Rosario, J.; Doppalapudi, V.R.; Pirie-Shepherd, S.; Levin, N.; Bradshaw, C.; et al. Specifically targeting angiopoietin-2 inhibits angiogenesis, tie2-expressing monocyte infiltration, and tumor growth. Clin. Cancer Res. 2011, 17, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Harney, A.S.; Karagiannis, G.S.; Pignatelli, J.; Smith, B.D.; Kadioglu, E.; Wise, S.C.; Hood, M.M.; Kaufman, M.D.; Leary, C.B.; Lu, W.P.; et al. The selective tie2 inhibitor rebastinib blocks recruitment and function of tie2(hi) macrophages in breast cancer and pancreatic neuroendocrine tumors. Mol. Cancer Ther. 2017, 16, 2486–2501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, S. T cells in fibrosis and fibrotic diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Contribution of regulatory t cells to cancer: A review. J. Cell Physiol 2019, 234, 7983–7993. [Google Scholar] [CrossRef] [PubMed]
- Nunoya, J.; Washburn, M.L.; Kovalev, G.I.; Su, L. Regulatory t cells prevent liver fibrosis during hiv type 1 infection in a humanized mouse model. J. Infect. Dis. 2014, 209, 1039–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; Satelli, A.; Yan, J.; Xueqing, X.; Gagea, M.; Hunter, C.A.; Mishra, L.; Li, S. Il-30 (il27p28) attenuates liver fibrosis through inducing nkg2d-rae1 interaction between nkt and activated hepatic stellate cells in mice. Hepatology 2014, 60, 2027–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine receptor cxcr6-dependent hepatic nk t cell accumulation promotes inflammation and liver fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.H.; Aloman, C. Dendritic cells and liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- An, Y.; Liu, F.; Chen, Y.; Yang, Q. Crosstalk between cancer-associated fibroblasts and immune cells in cancer. J. Cell Mol. Med. 2020, 24, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Zhou, L.; Lu, J.; Wang, Y.; Liu, C.; You, L.; Guo, J. Stroma-targeting therapy in pancreatic cancer: One coin with two sides? Front. Oncol. 2020, 10, 576399. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Therapeutic | Trial Phase | Trial Status | NCT Number |
|---|---|---|---|---|
| Disrupt CAF Signaling | Tocilizumab | 1b/2 | Recruiting | NCT03193190 |
| Tocilizumab | 2 | Recruiting | NCT02767557 | |
| Tocilizumab | 2 | Active | NCT04258150 | |
| Siltuximab | 1,2 | Recruiting | NCT04191421 | |
| Canakinumab | 1 | Recruiting | NCT04581343 | |
| Plerixafor | 2 | Recruiting | NCT04177810 | |
| Plerixafor | 1 | Completed | NCT02179970 | |
| BL-8040 | 2 | Active | NCT02826486 | |
| Reprogramming to Quiescence | ATRA 1 | 1 | Completed | NCT03307148 |
| ATRA 1 | 2 | Not yet recruiting | NCT04241276 | |
| Vitamin D3 | 3 | Recruiting | NCT03472833 | |
| Paricalcitrol | 2 | Completed | NCT03331562 | |
| Paricalcitrol | 1 | Recruiting | NCT03519308 | |
| Paricalcitrol | 2 | Recruiting | NCT04617067 | |
| Paricalcitrol | 1 | Active | NCT03883919 | |
| Paricalcitrol | 2 | Recruiting | NCT04524702 |
| Therapeutic | Trial Phase | Trail Status | Context | Trial ID |
|---|---|---|---|---|
| rhDNAse I | 1 | Recruiting | Severe SARS CoV-2 1 | NCT04409925 |
| 3 | Recruiting | Moderate to Severe ARDS 2 | NCT03368092 | |
| Danirixin | 2 | Terminated | COPD 3 | NCT03250689 |
| NucleoCapture Device | N/A | Recruiting | SA-AKI 4 | NCT04749238 |
| Target | Therapeutic | Trial Phase | Trial Status | Additional Interventions | Trial ID |
|---|---|---|---|---|---|
| CSF1-R | IMC-CS4 (LY3022855) | 1 | Recruiting | Cyclophosphamide, GVAX, Pembrolizumab | NCT03153410 |
| Cabiralizumab (FPA008) | 1a/1b | Completed | Nivolumab | NCT02526017 | |
| Cabiralizumab (FPA008) | 2 | Completed | Nivolumab +/− Chemotherapy | NCT03336216 | |
| Pexidartinib | 1 | Completed | Durvalumab | NCT02777710 | |
| CSF1 | MCS110 | 1b/2 | Completed | PDR001 | NCT02807844 |
| CCR2 | PF-04136309 | 1 | Completed | FOLFIRINOX | NCT01413022 |
| PF-04136309 | 1b | Completed | nab-paclitaxel and gemcitabine | NCT02732938 | |
| CCX872-B | 1 | Active | FOLFIRINOX | NCT02345408 | |
| CXCR4 | BL-8040 | 2b | Active | Pembrolizumab | NCT02907099 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, R.S.; Eubank, T.D.; Lukomski, S.; Boone, B.A. Immune Cell Modulation of the Extracellular Matrix Contributes to the Pathogenesis of Pancreatic Cancer. Biomolecules 2021, 11, 901. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060901
Ahmad RS, Eubank TD, Lukomski S, Boone BA. Immune Cell Modulation of the Extracellular Matrix Contributes to the Pathogenesis of Pancreatic Cancer. Biomolecules. 2021; 11(6):901. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060901
Chicago/Turabian StyleAhmad, Ramiz S., Timothy D. Eubank, Slawomir Lukomski, and Brian A. Boone. 2021. "Immune Cell Modulation of the Extracellular Matrix Contributes to the Pathogenesis of Pancreatic Cancer" Biomolecules 11, no. 6: 901. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060901