
The Right-Handed Parallel β-Helix Topology of Erwinia chrysanthemi Pectin Methylesterase Is Intimately Associated with Both Sequential Folding and Resistance to High Pressure †


Abstract
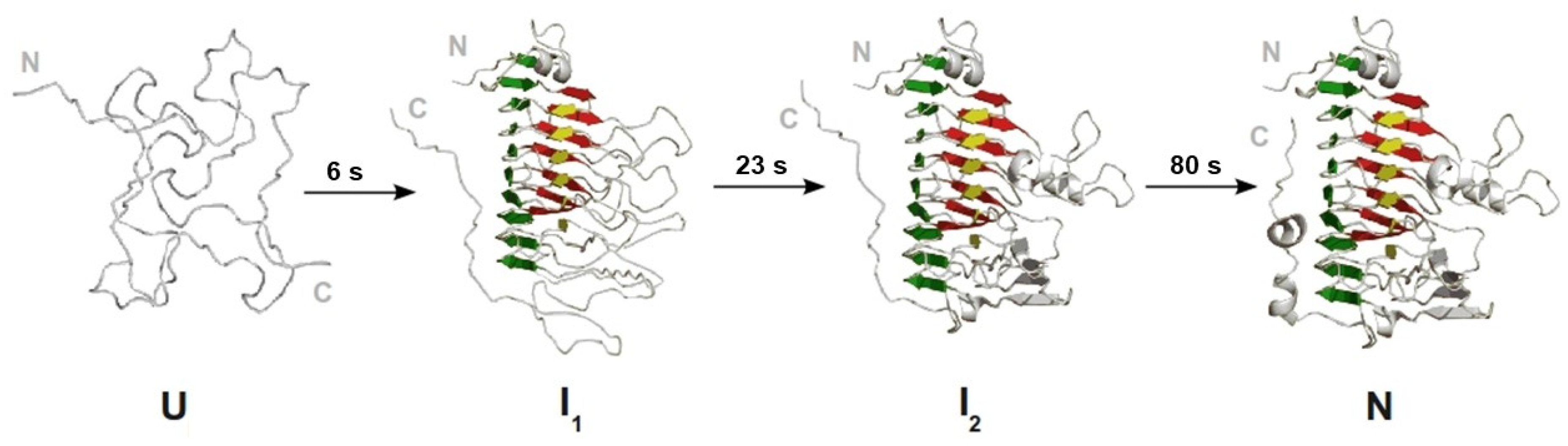
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Molecular Biology
2.3. Enzyme Expression and Purification
2.4. PemA Activity Assay
2.5. Circular Dichroism Measurements
2.6. Chemical-Induced Unfolding Transitions
2.7. Quenching of Intrinsic Fluorescence by Acrylamide
2.8. Pressure-Induced Unfolding
2.9. Kinetics of Unfolding and Refolding
2.10. Stopped-Flow Experiments
2.11. Thermodynamic Analysis
2.12. Kinetic Analysis
2.13. Analysis of the Folding Kinetics
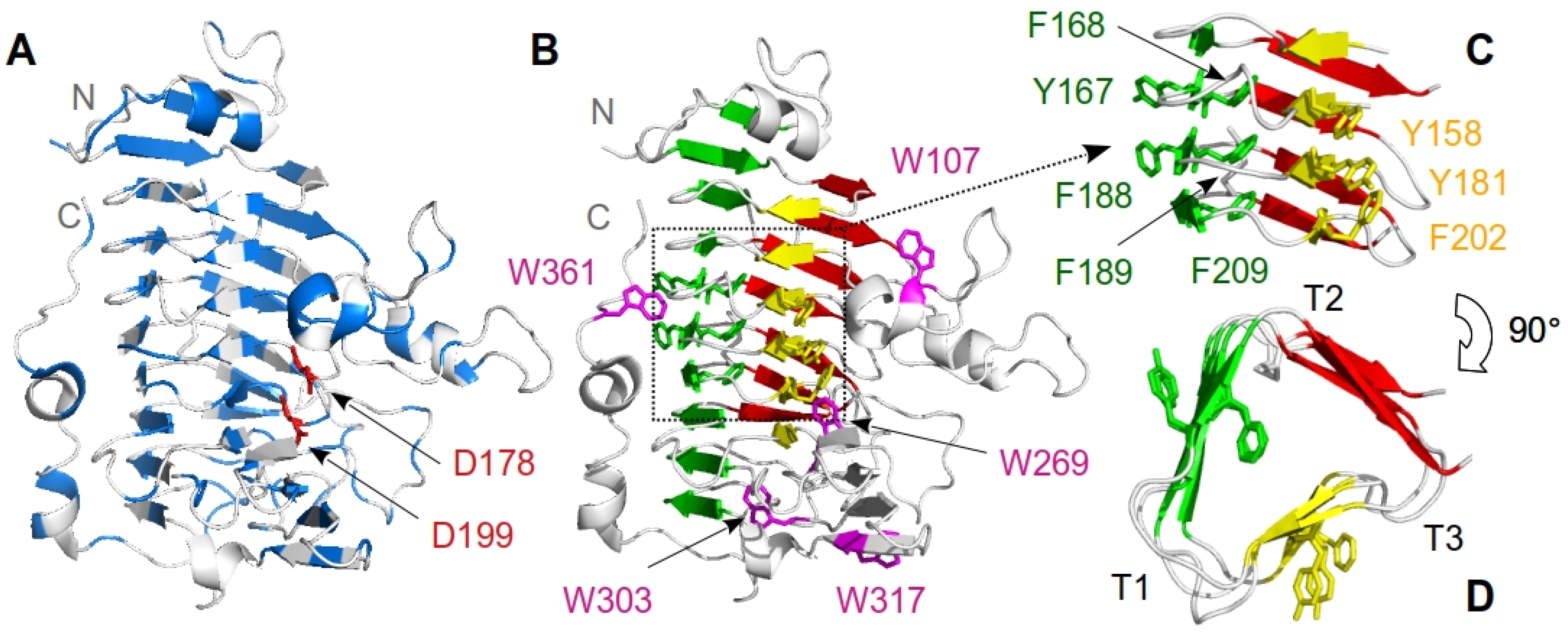
3. Results
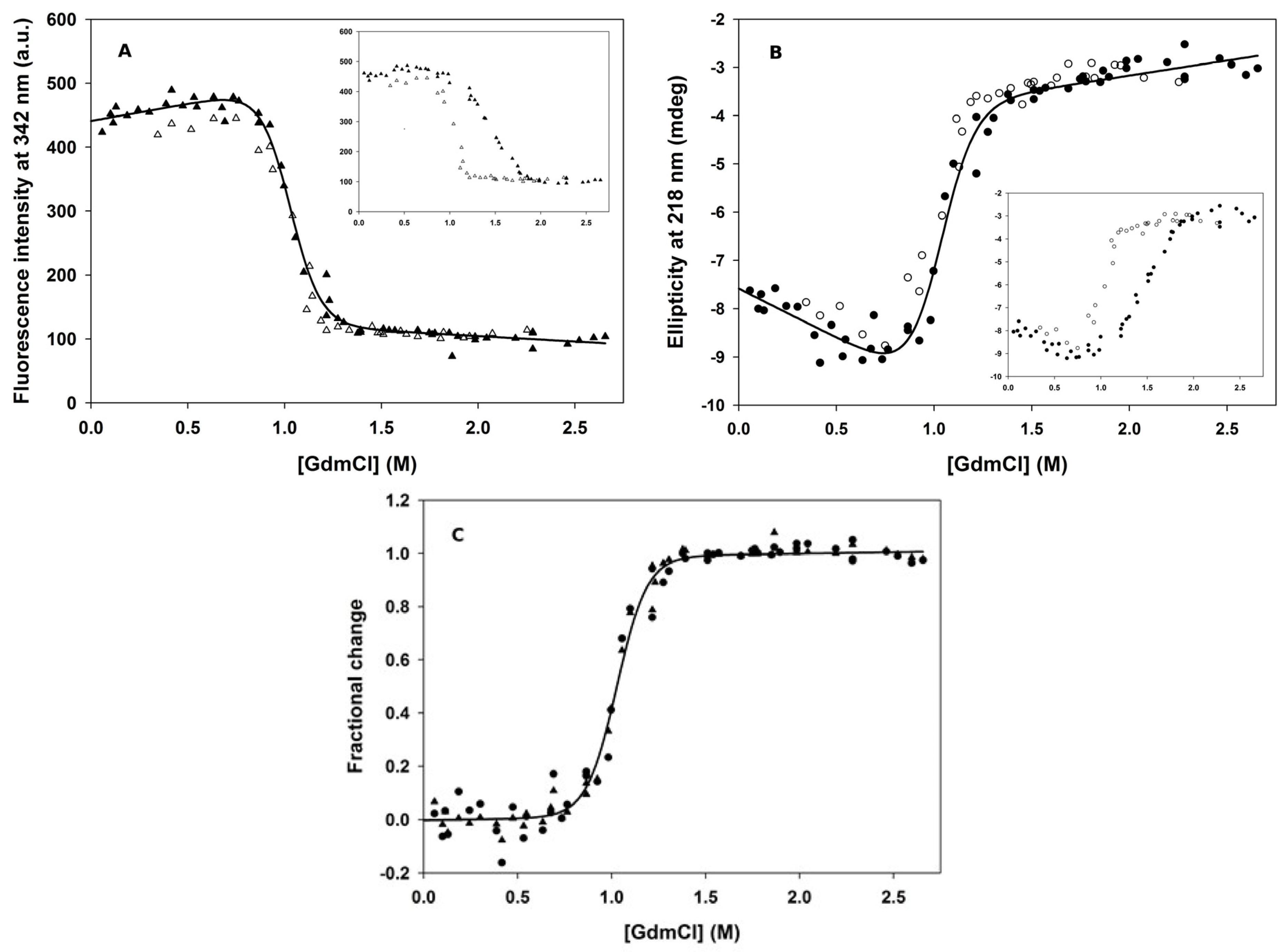
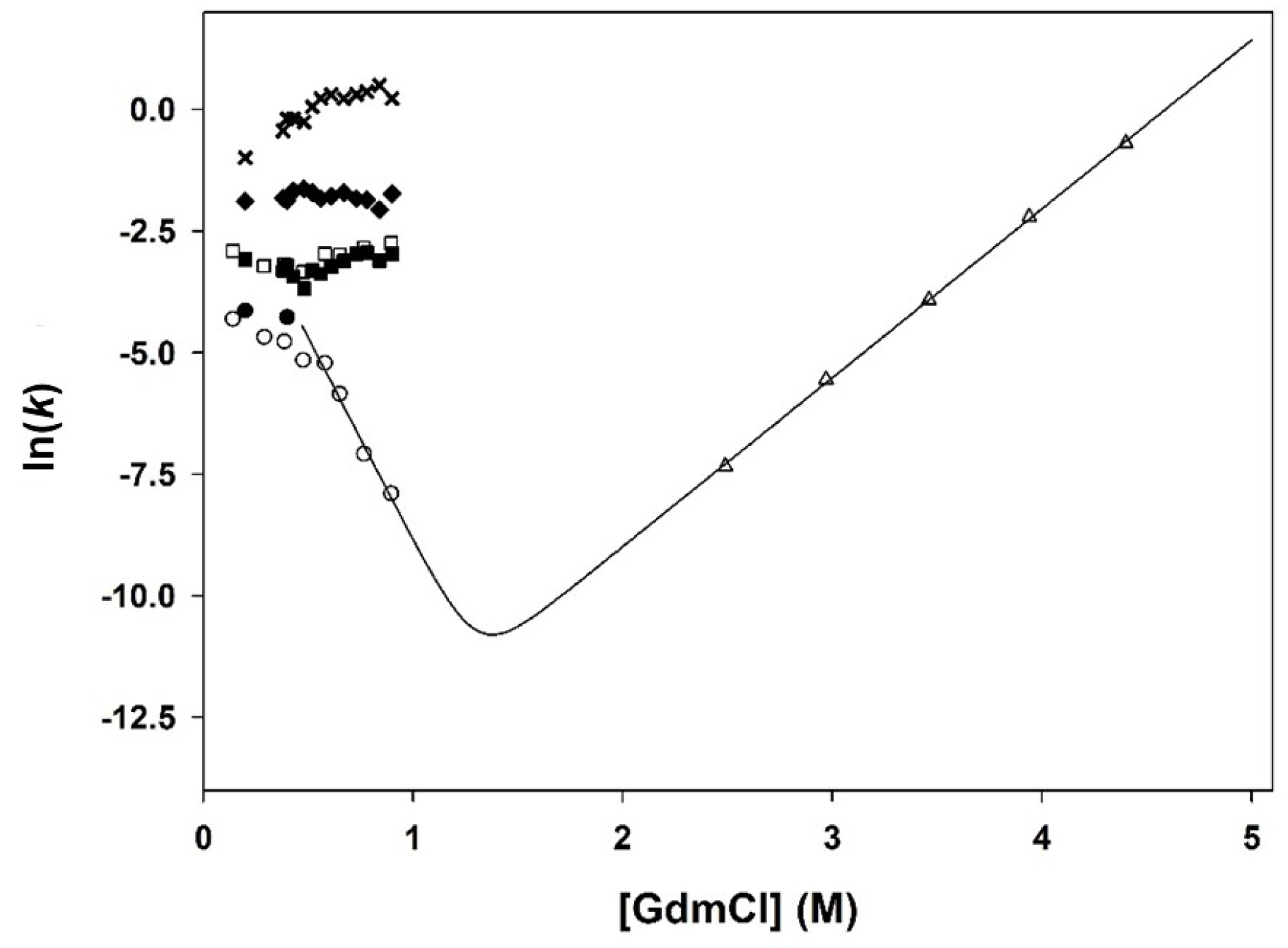
3.1. Chemical-Induced Unfolding
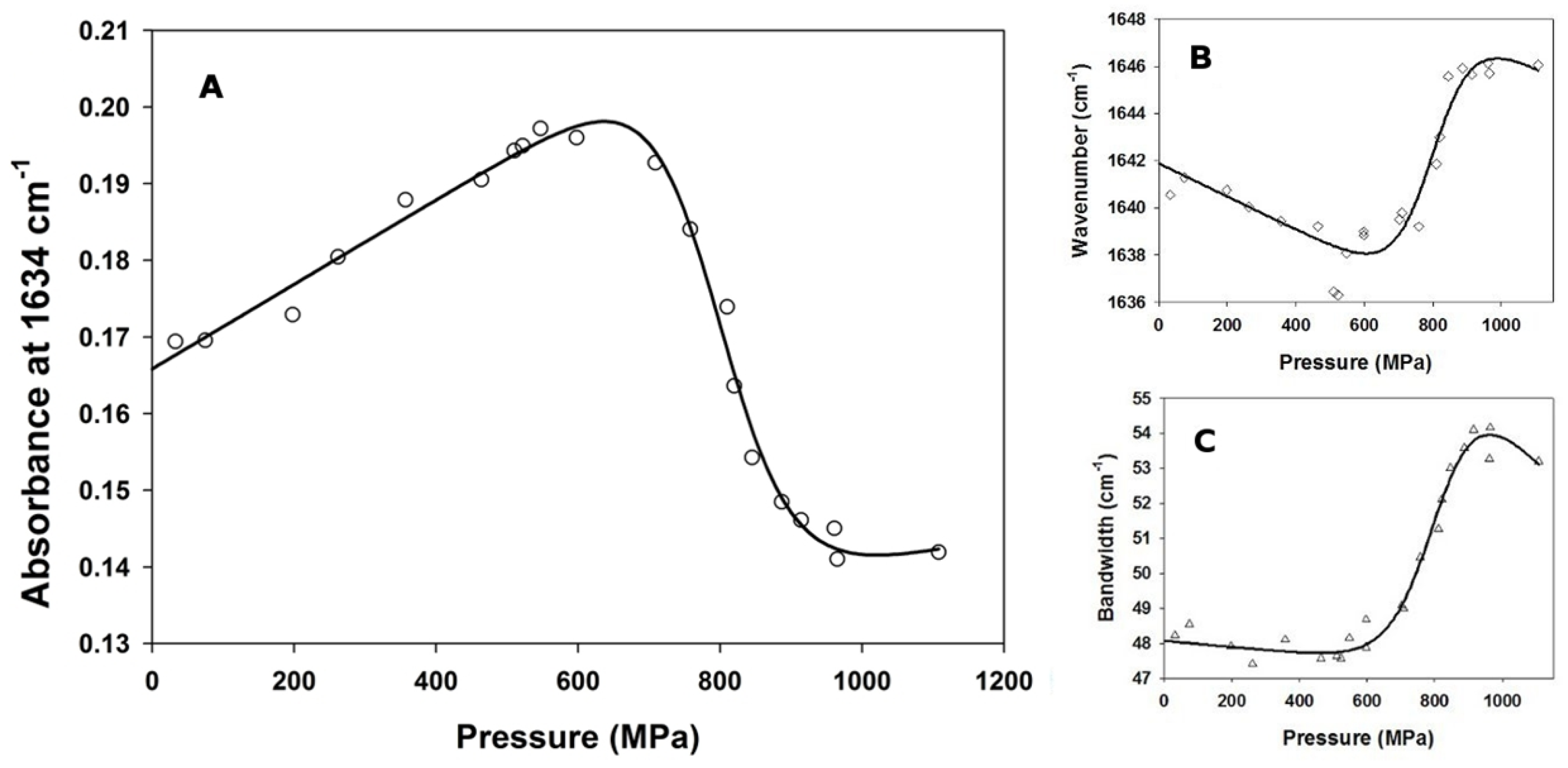
3.2. Pressure-Induced Unfolding
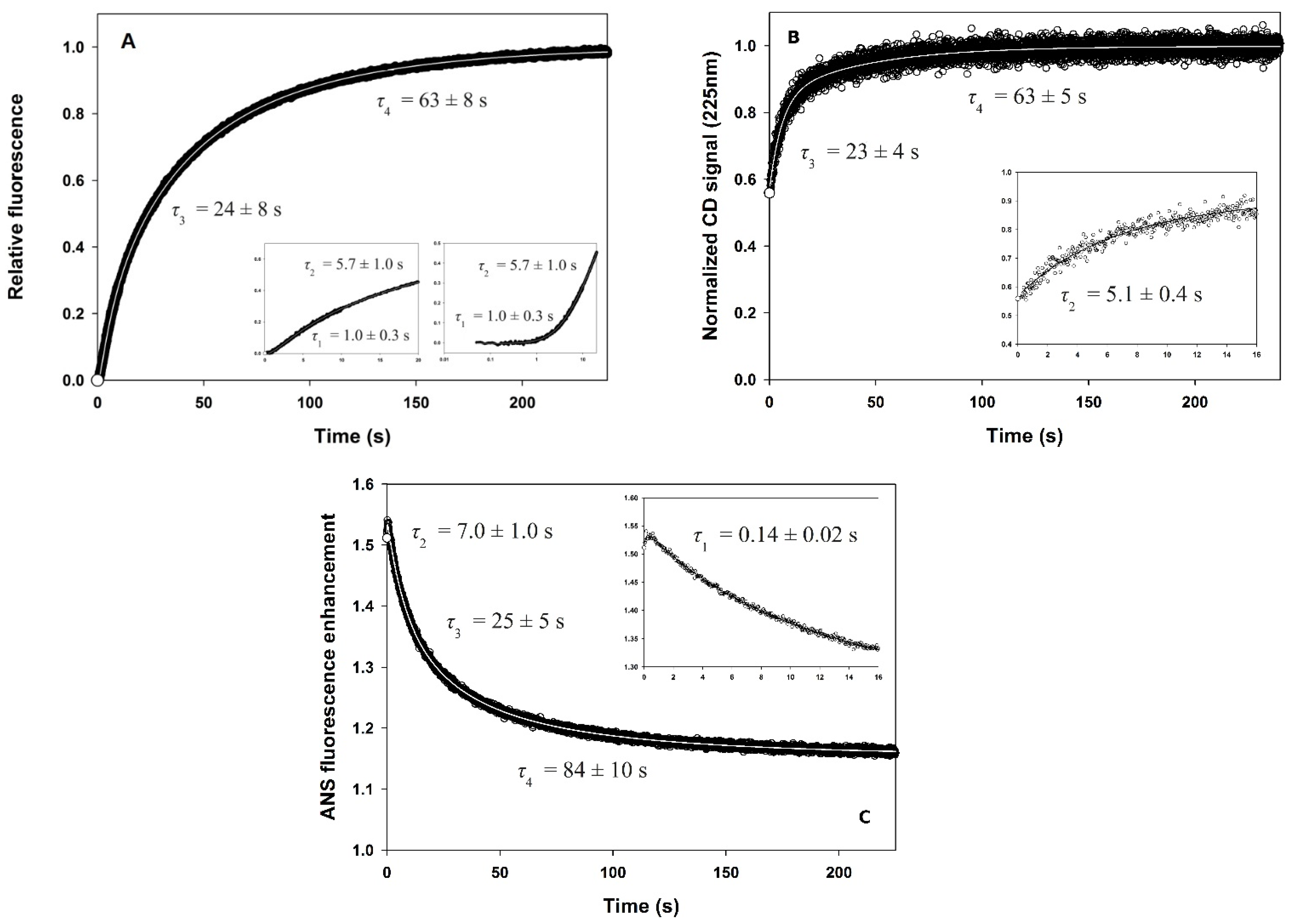
3.3. Fluorescence- and CD-Detected Folding Kinetics
3.4. ANS Binding
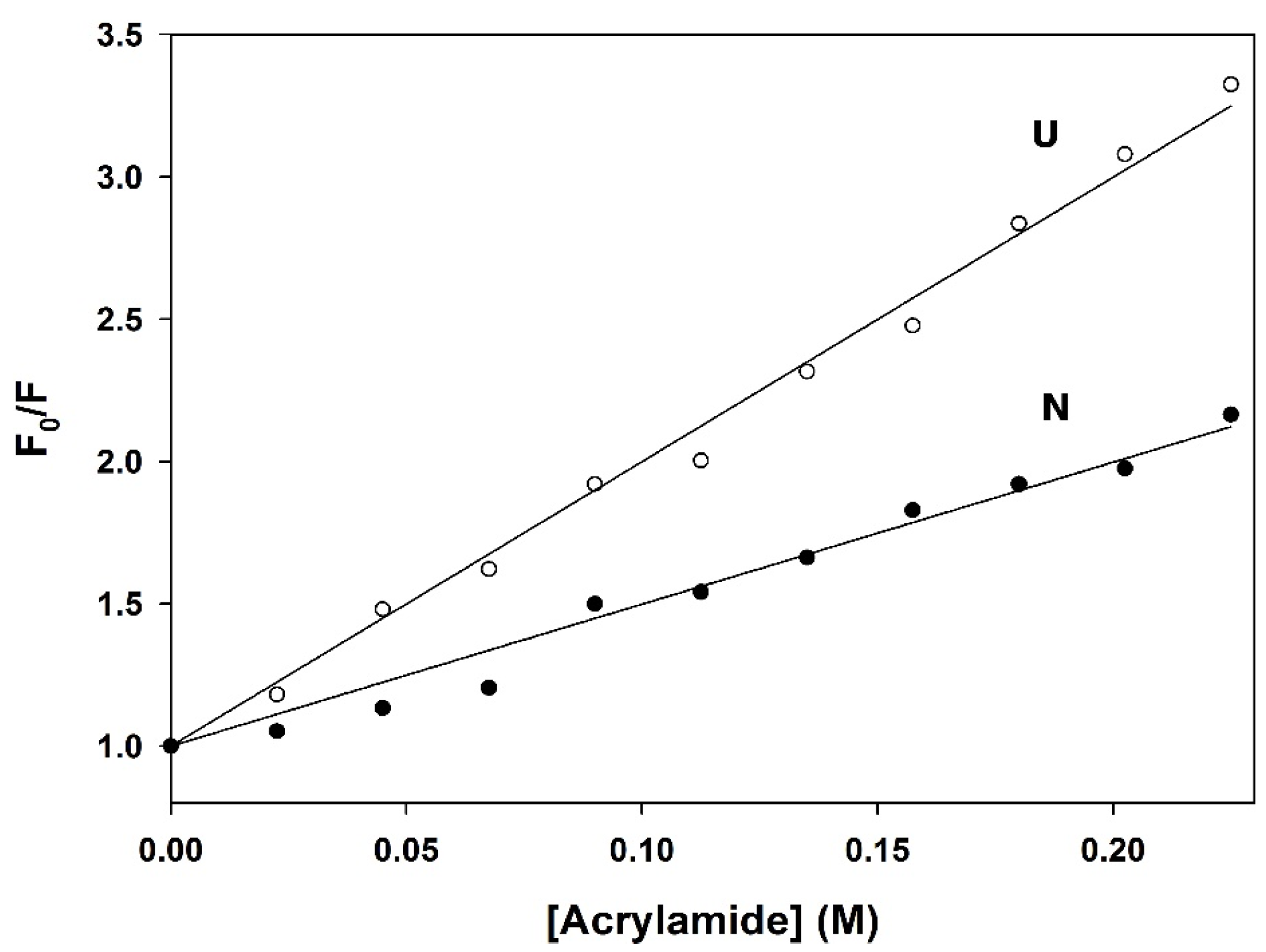
3.5. Quenching of Fluorescence by Acrylamide
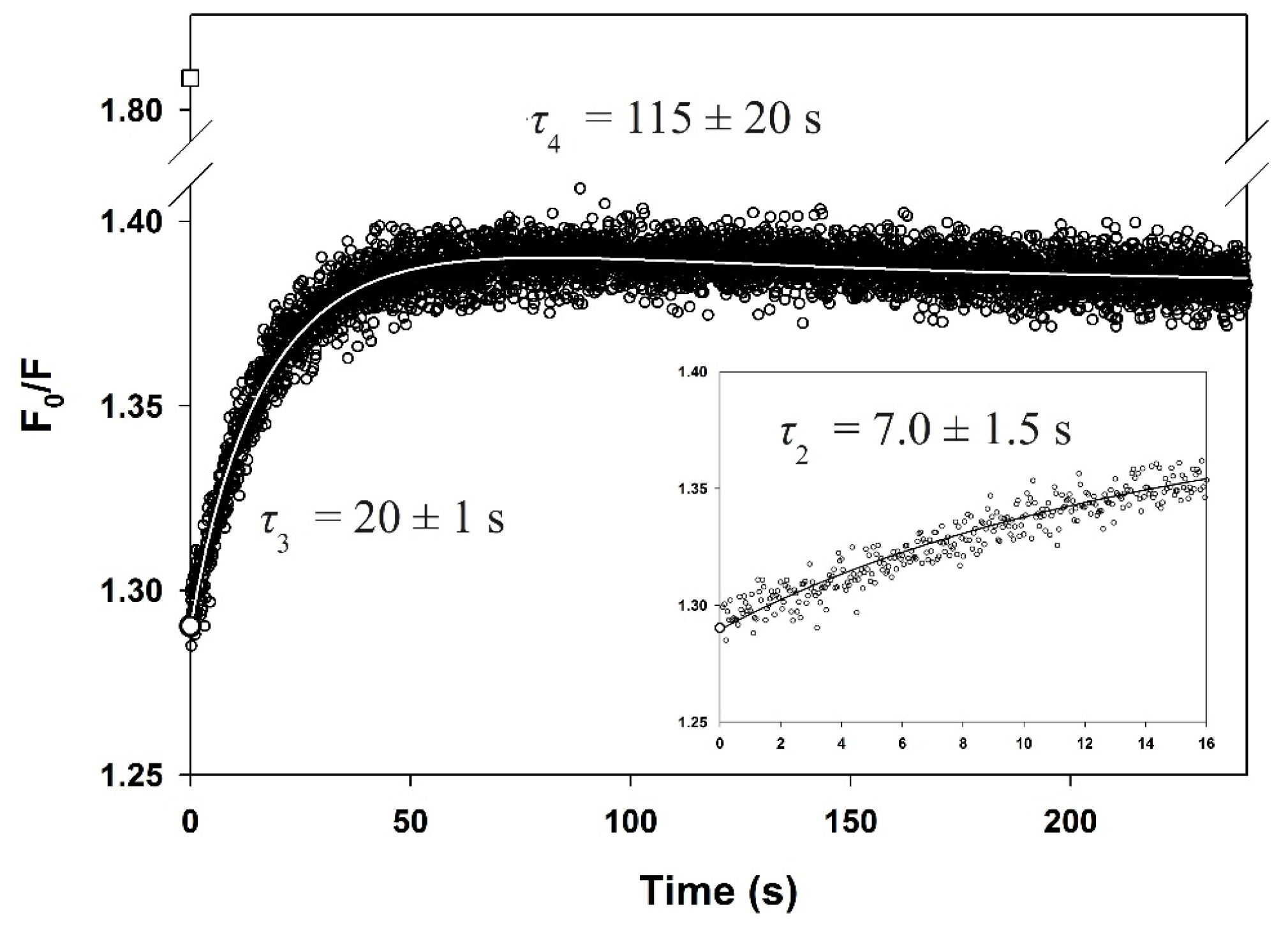
3.6. Slow Phases in PemA Folding
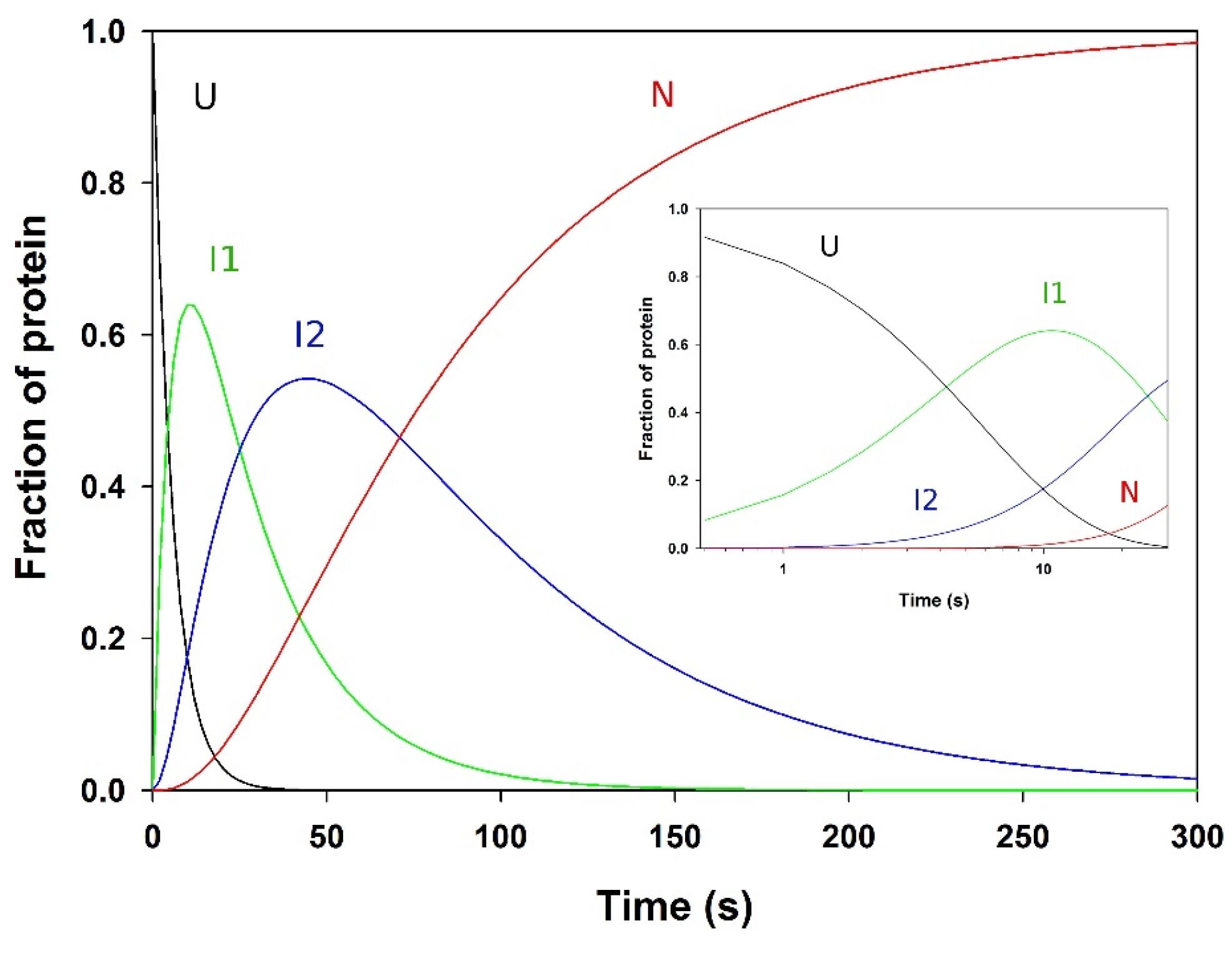
3.7. Analysis of the Folding Kinetics
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.J.; Dobson, C.M.; Hartl, U. Sequence does specify protein conformation. Trends Biochem. Sci. 1998, 23, 468. [Google Scholar] [CrossRef]
- Gierasch, L.M.; Gershenson, A. Post-reductionist protein science, or putting Humpty Dumpty back together again. Nat. Chem. Biol. 2009, 5, 774–777. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Villaverde, A. Protein quality in bacterial inclusion bodies. Trends. Biotechnol. 2006, 24, 179–185. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Finding the right fold. Nat. Struct. Biol. 1995, 2, 513–517. [Google Scholar] [CrossRef]
- Jackson, S.E.; Fersht, A.R. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 1991, 31, 10428–10435. [Google Scholar] [CrossRef]
- Viguera, A.R.; Martínez, J.C.; Filimonov, V.V.; Mateo, P.L.; Serrano, L. Thermodynamic and kinetic analysis of the SH3 domain of spectrin shows a two-state folding transition. Biochemistry 1994, 33, 2142–2150. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.S.; Oas, T.G. Submillisecond folding of monomeric lambda repressor. Proc. Natl. Acad. Sci. USA 1995, 95, 6878–6882. [Google Scholar] [CrossRef] [Green Version]
- Schindler, T.; Herrler, M.; Marahiel, M.A.; Schmid, F.X. Extremely rapid protein folding in the absence of intermediates. Nat. Struct. Biol. 1995, 2, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Kragelund, B.B.; Robinson, C.V.; Knudsen, J.; Dobson, C.M.; Poulsen, F.M. Folding of a four-helix bundle: Studies of acyl-coenzyme a binding protein. Biochemistry 1995, 34, 7217–7224. [Google Scholar] [CrossRef]
- Van Nuland, N.A.; Meijberg, W.; Warner, J.; Forge, V.; Scheek, R.M.; Robillard, G.T.; Dobson, C.M. Slow cooperative folding of a small globular protein HPr. Biochemistry 1998, 37, 622–637. [Google Scholar] [CrossRef] [Green Version]
- Friel, C.T.; Beddard, G.S.; Radford, S.E. Switching two-state to three-state kinetics in the helical protein Im9 via the optimisation of stabilising non-native interactions by design. J. Mol. Biol. 2004, 342, 261–273. [Google Scholar] [CrossRef]
- Radford, S.E.; Dobson, C.M.; Evans, P.A. The folding pathway of hen lysozyme involves partially structured intermediates and multiple pathways. Nature 1992, 358, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Forge, V.; Wijesinha, R.T.; Brew, K.; Robinson, C.V.; Redfield, C.; Dobson, C.M. Rapid collapse and slow structural reorganisation during the refolding of bovine α-lactalbumin. J. Mol. Biol. 1999, 288, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A.R. The sixth Datta Lecture. Protein folding and stability: The pathway of folding of barnase. FEBS Lett. 1993, 325, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Khorasanizadeh, S.; Peters, I.D.; Roder, H. Evidence for a three-state model of protein folding from kinetic analysis of ubiquitin variants with altered core residues. Nat. Struct. Biol. 1996, 3, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Friel, C.T.; Smith, D.A.; Vendruscolo, M.; Gsponer, J.; Radford, S.E. The mechanism of folding of Im7 reveals competition between functional and kinetic evolutionary constraints. Nat. Struct. Mol. Biol. 2009, 16, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Iwakura, M.; Matthews, C.R.; Bilsel, O. Microsecond subdomain folding in dihydrofolate reductase. J. Mol. Biol. 2011, 410, 329–342. [Google Scholar] [CrossRef]
- Houwman, J.A.; Westphal, A.H.; Visser, J.W.G.; Borst, J.W.; van Mierlo, C.P. Concurrent presence of on- and off-pathway folding intermediates of apoflavodoxin at physiological ionic strength. Phys. Chem. Chem. Phys. 2018, 20, 7059–7072. [Google Scholar] [CrossRef]
- Kim, P.S.; Baldwin, R.L. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem. 1990, 59, 631–660. [Google Scholar] [CrossRef]
- Matthews, C.R. Pathways of protein folding. Annu. Rev. Biochem. 1993, 62, 653–683. [Google Scholar] [CrossRef]
- Dobson, C.M.; Šali, A.; Karplus, M. Protein folding: A perspective from theory and experiment. Angew. Chem. Int. Ed. Engl. 1998, 37, 868–893. [Google Scholar] [CrossRef]
- Chan, H.S.; Dill, K.A. Protein folding in the landscape perspective: Chevron plots and non-Arrhenius kinetics. Proteins 1998, 30, 2–33. [Google Scholar] [CrossRef]
- Radford, S.E. Protein folding: Progress made and promises ahead. Trends Biochem. Sci. 2000, 25, 611–618. [Google Scholar] [CrossRef]
- Szilágyi, A.; Kardos, J.; Osváth, S.; Barna, L.; Závodszky, P. Protein Folding. In Handbook of Neurochemistry and Molecular Neurobiology, 3rd ed.; Lajtha, A., Banik, N., Eds.; Springer: Boston, MA, USA, 2007; pp. 303–343. [Google Scholar]
- Udgaonkar, J.B. Multiple routes and structural heterogeneity in protein folding. Annu. Rev. Biophys. 2008, 489–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The Protein Folding Problem. Annu. Rev. Biophys. 2008, 37, 289–316. [Google Scholar] [CrossRef] [PubMed]
- Gruebele, M.; Dave, K.; Sukenik, S. Globular protein folding in vitro and in vivo. Annu. Rev. Biophys. 2016, 45, 233–251. [Google Scholar] [CrossRef]
- Daggett, V.; Fersht, A.R. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 2003, 28, 18–25. [Google Scholar] [CrossRef]
- Plaxco, K.W.; Simons, K.T.; Ruczinski, I.; Baker, D. Topology, Stability, Sequence, and Length: Defining the Determinants of Two-State Protein Folding Kinetics. Biochemistry 2000, 39, 11177–11183. [Google Scholar] [CrossRef]
- Ivankov, D.N.; Garbuzynskiy, S.O.; Alm, E.; Plaxco, K.W.; Baker, D.; Finkelstein, A.V. Contact order revisited: Influence of protein size on the folding rate. Prot. Sci. 2003, 12, 2057–2062. [Google Scholar] [CrossRef] [Green Version]
- Walters, B.T.; Mayne, L.; Hinshaw, T.R.; Englander, S.W. Folding of a large protein at high structural resolution. Proc. Natl. Acad. Sci. USA 2013, 110, 18898–18903. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.; Pappu, R.V. Protein Folding and Binding, Complexity Comes of Age. Curr. Opin. Struct. Biol. 2017, 42, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Kajava, A.V. Tandem repeats in proteins: From sequence to structure. J. Struct. Biol 2012, 179, 279–288. [Google Scholar] [CrossRef]
- Mello, C.C.; Barrick, D. An experimentally determined protein folding energy landscape. Proc. Natl. Acad. Sci. USA 2004, 101, 14102–14107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreiro, D.U.; Cho, S.S.; Komives, E.A.; Wolynes, P.G. The energy landscape of modular repeat proteins: Topology determines folding mechanism in the ankyrin family. J. Mol. Biol. 2005, 354, 679–692. [Google Scholar] [CrossRef]
- Main, E.R.G.; Lowe, A.R.; Mochrie, S.G.J.; Jackson, S.E.; Regan, L. A recurring theme in protein engineering: The design, stability and folding of repeat proteins. Curr. Opin. Struct. Biol. 2005, 15, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Löw, C.; Weininger, U.; Neumann, P.; Klepsch, M.; Lilie, H.; Stubbs, M.L.; Balbach, J. Structural insights into an equilibrium folding intermediate of an archaeal ankyrin repeat protein. Proc. Natl. Acad. Sci. USA 2008, 105, 3779–3784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrick, D.; Ferreiro, D.U.; Komives, E.A. Folding landscapes of ankyrin repeat proteins: Experiments meet theory. Curr. Opin. Struct. Biol. 2008, 18, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Kloss, E.; Courtemanche, N.; Barrick, D. Repeat-protein folding: New insights into origins of cooperativity, stability, and topology. Arch. Biochem. Biophys. 2008, 469, 83–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzel, S.K.; Settanni, G.; Kenig, M.; Binz, H.K.; Plückthun, A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J. Mol. Biol. 2008, 376, 241–257. [Google Scholar] [CrossRef] [Green Version]
- Javadi, Y.; Itzhaki, L. Tandem-repeat proteins: Regularity plus modularity equals design-ability. Curr. Opin. Struct. Biol. 2013, 23, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Rowling, P.J.E.; Sivertsson, E.M.; Perez-Riba, A.; Main, E.R.G.; Itzhaki, L.S. Dissecting and reprogramming the folding and assembly of tandem-repeat proteins. Biochem. Soc. Trans. 2015, 43, 881–888. [Google Scholar] [CrossRef]
- Perez-Riba, A.; Synakewicz, M.; Itzhaki, L.S. Folding cooperativity and allosteric function in the tandem-repeat protein class. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Toth, I.K.; Bell, K.S.; Holeva, M.C.; Birch, P.R.J. Soft rot erwiniae: From genes to genomes. Mol. Plant Pathol. 2003, 4, 17–30. [Google Scholar] [CrossRef]
- Kazemi-Pour, N.; Condemine, G.; Hugouvieux-Cotte-Pattat, N. The secretome of the plant pathogenic bacterium Erwinia chrysanthemi. Proteomics 2004, 4, 3177–3186. [Google Scholar] [CrossRef] [PubMed]
- Fries, M.; Ihrig, J.; Brocklehurst, K.; Shevchik, V.E.; Pickersgill, R.W. Molecular basis of the activity of the phytopathogen pectin methylesterase. EMBO J. 2007, 26, 3879–3887. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, J.; Mayans, O.; Smith, D.; Worboys, K.; Pickersgill, R.W. Three-dimensional structure of Erwinia chrysanthemi pectin methylesterase reveals a novel esterase active site. J. Mol. Biol. 2001, 305, 951–960. [Google Scholar] [CrossRef]
- McDonnel, A.V.; Menke, M.; Palmer, N.; King, J.; Cowen, L.; Berger, B. Fold recognition and accurate sequence-structure alignment of sequences directing β-sheet proteins. Proteins 2006, 63, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Kajava, A.V.; Steven, A.C. β-Rolls, β-helices, and other β-solenoid proteins. Adv. Protein Chem. 2006, 73, 55–96. [Google Scholar]
- Jenkins, J.; Pickersgill, R. The architecture of parallel β-helices and related folds. Prog. Biophys. Mol. Biol. 2001, 77, 111–175. [Google Scholar] [CrossRef]
- Wetzel, R. Ideas of order for amyloid fibrils. Structure 2002, 10, 1031–1036. [Google Scholar] [CrossRef] [Green Version]
- Wille, H.; Michelitsch, M.; Guenebaut, V.; Supattapone, S.; Serban, A.; Cohen, F.; Agard, D.; Prusiner, S. Structural studies of the scrapie prion protein by electron crystallography. Proc. Natl. Acad. Sci. USA 2002, 99, 3563–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickersgill, R.W. A primordial structure underlying amyloid. Structure 2003, 11, 137–138. [Google Scholar] [CrossRef] [Green Version]
- Jahn, T.R.; Makin, O.S.; Morris, K.L.; Marshall, K.E.; Tian, P.; Sikorski, P.; Serpell, L.C. The Common Architecture of Cross-β Amyloid. J. Mol. Biol. 2010, 395, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.D.; Lietzke, S.E.; Jurnak, F. Unusual structural features in the parallel β-helix in pectate lyases. Structure 1993, 1, 241–251. [Google Scholar] [CrossRef]
- Yoder, M.D.; Keen, N.T.; Jurnak, F. New domain motif: The structure of pectate lyase C, a secreted plant virulence factor. Science 1993, 260, 1503–1507. [Google Scholar] [CrossRef]
- Steinbacher, S.; Miller, S.; Baxa, U.; Budisa, N.; Weintraub, A.; Seckler, R.; Huber, R. Phage P22 tailspike protein: Crystal structure of the head-binding domain at 2.3 Å, fully refined structure of the endorhamnosidase at 1.56 Å resolution, and the molecular basis of O-antigen recognition and cleavage. J. Mol. Biol. 1997, 267, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Charles, I.G.; Fairweather, N.F.; Isaacs, N.W. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature 1996, 381, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.W., Jr.; Starner-Kreinbrink, J.L.; Hosur, R.; Clark, P.L.; Berger, B. Structure-based prediction reveals capping motifs that inhibit β-helix aggregation. Proc. Natl. Acad. Sci. USA 2011, 108, 11099–11104. [Google Scholar] [CrossRef] [Green Version]
- McGaughey, G.B.; Gagné, M.; Rappé, A.K. Pi-Stacking interactions. Alive and well in proteins. J Biol Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef] [Green Version]
- Betts, S.; Haase-Pettingell, C.; Cook, K.; King, J. Buried hydrophobic side-chains essential for the folding of the parallel β-helix domains of the P22 tailspike. Protein Sci. 2004, 13, 2291–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simkovsky, R.; King, J. An elongated spine of buried core residues necessary for in vivo folding of the parallel β-helix of P22 tailspike adhesin. Proc. Natl. Acad. Sci. USA 2006, 103, 3575–3580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts, S.; King, J. There’s a right way and a wrong way: In vivo and in vitro folding, misfolding and subunit assembly of the P22 tailspike. Structure 1999, 7, R131–R139. [Google Scholar] [CrossRef] [Green Version]
- King, J.; Haase-Pettingell, C.; Robinson, A.S.; Speed, M.; Mitraki, A. Thermolabile folding intermediates: Inclusion body precursors and chaperonin substrates. FASEB J. 1996, 10, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Haase-Pettingell, C.; King, J. Prevalence of temperature sensitive folding mutations in the parallel β coil domain of the phage P22 tailspike endorhamnosidase. J. Mol. Biol. 1997, 267, 88–102. [Google Scholar] [CrossRef]
- Seckler, R. Folding and function of repetitive structure in the homotrimeric phage P22 tailspike protein. J. Struct. Biol. 1998, 122, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.; Seckler, R. P22 tailspike folding mutants revisited: Effects on the thermodynamic stability of the isolated β-helix domain. J. Mol. Biol. 1998, 281, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, C.B.; King, J.; Clark, P.L. Characterization of the protrimer intermediate in the folding pathway of the interdigitated β-helix tailspike protein. Biochemistry 2002, 41, 5093–5103. [Google Scholar] [CrossRef]
- Jain, M.; Evans, M.S.; King, J.; Clark, P.L. Monoclonal antibody epitope mapping describes tailspike β-helix folding and aggregation intermediates. J. Biol. Chem. 2005, 280, 23032–23040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.S.; Sander, I.M.; Clark, P.L. Cotranslational folding promotes β -helix formation and avoids aggregation in vivo. J. Mol. Biol. 2008, 383, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Kamen, D.E.; Griko, Y.; Woody, R.W. The Stability, Structural Organization, and Denaturation of Pectate Lyase C, a Parallel β-Helix Protein. Biochemistry 2000, 39, 15932–15943. [Google Scholar] [CrossRef] [PubMed]
- Kamen, D.E.; Woody, R.W. Folding kinetics of the protein pectate lyase C reveal fast-forming intermediates and slow proline isomerization. Biochemistry 2002, 41, 4713–4723. [Google Scholar] [CrossRef] [PubMed]
- Kamen, D.E.; Woody, R.W. Identification of proline residues responsible for the slow folding kinetics in pectate lyase C by mutagenesis. Biochemistry 2002, 41, 4724–4732. [Google Scholar] [CrossRef]
- Junker, M.; Schuster, C.C.; McDonnell, A.V.; Sorg, K.A.; Finn, M.C.; Berger, B.; Clark, P.L. Pertactin β-helix folding mechanism suggests common themes for the secretion and folding of autotransporter proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 4918–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junker, M.; Clark, P.L. Slow formation of aggregation-resistant β-sheet folding intermediates. Proteins 2010, 78, 812–824. [Google Scholar] [CrossRef]
- Sambrook, J.; Russel, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Vandevenne, M.; Filée, P.; Scarafone, N.; Cloes, B.; Gaspard, G.; Yilmaz, N.; Dumoulin, M.; François, J.-M.; Frère, J.-M.; Galleni, M. The Bacillus licheniformis BlaP β-lactamase as a model protein scaffold to study the insertion of protein fragments. Protein Sci. 2007, 16, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, P.; Johnson, W.C., Jr. Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 1987, 167, 76–85. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from CD spectra: Comparison of CONTIN, SELCON and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef]
- Nozaki, Y. The preparation of guanidine hydrochloride. Methods Enzymol. 1972, 26, 43–50. [Google Scholar]
- Lakowicz, J.R. Quenching of Fluorescence. In Principles of Fluorescence Spectroscopy, 2nd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999; pp. 237–265. [Google Scholar]
- Dumoulin, M.; Conrath, K.; Van Meirhaeghe, A.; Meersman, F.; Heremans, K.; Frenken, L.G.; Muyldermans, S.; Wyns, L.; Matagne, A. Single-domain antibody fragments with high conformational stability. Protein Sci. 2002, 11, 500–515. [Google Scholar] [CrossRef]
- Dirix, C.; Duvetter, T.; Loey, A.V.; Hendrickx, M.; Heremans, K. The in situ observation of the temperature and pressure stability of recombinant Aspergillus aculeatus pectin methylesterase with Fourier transform IR spectroscopy reveals an unusual pressure stability of β-helices. Biochem. J. 2005, 392, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paolo, A.; Balbeur, D.; De Pauw, E.; Redfield, C.; Matagne, A. Rapid Collapse into a Molten Globule Is Followed by Simple Two-State Kinetics in the Folding of Lysozyme from Bacteriophage λ. Biochemistry 2010, 49, 8646–8657. [Google Scholar] [CrossRef]
- Vandenameele, J.; Lejeune, A.; Di Paolo, A.; Brans, A.; Frère, J.-M.; Schmid, F.X.; Matagne, A. Folding of class A β-lactamases is rate-limited by peptide bond isomerization and occurs via parallel pathways. Biochemistry 2010, 49, 4264–4275. [Google Scholar] [CrossRef] [PubMed]
- Tanford, C. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv. Protein Chem. 1970, 24, 1–95. [Google Scholar]
- Baldwin, R.L. On-pathway versus off-pathway folding intermediates. Fold. Des. 1996, 1, R1–R8. [Google Scholar] [CrossRef] [Green Version]
- Fersht, A.R. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; WH. Freeman and Co.: New York, NY, USA, 1998. [Google Scholar]
- Moore, J.W.; Pearson, R.G. Complex reactions. In Kinetics and Mechanism, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 1981; pp. 284–333. [Google Scholar]
- Doetsch, G. Introduction to the Theory and Application of the Laplace Transformation; Springer: Berlin/Heidelberg, Germany, 1974. [Google Scholar]
- Johnson, W.C., Jr. Protein secondary structure and circular dichroism: A practical guide. Proteins 1990, 7, 205–214. [Google Scholar] [CrossRef]
- Ptitsyn, O.B.; Pain, R.H.; Semisotnov, G.V.; Zerovnik, E.; Razgulyaev, O.I. Evidence for a molten globule state as a general intermediate in protein folding. FEBS Lett. 1990, 262, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Semisotnov, G.V.; Rodionova, N.A.; Razgulyaev, O.I.; Uversky, V.N.; Gripas, A.F.; Gilmanshin, R.I. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 1991, 31, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Jennings, P.A.; Pierre, R.A.; Matthews, C.R. Development of nonpolar surfaces in the folding of Escherichia coli dihydrofolate reductase detected by 1-anilinonaphthalene-8-sulfonate binding. Biochemistry 1994, 33, 15250–15258. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, L.S.; Evans, P.A.; Dobson, C.M.; Radford, S.E. Tertiary interactions in the folding pathway of hen lysozyme: Kinetic studies using fluorescent probes. Biochemistry 1994, 33, 5212–5220. [Google Scholar] [CrossRef]
- Engelhard, M.; Evans, P.A. Kinetics of interaction of partially folded proteins with a hydrophobic dye: Evidence that molten globule character is maximal in early folding intermediates. Protein Sci. 1995, 4, 1553–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhove, M.; Lejeune, A.; Guillaume, G.; Virden, R.; Pain, R.H.; Schmid, F.X.; Frère, J.-M. A collapsed intermediate with nonnative packing of hydrophobic residues in the folding of TEM-1 β-lactamase. Biochemistry 1998, 37, 1941–1950. [Google Scholar] [CrossRef]
- Khurana, R.; Fink, A.L. Do parallel βhelix proteins have a unique Fourier transform infrared spectrum? Biophys. J. 2000, 78, 994–1000. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.; Mantsch, H.H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [Google Scholar] [CrossRef]
- Sandroff, C.J.; King, H.E.; Herschbach, D.R. High pressure study of the liquid/solid interface: Surface enhanced raman scattering from adsorbed molecules. J. Phys. Chem. 1984, 88, 5647–5653. [Google Scholar] [CrossRef]
- Haris, P.I.; Chapman, D. The conformational analysis of peptides using Fourier transform IR spectroscopy. Biopolymers 1995, 37, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Creighton, T.E. Protein folding. Biochem. J. 1990, 270, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Walkenhorst, W.F.; Green, S.M.; Roder, H. Kinetic evidence for folding and unfolding intermediates in staphylococcal nuclease. Biochemistry 1997, 36, 5795–5805. [Google Scholar] [CrossRef]
- Heidary, D.K.; O’Neill, J.C., Jr.; Roy, M.; Jennings, P.A. An essential intermediate in the folding of dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 2000, 97, 5866–5870. [Google Scholar] [CrossRef] [Green Version]
- Garvey, E.P.; Swank, J.; Matthews, C.R. A hydrophobic cluster forms early in the folding of dihydrofolate reductase. Proteins 1989, 6, 259–266. [Google Scholar] [CrossRef]
- Hubbard, S.J.; Thornton, J.M. ‘NACCESS’, Computer Program; Department Biochemistry and Molecular Biology, University College: London, UK, 1993. [Google Scholar]
- Schmid, F.X.; Baldwin, R.L. Acid catalysis of the formation of the slow-folding species of RNase A: Evidence that the reaction is proline isomerization. Proc. Nat. Acad. Sci. USA 1978, 75, 4764–4768. [Google Scholar] [CrossRef] [Green Version]
- Kiefhaber, T.; Kohler, H.H.; Schmid, F.X. Kinetic coupling between protein folding and prolyl isomerization. I. Theoretical models. J. Mol. Biol. 1992, 224, 217–229. [Google Scholar] [CrossRef]
- Schmid, F.X. Prolyl isomerase: Enzymatic catalysis of slow protein-folding reactions. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Brandts, J.F.; Halvorson, H.R.; Brennan, M. Consideration of the Possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 1975, 14, 4953–4963. [Google Scholar] [CrossRef]
- Maier, R.; Scholz, C.; Schmid, F.X. Dynamic association of trigger factor with protein substrates. J. Mol. Biol. 2001, 314, 1181–1190. [Google Scholar] [CrossRef]
- Weininger, U.; Haupt, C.; Schweimer, K.; Graubner, W.; Kovermann, M.; Brüser, T.; Scholz, C.; Schaarschmidt, P.; Žoldák, G.; Schmid, F.X.; et al. NMR solution structure of SlyD from Escherichia coli: Spatial separation of prolyl isomerase and chaperone function. J. Mol. Biol. 2009, 387, 295–305. [Google Scholar] [CrossRef]
- Sterner, R.; Liebl, W. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 39–106. [Google Scholar] [CrossRef]
- Feller, G. Protein folding at extreme temperatures: Current issues. Semin. Cell Dev. Biol. 2018, 84, 129–137. [Google Scholar] [CrossRef]
- Myers, J.K.; Pace, C.N.; Scholtz, J.M. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4, 2138–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, C.N. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986, 131, 266–280. [Google Scholar]
- Spudich, G.; Marqusee, S. A change in the apparent m value reveals a populated intermediate under equilibrium conditions in Escherichia coli ribonuclease HI. Biochemistry 2000, 39, 11677–11683. [Google Scholar] [CrossRef] [PubMed]
- Renn, J.P.; Clark, P.L. A conserved stable core structure in the passenger domain beta-helix of autotransporter virulence proteins. Biopolymers 2008, 89, 420–427. [Google Scholar] [CrossRef]
- Miller, S.; Schuler, B.; Seckler, R. A reversibly unfolding fragment of P22 tailspike protein with native structure: The isolated beta-helix domain. Biochemistry 1998, 37, 9160–9168. [Google Scholar] [CrossRef]
- Meza-Aguilar, J.D.; Fromme, P.; Torres-Larios, A.; Mendoza-Hernández, G.; Hernandez-Chiñas, U.; Monteros, R.A.A.-E.D.L.; Campos, C.A.E.; Fromme, R. X-ray crystal structure of the passenger domain of plasmid encoded toxin(Pet), an autotransporter enterotoxin from enteroaggregative Escherichia coli (EAEC). Biochem. Biophys. Res. Commun. 2014, 445, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Delhaise, P.; Bardiaux, M.; De Maeyer, M.; Prévost, M.; Vanbelle, D.; Donneux, J.; Lasters, I.; Vancustem, E.; Alard, P.; Wodak, S.J. The brugel package—Toward computer-aided-design of macromolecules. J. Mol. Graph. 1988, 6, 219. [Google Scholar] [CrossRef]
- Gross, M.; Jaenicke, R. Proteins under pressure. The influence of high hydrostatic pressure on structure, function and assembly of proteins and protein complexes. Eur. J. Biochem. 1994, 221, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.A. Revisiting volume changes in pressure-induced protein unfolding. Biochim. Biophys. Acta. 2002, 1595, 201–209. [Google Scholar] [CrossRef]
- Roche, J.; Caro, J.A.; Norberto, D.R.; Barhe, P.; Roumestand, C.; Schlessman, J.L.; Garcia, A.E.; Garcia-Moreno, B.E.; Royer, C. Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 6945–6950. [Google Scholar] [CrossRef] [Green Version]
- Rouget, J.B.; Aksel, T.; Roche, J.; Saldana, J.L.; Garcia, A.E.; Barrick, D.; Royer, C.A. Size and sequence and the volume change of protein folding. J. Am. Chem. Soc. 2011, 133, 6020–6027. [Google Scholar] [CrossRef] [Green Version]
- Ando, N.; Barstow, B.U.; Baase, W.A.; Fields, A.; Matthews, B.W.; Gruner, S.M. Structural and Thermodynamic Characterization of T4 Lysozyme Mutants and the Contribution of Internal Cavities to Pressure Denaturation. Biochemistry 2008, 47, 11097–11109. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Foguel, D.; Royer, C.A. Pressure provides new insights into protein folding, dynamics and structure. Trends Biochem. Sci. 2001, 26, 612–618. [Google Scholar] [CrossRef]
- Meersman, F.; Dobson, C.M.; Heremans, K. Protein unfolding, amyloid fibril formation and configurational energy landscapes under high pressure conditions. Chem. Soc. Rev. 2006, 35, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins 1989, 6, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.; Pain, R.H. Molten globule intermediates and protein folding. Eur. Biophys. J. 1991, 19, 221–229. [Google Scholar] [CrossRef]
- Ptitsyn, O.B. Molten globule and protein folding. Adv. Protein. Chem. 1995, 47, 83–229. [Google Scholar]
- Redfield, C. Molten Globules. Curr. Biol. 1999, 9, R313. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, R.L.; Frieden, C.; Rose, G.D. Dry molten globule intermediates and the mechanism of protein unfolding. Proteins 2010, 78, 2725–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matagne, A.; Jamin, M.; Chung, E.W.; Radford, S.E.; Robinson, C.V.; Dobson, C.M. Thermal unfolding of an intermediate is associated with non-Arrhenius kinetics in the folding of hen lysozyme. J. Mol. Biol. 2000, 297, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.A.; Matthews, C.R. Sequential vs. parallel protein-folding mechanisms: Experimental tests for complex folding reactions. Biophys. Chem. 2002, 101–102, 113–131. [Google Scholar] [CrossRef]
- Mello, C.C.; Bradley, C.M.; Tripp, K.W.; Barrick, D. Experimental characterization of the folding kinetics of the notch ankyrin domain. J. Mol. Biol. 2005, 352, 266–281. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Burst Phase | Phase 1 | Phase 2 | Phase 3 | Phase 4 |
|---|---|---|---|---|---|
| Int. fluo. a | |||||
| τ (s) e | <0.003 g | 1.0 ± 0.3 | 5.7 ± 1.0 | 24 ± 8 | 63 ± 8 |
| amplitude f | 0 | −0.06 ± 0.01 | 0.20 ± 0.01 | 0.31 ± 0.02 | 0.55 ± 0.04 |
| CD225nm b | |||||
| τ (s) e | <0.007 | nd h | 5.1 ± 0.4 | 23 ± 4 | 63 ± 5 |
| amplitude f | 0.56 ± 0.09 | nd | 0.25 ± 0.01 | 0.12 ± 0.02 | 0.07 ± 0.02 |
| ANS fluo.c | |||||
| τ (s) e | <0.003 | 0.14 ± 0.02 | 7.0 ± 1.0 | 25 ± 5 | 84 ± 10 |
| amplitude f | 0.52 ± 0.02 | 0.032 ± 0.003 | −0.13 ± 0.02 | −0.15 ± 0.01 | −0.10 ± 0.02 |
| Acryl. d | |||||
| τ (s) e | <0.003 | nd | 7.0 ± 1.5 | 20 ± 1 | 115 ± 20 |
| amplitude f | −0.54 ± 0.05 | nd | 0.014 ± 0.006 | 0.10 ± 0.02 | −0.018 ± 0.002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guillerm, J.; Frère, J.-M.; Meersman, F.; Matagne, A. The Right-Handed Parallel β-Helix Topology of Erwinia chrysanthemi Pectin Methylesterase Is Intimately Associated with Both Sequential Folding and Resistance to High Pressure. Biomolecules 2021, 11, 1083. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081083
Guillerm J, Frère J-M, Meersman F, Matagne A. The Right-Handed Parallel β-Helix Topology of Erwinia chrysanthemi Pectin Methylesterase Is Intimately Associated with Both Sequential Folding and Resistance to High Pressure. Biomolecules. 2021; 11(8):1083. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081083
Chicago/Turabian StyleGuillerm, Jessica, Jean-Marie Frère, Filip Meersman, and André Matagne. 2021. "The Right-Handed Parallel β-Helix Topology of Erwinia chrysanthemi Pectin Methylesterase Is Intimately Associated with Both Sequential Folding and Resistance to High Pressure" Biomolecules 11, no. 8: 1083. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081083