
How Inflammation Pathways Contribute to Cell Death in Neuro-Muscular Disorders

 , , , and
, , , and
Abstract
:
1. Introduction
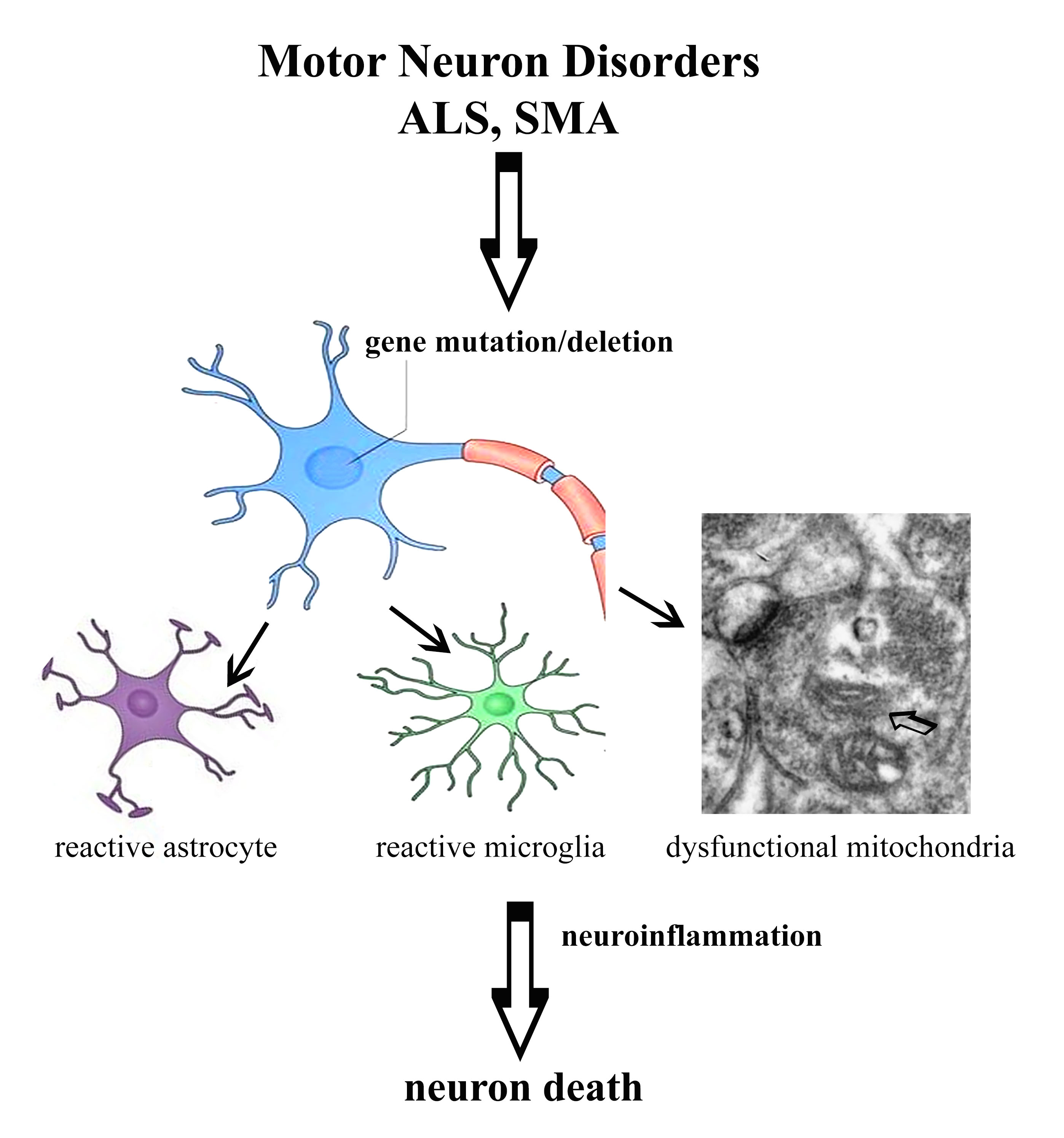
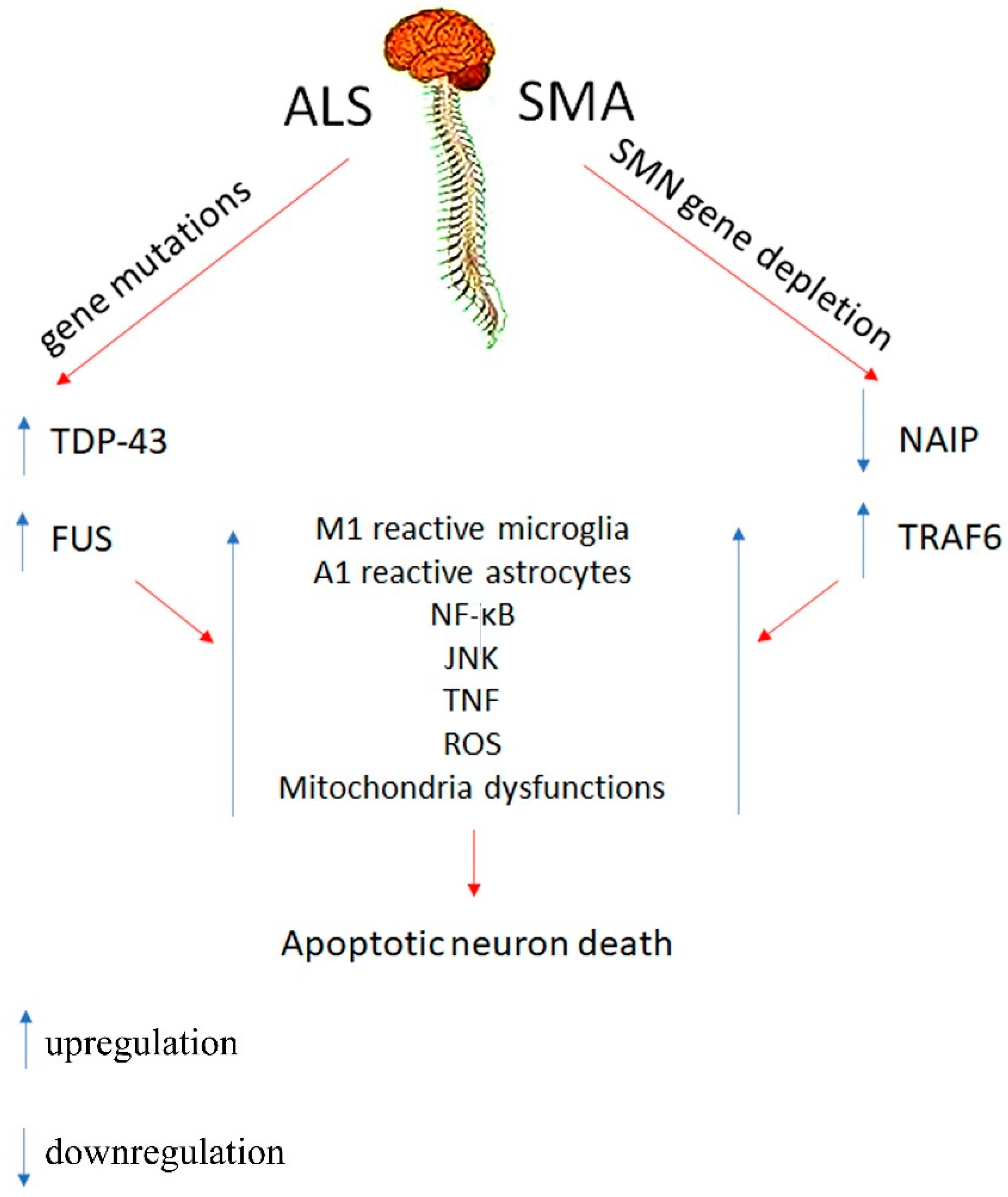
2. Motor Neuron Disorders
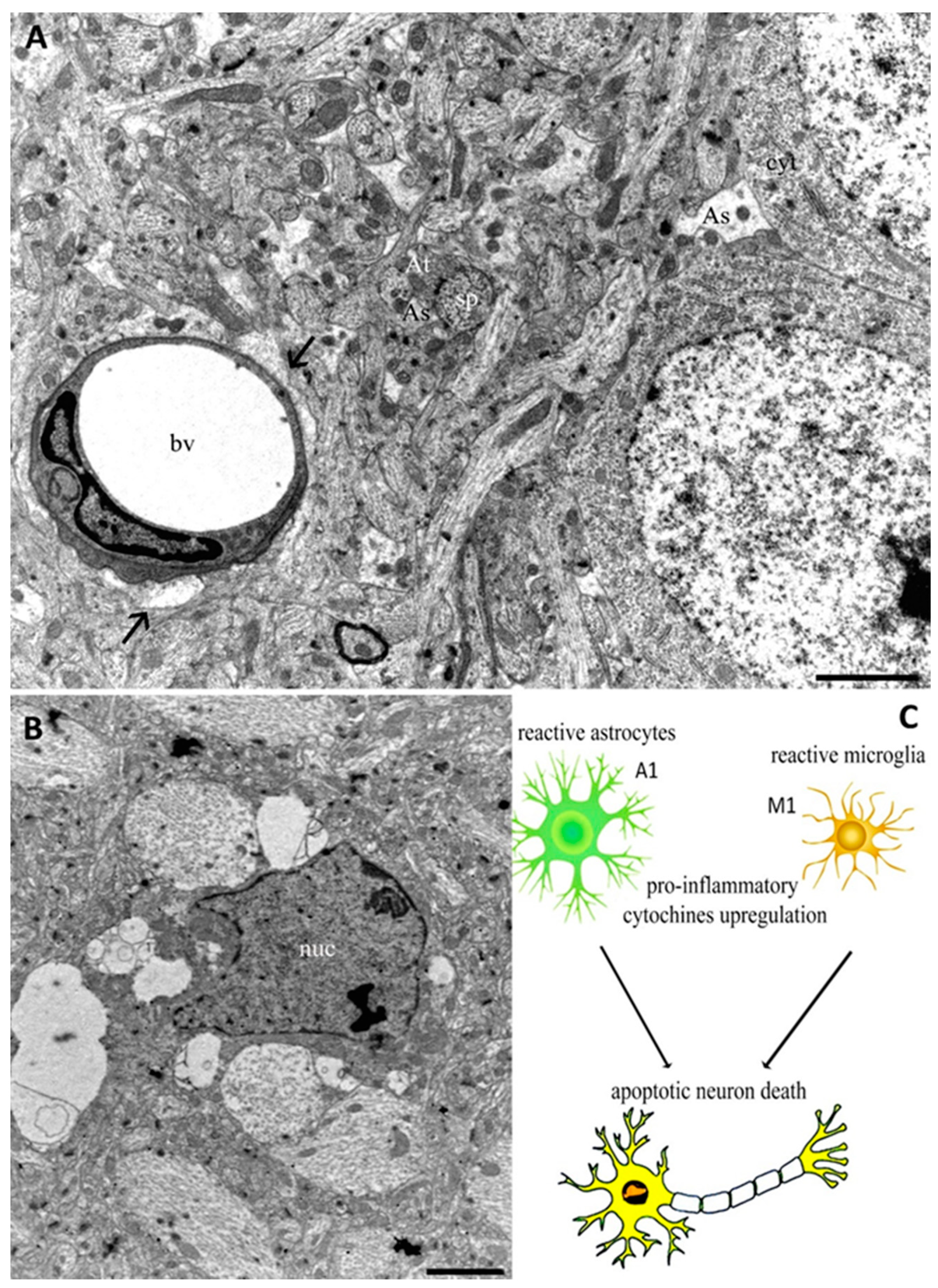
2.1. Innate Immune System
2.2. Cellular Mechanisms
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meacci, E.; Garcia-Gil, M. S1P/S1P Receptor Signaling in Neuromuscolar Disorders. Int. J. Mol. Sci. 2019, 20, 6364. [Google Scholar] [CrossRef] [Green Version]
- Cowling, B.S.; Thielemans, L. Translational medicine in neuromuscular disorders: From academia to industry. Dis. Model. Mech. 2019, 13, 41434. [Google Scholar] [CrossRef] [Green Version]
- Dowling, J.J.; D. Gonorazky, H.; Cohn, R.D.; Campbell, C. Treating pediatric neuromuscular disorders: The future is now. Am. J. Med. Genet. 2018, 176, 804–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, J.M. The Epidemiology of Neuromuscular Diseases. Neurol. Clin. 2016, 34, 999–1021. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.M. Neuromuscular Diseases. Semin. Neurol. 2016, 36, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Salucci, S.; Falcieri, E. Polyphenols and their potential role in preventing skeletal muscle atrophy. Nutr. Res. 2020, 74, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Grande, V.; Hathazi, D.; O’Connor, E.; Marteau, T.; Schara-Schmidt, U.; Hentschel, A.; Genevieve, G.; Nikolenko, N.; Lochmüller, H.; Roos, A. Dysregulation of GSK3β-Target Proteins in Skin Fibroblasts of Myotonic Dystrophy Type 1 (DM1) Patients. J. Neuromuscul. Dis. 2021. [Google Scholar] [CrossRef]
- Azotla-Vilchis, C.N.; Sanchez-Celis, D.; Agonizantes-Juárez, L.E.; Suárez-Sánchez, R.; Hernández-Hernández, J.M.; Peña, J.; Vázquez-Santillán, K.; Leyva-García, N.; Ortega, A.; Maldonado, V.; et al. Transcriptome Analysis Reveals Altered Inflammatory Pathway in an Inducible Glial Cell Model of Myotonic Dystrophy Type 1. Biomolecules 2021, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Di Benedetto, S.; Pawelec, G. The Immune System and Its Dysregulation with Aging. Rev. Subcell. Biochem. 2019, 91, 21–43. [Google Scholar]
- Comley, L.H.; Nijssen, J.; Frost-Nylen, J.; Hedlund, E. Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J. Comp. Neurol. 2016, 524, 1424–1442. [Google Scholar] [CrossRef] [Green Version]
- Perez-Garcia, M.J.; Kong, L.; Sumner, C.J.; Tizzano, E.F. Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.P., Eds.; Elsevier: London, UK, 2017; pp. 21–40. [Google Scholar]
- Chi, B.; O’Connell, J.D.; Iocolano, A.D.; Coady, J.A.; Yu, Y.; Gangopadhyay, J.; Gygi, S.P.; Reed, R. The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex. Nucleic Acids Res. 2018, 46, 11939–11951. [Google Scholar] [CrossRef] [PubMed]
- Šoltić, D.; Bowerman, M.; Stock, J.; Shorrock, H.K.; Gillingwater, T.H.; Fuller, H.R. Multi-Study Proteomic and Bioinformatic Identification of Molecular Overlap between Amyotrophic Lateral Sclerosis (ALS) and Spinal Muscular Atrophy (SMA). Brain Sci. 2018, 8, 212. [Google Scholar] [CrossRef] [Green Version]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neuroscientist 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Bonafede, R.; Mariotti, R. ALS Pathogenesis and Therapeutic Approaches: The Role of Mesenchymal Stem Cells and Extracellular Vesicles. Front. Cell. Neurosci. 2017, 11, 80. [Google Scholar] [CrossRef]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Droppelmann, C.A.; Campos-Melo, D.; Ishtiaq, M.; Volkening, K.; Strong, M.J. RNA metabolism in ALS: When normal processes become pathological. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 321–326. [Google Scholar] [CrossRef]
- Serio, A.; Patani, R. Concise review: The cellular conspiracy of amyotrophic lateral sclerosis. Stem Cells 2018, 36, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Faenza, I.; Blalock, W.; Bavelloni, A.; Schoser, B.; Fiume, R.; Pacella, S.; Piazzi, M.; D’Angelo, A.; Cocco, L. A role for PLCβ1 in myotonic dystrophies type 1 and 2. FASEB J. 2012, 26, 3042–3048. [Google Scholar] [CrossRef]
- Staats, K.A.; Van Helleputte, L.; Jones, A.R.; Bento-Abreu, A.; Van Hoecke, A.; Shatunov, A.; Simpson, C.L.; Lemmens, R.; Jaspers, T.; Fukami, K.; et al. Genetic ablation of phospholipase C delta 1 increases survival in SOD1(G93A) mice. Neurobiol. Dis. 2013, 60, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.R.; Aslinia, F.; Yale, S.H.; Mazza, J.J. Jean-Martin Charcot: The father of neurology. Clin. Med. Res. 2011, 9, 46–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.S.; Dhull, D.K.; Nalini, A.; Vijayalakshmi, K.; Sathyaprabha, T.N.; Alladi, P.A.; Raju, T.R. Astroglia acquires a toxic neuroinflammatory role in response to the cerebrospinal fluid from amyotrophic lateral sclerosis patients. J. Neuroinflamm. 2016, 13, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis; CRC Press: Boca Raton, FL, USA, 2017; Volume 5, p. 17071. [Google Scholar]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics:Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350. [Google Scholar] [CrossRef]
- Herrando-Grabulosa, M.; Gaja-Capdevila, N.; Vela, J.M.; Navarro, X. Sigma 1 receptor as a therapeutic target for amyotrophic lateral sclerosis. Br. J. Pharmacol. 2021, 178, 1336–1352. [Google Scholar] [CrossRef]
- Darras, B.T.; Farrar, M.A.; Mercuri, E.; Finkel, R.S.; Foster, R.; Hughes, S.G.; Bhan, I.; Farwell, W.; Gheuens, S. An Integrated Safety Analysis of Infants and Children with Symptomatic Spinal Muscular Atrophy (SMA) Treated with Nusinersen in Seven Clinical Trials. CNS Drugs 2019, 33, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Donlin-Asp, P.G.; Fallini, C.; Campos, J.; Chou, C.C.; Merritt, M.E.; Phan, H.C.; Bassell, G.J.; Rossoll, W. The survival of motor neuron protein acts as a molecular chaperone for mRNP assembly. Cell Rep. 2017, 18, 1660–1673. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaidy, S.A.; Mendell, J.R. From Clinical Trials to Clinical Practice: Practical Considerations for Gene Replacement Therapy in SMA Type 1. Pediatric Neurol. 2019, 100, 3–11. [Google Scholar] [CrossRef]
- Ross, L.F.; Kwon, J.M. Spinal Muscular Atrophy: Past, Present, and Future. Neoreviews 2019, 20, e437–e451. [Google Scholar] [CrossRef]
- Tisdale, S.; Lotti, F.; Saieva, L.; Van Meerbeke, J.P.; Crawford, T.O.; Sumner, C.J.; Mentis, G.Z.; Pellizzoni, L. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′-end formation of histone mRNAs. Cell Rep. 2013, 5, 1187–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, B.R.; Wan, L.; Zhang, Z.; Li, P.; Babiash, E.; Duan, J.; Younis, I.; Dreyfuss, G. A U1 snRNP-specific assembly pathway reveals the SMN complex as a versatile hub for RNP exchange. Nat. Struct. Mol. Biol. 2016, 23, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Nash, L.A.; Burns, J.K.; Chardon, J.W.; Kothary, R.; Parks, R.J. Spinal Muscular Atrophy: More than a Disease of Motor Neurons? Curr. Mol. Med. 2016, 16, 779–792. [Google Scholar] [CrossRef]
- Wan, B.; Feng, P.; Guan, Z.; Sheng, L.; Liu, Z.; Hua, Y. A severe mouse model of spinal muscular atrophy develops early systemic inflammation. Hum. Mol. Genet. 2018, 27, 4061–4076. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, D.; Le Verche, V.; Jacquier, A.; Ikiz, B.; Przedborski, S.; Re, D.B. Inflammation in ALS and SMA: Sorting out the good from the evil. Neurobiol. Dis. 2010, 37, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguise, M.O.; Kothary Deguise, M.O.; Kothary, R. New insights into SMA pathogenesis: Immune dysfunction and neuroinflammation. Ann. Clin. Transl. Neurol. 2017, 4, 522–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, S.; Osanai, D.; Takahashi, K.; Nakamura, S.; Shimazawa, M.; Hara, H. Survival motor neuron protein regulates oxidative stress and inflammatory response in microglia of the spinal cord in spinal muscular atrophy. J. Pharmacol. Sci. 2020, 144, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Schellino, R.; Boido, M.; Borsello, T.; Vercelli, A. Pharmacological c-Jun NH(2)-Terminal Kinase (JNK) Pathway Inhibition Reduces Severity of Spinal Muscular Atrophy Disease in Mice. Front. Mol. Neurosci. 2018, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Genabai, N.K.; Ahmad, S.; Zhang, Z.; Jiang, X.; Gabaldon, C.A.; Gangwani, L. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum. Mol. Genet. 2015, 24, 6986–7004. [Google Scholar] [CrossRef] [Green Version]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Cowan, M.; Petri, W.A., Jr. Microglia: Immune Regulators of Neurodevelopment. Front. Immunol. 2018, 9, 2576. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Phani, S.; Berengere Re, D.; Przedborski, S. The Role of the Innate Immune System in ALS. Front Pharmacol. 2012, 3, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Singh, N.N. Mechanism of Splicing Regulation of Spinal Muscular Atrophy Genes. Adv. Neurobiol. 2018, 20, 31–61. [Google Scholar] [PubMed]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; Shepherd, C.E.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia—Amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef]
- Keinath, M.C.; Prior, D.E.; Prior, T.W. Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance. Appl. Clin. Genet. 2021, 14, 11–25. [Google Scholar] [CrossRef]
- McCombe, P.A.; Henderson, R.D. The Role of Immune and Inflammatory Mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254. [Google Scholar] [CrossRef]
- Deguise, M.O.; De Repentigny, Y.; McFall, E.; Auclair, N.; Sad, S.; Kothary, R. Immune dysregulation may contribute to disease pathogenesis in spinal muscular atrophy mice. Hum. Mol. Genet. 2017, 26, 801–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain. 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weskamp, K.; Barmada, S.J. TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res. 2018, 1693, 67–74. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Huntley, M.L.; Perry, G.; Wang, X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018, 146, 7–20. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Dutta, K.; Phaneuf, D.; Sobue, G.; Julien, J.P. Ubiquilin-2 drives NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol. Brain 2015, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, H. Identification of a new causative gene of amyotrophic lateral sclerosis; optineurin. Rinsho Shinkeigaku 2012, 52, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Suzuki, S.; Okubo, S.; Ohuchi, K.; Takahashi, K.; Nakamura, S.; Shimazawa, M.; Fuji, K.; Hara, H. Discovery of a CNS penetrant small molecule SMN2 splicing modulator with improved tolerability for spinal muscular atrophy. Sci. Rep. 2020, 10, 17472. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. SMN1 functions as a novel inhibitor for TRAF6-mediated NF-κB signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E. TNuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Pilato, C.M.; Park, J.H.; Kong, L.; D’Ydewalle, C.; Valdivia, D.; Chen, K.S.; Griswold-Prenner, I.; Sumner, C.J. Motor neuron loss in SMA is not associated with somal stress-activated JNK/c-Jun signaling. Hum. Mol. Genet. 2019, 28, 3282–3292. [Google Scholar] [CrossRef] [PubMed]
- Tortarolo, M.; Vallarola, A.; Lidonnici, D.; Battaglia, E.; Gensano, F.; Spaltro, G.; Fiordaliso, F.; Corbelli, A.; Garetto, S.; Martini, E.; et al. Lack of TNF-alpha receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J. Neurochem. 2015, 135, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Götz, R. Regulation of neuronal cell death and differentiation by NGF and IAP family members. Adv. Res. Neurodegener. 2000, 60, 247–259. [Google Scholar]
- Moisse, K.; Strong, M.J. Innate immunity in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Abati, E.; Citterio, G.; Bresolin, N.; Comi, G.P.; Corti, S. Glial cells involvement in spinal muscular atrophy: Could SMA be a neuroinflammatory disease? Neurobiol. Dis. 2020, 140, 104870. [Google Scholar] [CrossRef]
- Cervero, C.; Blasco, A.; Tarabal, O.; Casanovas, A.; Piedrafita, L.; Navarro, X.; Esquerda, J.E.; Calderó, J. Glial activation and central synapse loss, but not motoneuron degeneration, are prevented by the sigma-1 receptor agonist pre-084 in the SMN2B/mouse model of spinal muscular atrophy. J. Neuropathol. Exp. Neurol. 2018, 77, 577–597. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosi, N.; Rossi, S.; Gerbino, V.; Cozzolino, M. Rac1 at the crossroad of actin dynamics and neuroinflammation in Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2014, 8, 279. [Google Scholar]
- Grottelli, S.; Mezzasoma, L.; Scarpelli, P.; Cacciatore, I.; Cellini, B.; Bellezza, I. Cyclo(His-Pro) inhibits NLRP3 inflammasome cascade in ALS microglial cells. Mol. Cell Neurosci. 2019, 94, 23–31. [Google Scholar] [CrossRef]
- de Araújo Boleti, A.P.; de Oliveira Flores, T.M.; Moreno, S.E.; Anjos, L.D.; Mortari, M.R.; Migliolo, L. Neuroinflammation: An overview of neurodegenerative and metabolic diseases and of biotechnological studies. Neurochem. Int. 2020, 136, 104714. [Google Scholar] [CrossRef]
- Thelen, M.P.; Wirth, B.; Kye, M.J. Mitochondrial defects in the respiratory complex I contribute to impaired translational initiation via ROS and energy homeostasis in SMA motor neurons. Acta Neuropathol. Commun. 2020, 8, 223. [Google Scholar] [CrossRef]
- Gobbi, P.; Castaldo, P.; Minelli, A.; Salucci, S.; Magi, S.; Corcione, E.; Amoroso, S. Mitochondrial localization of Na+/Ca2+ exchangers NCX1-3 in neurons and astrocytes of adult rat brain in situ. Pharmacol. Res. 2007, 56, 556–565. [Google Scholar] [CrossRef]
- Salucci, S.; Ambrogini, P.; Lattanzi, D.; Betti, M.; Gobbi, P.; Galati, C.; Galli, F.; Cuppini, R.; Minelli, A. Maternal dietary loads of alpha-tocopherol increase synapse density and glial synaptic coverage in the hippocampus of adult offspring. Eur. J. Histochem. 2014, 58, 2355. [Google Scholar] [CrossRef] [Green Version]
- Kacerovsky, J.B.; Murai, K.K. Stargazing: Monitoring subcellular dynamics of brain astrocytes. Neuroscience 2016, 323, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Sargsyan, S.A.; Monk, P.N.; Shaw, P.J. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia 2005, 51, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Heneka MT, Kummer MP, Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Zondler, L.; Müller, K.; Khalaji, S.; Bliederhäuser, C.; Ruf, W.P.; Grozdanov, V.; Thiemann, M.; Fundel-Clemes, K.; Freischmidt, A.; Holzmann, K.; et al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol. 2016, 132, 391–411. [Google Scholar] [CrossRef]
- Vukojicic, A.; Delestrée, N.; Fletcher, E.V.; Pagiazitis, J.G.; Sankaranarayanan, S.; Yednock, T.A.; Barres, B.A.; Mentis, G.Z. The classical complement pathway mediates microglia-dependent remodeling of spinal motor circuits during development and in SMA. Cell Rep. 2019, 29, 3087–3100. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, M.; Keyhanian, K.; Douthwright, C. Glial Cell Dysfunction in C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Cells 2021, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Harland, M.; Torres, S.; Liu, J.; Wang, F. Neuronal Mitochondria Modulation of LPS-Induced Neuroinflammation. J Neurosci. 2020, 40, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.C. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef] [PubMed]
- Bader, V.; Winklhofer, K.F. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 2020, 99, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, E.; Beltrán, S.; Labrador, L.; Manque, P.; Nassif, M.; Woehlbier, U. Implications of Selective Autophagy Dysfunction for ALS Pathology. Cells 2020, 9, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teyssou, E.; Takeda, T.; Lebon, V.; Boillée, S.; Doukouré, B.; Bataillon, G.; Sazdovitch, V.; Cazeneuve, C.; Meininger, V.; LeGuern, E.; et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: Genetics and neuropathology. Acta Neuropathol. 2013, 125, 511–522. [Google Scholar] [CrossRef]
- Chantal Sellier, C.; Campanari, M.L.; Corbier, C.J.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef]
- Corcia, P.; Couratier, P.; Blasco, H.; Andres, C.R.; Beltran, S.; Meininger, V.; Vourc’h, P. Genetics of amyotrophic lateral sclerosis. Rev. Neurol. 2017, 173, 254–262. [Google Scholar] [CrossRef]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef]
- Rodriguez-Muela, N.; Parkhitko, A.; Grass, T.; Gibbs, R.M.; Norabuena, E.M.; Perrimon, N.; Singh, R.; Rubin, L.L. Blocking p62-dependent SMN degradation ameliorates spinal muscular atrophy disease phenotypes. J. Clin. Investig. 2018, 128, 3008–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salucci, S.; Baldassarri, V.; Canonico, B.; Burattini, S.; Battistelli, M.; Guescini, M.; Papa, S.; Stocchi, V.; Falcieri, E. Melatonin behavior in restoring chemical damaged C2C12 myoblasts. Microsc. Res. Tech. 2016, 79, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Salucci, S.; Battistelli, M.; Baldassarri, V.; Burini, D.; Falcieri, E.; Burattini, S. Melatonin prevents mitochondrial dysfunctions and death in differentiated skeletal muscle cells. Microsc. Res. Tech. 2017, 80, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Garabadu, D.; Agrawal, N.; Sharma, A.; Sharma, S. Mitochondrial metabolism: A common link between neuroinflammation and neurodegeneration. Behav Pharmacol. 2019, 30, 642–652. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Pichaud, N.; Berube, R.; Cote, G.; Belzile, C.; Dufresne, F.; Morrow, G.; Tanguay, R.M.; Rand, D.M.; Blier, P.U. Age dependent dysfunction of mitochondrial and ROS metabolism induced by mitonuclear mismatch. Front. Genet. 2019, 10, 130. [Google Scholar] [CrossRef]
- Valera-Alberni, M.; Canto, C. Mitochondrial stress management: A dynamic journey. Cell Stress 2018, 2, 253–274. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Yadav, A.; Tiwari, S.K.; Seth, B.; Chauhan, L.K.; Khare, P.; Ray, R.S.; Chaturvedi, R.K. Dynamin-related protein 1 inhibition mitigates bisphenol A-mediated alterations in mitochondrial dynamics and neural stem cell proliferation and differentiation. J. Biol. Chem. 2016, 291, 15923–15939. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.; Chaturvedi, R.K. Mitochondrial Protein Import Dysfunction in Pathogenesis of Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 1418–1437. [Google Scholar] [CrossRef]
- Stanga, S.; Boido, M.; Kienlen-Campard, P. How to Build and to Protect the Neuromuscular Junction: The Role of the Glial Cell Line-Derived Neurotrophic Factor. Int. J. Mol. Sci. 2020, 22, 136. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Ince, P.G.; Shaw, P.J. Mitochondrial involvement in amyotrophic lateral sclerosis. Neurochem. Int. 2002, 40, 543–551. [Google Scholar] [CrossRef]
- Bahadorani, S.; Mukai, S.T.; Rabie, J.; Beckman, J.S.; Phillips, J.P.; Hilliker, A.J. Expression of zinc-deficient human superoxide dismutase in Drosophila neurons produces a locomotor defect linked to mitochondrial dysfunction. Neurobiol. Aging 2013, 34, 2322–2330. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 2014, 1842, 1295–1301. [Google Scholar] [CrossRef] [Green Version]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Lotti, F.; Imlach, W.L.; Saieva, L.; Beck, E.S.; Hao, L.T.; Li, D.K.; Jiao, W.; Mentis, G.Z.; Beattie, C.E.; McCabe, B.D.; et al. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 2012, 151, 440–454. [Google Scholar] [CrossRef] [Green Version]
- Boido, M.; Vercelli, A. Neuromuscular junctions as key contributors and therapeutic targets in spinal muscular atrophy. Front. Neuroanat. 2016, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, L.G.; Angelo, Y.S.; Iglesias, A.H.; Peron, J.P.S. Unraveling the Link Between Mitochondrial Dynamics and Neuroinflammation. Front. Immunol. 2021, 2, 624919. [Google Scholar] [CrossRef] [PubMed]
- Hellbach, N.; Peterson, S.; Haehnke, D.; Shankar, A.; LaBarge, S.; Pivaroff, C.; Thomas, C.; McCarthy, K.; Ebeling, M.; Metzger, F.; et al. Impaired myogenic development, differentiation and function in hESC-derived SMA myoblasts and myotubes. PLoS ONE 2018, 13, e1006744. [Google Scholar] [CrossRef]
- James, R.; Chaytow, H.; Ledahawsky, L.M.; Gillingwater, T.H. Revisiting the role of mitochondria in spinal muscular atrophy. Cell Mol. Life Sci. 2021, 78, 4785–4804. [Google Scholar] [CrossRef] [PubMed]
- Anderton, R.S.; Meloni, B.P.; Mastaglia, F.L.; Boulos, S. Spinal muscular atrophy and the anti-apoptotic role of survival of motor neuron (SMN) protein. Mol. Neurobiol. 2013, 47, 821–832. [Google Scholar] [CrossRef]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Watihayati, M.S.; Fatemeh, H.; Marini, M.; Atif, A.B.; Zahiruddin, W.M.; Teguh Haryo Sasongko, T.H.; Tang, T.H.; Zabidi-Hussin, Z.A.; Nishio, H.; Zilfalil, B.A. Combination of SMN2 copy number and NAIP deletion predicts disease severity in spinal muscular atrophy. Brain Dev. 2009, 31, 42–45. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Vance, R.E. NAIPs: Building an innate immune barrier against bacterial pathogens: NAIPs function as sensors that initiate innate immunity by detection of bacterial proteins in the host cell cytosol. BioEssays 2012, 34, 589–598. [Google Scholar] [CrossRef]
- Sareen, D.; Ebert, A.D.; Heins, B.M.; McGivern, J.V.; Ornelas, L.; Svendsen, C.N. Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS ONE 2012, 7, e39113. [Google Scholar] [CrossRef]
- Maretina, M.A.; Zheleznyakova, G.Y.; Lanko, K.M.; Egorova, A.A.; Baranov, V.S.; Kiselev., A.V. Molecular Factors Involved in Spinal Muscular Atrophy Pathways as Possible Disease-modifying Candidates. Curr. Genomics 2018, 19, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Shibata, N.; Saito, K.; Kobayashi, M.; Osawa, M. New insights into the pathogenesis of spinal muscular atrophy. Brain Dev. 2011, 33, 321–331. [Google Scholar] [CrossRef]
- Bonafede, R.; Brandi, J.; Manfredi, M.; Scambi, I.; Schiaffino, L.; Merigo, F.; Turano, E.; Bonetti, B.; Marengo, E.; Cecconi, D.; et al. The Anti-Apoptotic Effect of ASC-Exosomes in an In Vitro ALS Model and Their Proteomic Analysis. Cells 2019, 8, 1087. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.L.; Wu, L.S.; Lee, M.; Chang, C.W.; Cheng, W.C.; Fang, Y.S.; Chen, Y.R.; Cheng, P.L.; Shen, C.J. A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol. Commun. 2020, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Wu, H.T.; Li, X.X.; Yu, Y.; Gu, R.Z.; Lan, R.; Qin, X.Y. Edaravone protects rat astrocytes from oxidative or neurotoxic inflammatory insults by restoring Akt/Bcl-2/Caspase-3 signaling axis. IBRO Rep. 2020, 8, 122–128. [Google Scholar] [CrossRef]
- Giordano, F.M.; Burattini, S.; Buontempo, F.; Canonico, B.; Martelli, A.M.; Papa, S.; Sampaolesi, M.; Falcieri, E.; Salucci, S. Diet Modulation Restores Autophagic Flux in Damaged Skeletal Muscle Cells. J. Nutr. Health Aging 2019, 23, 739–745. [Google Scholar] [CrossRef]
- Von Stockum, S.; Nardin, A.; Schrepfer, E.; Ziviani, E. Mitochondrial dynamics and mitophagy in parkinson’s disease: A fly point of view. Neurobiol. Dis. 2016, 90, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Piazzi, M.; Bavelloni, A.; Faenza, I.; Blalock, W. Glycogen synthase kinase (GSK)-3 and the double-strand RNA-dependent kinase, PKR: When two kinases for the common good turn bad. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118769. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Khoramian Tusi, S.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef] [PubMed]
- Ghadge, G.D.; Sonobe, Y.; Camarena, A.; Drigotas, C.; Rigo, F.; Ling, K.K.; Roos, R.P. Knockdown of GADD34 in neonatal mutant SOD1 mice ameliorates ALS. Neurobiol. Dis. 2020, 136, 104702. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, N.; Saxena, S. ER strikes again: Proteostasis Dysfunction in ALS. EMBO J. 2016, 35, 798–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Muela, N. Autophagy in motor neuron diseases. Prog. Mol. Biol. Transl. Sci. 2020, 172, 157–202. [Google Scholar] [PubMed]
- Ryter, S.W.; Bhatia, D.; Choi, M.E. Autophagy: A lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid. Redox Signal. 2019, 30, 138–159. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 2021, 17, 385–401. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Huang, C.; Yan, S.; Zhang, Z. Maintaining the Balance of TDP-43, Mitochondria, and Autophagy: A Promising Therapeutic Strategy for Neurodegenerative Diseases. Transl. Neurodegener. 2020, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Arakawa, H.; Wang, L.; Okolo, O.; Siedlak, S.L.; Jiang, Y.; Gao, J.; Xie, F.; Petersen, R.B.; Wang, X. Motor-Coordinative and Cognitive Dysfunction Caused by Mutant TDP-43 Could Be Reversed by Inhibiting Its Mitochondrial Localization. Mol. Ther. 2017, 25, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madruga, E.; Maestro, I.; Martínez, A. Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 2021, 22, 740. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Hoffman, S.; Reddi, P.P.; Singh, R.N. Spinal muscular atrophy: Broad disease spectrum and sex-specific phenotypes. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166063. [Google Scholar] [CrossRef] [PubMed]
- Acsadi, G.; Lee, I.; Li, X.; Khaidakov, M.; Pecinova, A.; Parker, G.C.; Hüttemann, M. Mitochondrial dysfunction in a neural cell model of spinal muscular atrophy. J. Neurosci. Res. 2009, 87, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Manzaneda, M.; Franco-Espin, J.; Tejero, R.; Cano, R.; Tabares, L. Calcium is reduced in presynaptic mitochondria of motor nerve terminals during neurotransmission in SMA mice. Hum. Mol. Genet. 2021, 30, 629–643. [Google Scholar] [CrossRef]
- Custer, S.K.; Androphy, E.J. Autophagy dysregulation in cell culture and animals models of spinal muscular atrophy. Mol. Cell Neurosci. 2014, 61, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Periyakaruppiah, A.; de la Fuente, S.; Arumugam, S.; Bahí, N.; Garcera, A.; Soler, R.M. Autophagy modulators regulate survival motor neuron protein stability in motoneurons. Exp. Neurol. 2016, 283, 287–297. [Google Scholar] [CrossRef]
- Piras, A.; Schiaffino, L.; Boido, M.; Valsecchi, V.; Guglielmotto, M.; De Amicis, E.; Puyal, J.; Garcera, A.; Tamagno, E.; Soler, R.M.; et al. Inhibition of autophagy delays motoneuron degeneration and extends lifespan in a mouse model of spinal muscular atrophy. Cell Death Dis. 2017, 8, 3223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, W.S.; Chad, D.A. Neuromuscular Disorders. Semin. Neurol. 2015, 35, 325. [Google Scholar]
- Ferlini, A.; Goyenvalle, A.; Muntoni, F. RNA-targeted drugs for neuromuscular diseases. Science 2021, 371, 29–31. [Google Scholar] [CrossRef]
- Corcia, P.; Vourc’h, P.; Blasco, H.; Couratier, P.; Dangoumau, A.; Bellance, R.; Desnuelle, C.; Viader, F.; Pautot, V.; Millecamps, S.; et al. Phenotypic and genotypic studies of ALS cases in ALS-SMA families. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Béland, L.C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in amyotrophic lateral sclerosis: Blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef] [PubMed]
- Ashford, B.A.; Boche, D.; Cooper-Knock, J.; Heath, P.R.; Simpson, J.E.; Highley, J.R. Review: Microglia in motor neuron disease. Neuropathol. Appl. Neurobiol. 2021, 47, 179–197. [Google Scholar] [CrossRef]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. The effects of statins on microglial cells to protect against neurodegenerative disorders: A mechanistic review. Biofactors 2020, 46, 309–325. [Google Scholar] [CrossRef]
- Boczonadi, V.; King, M.S.; Smith, A.C.; Olahova, M.; Bansagi, B.; Roos, A.; Eyassu, F.; Borchers, C.; Ramesh, V.; Lochmüller, H.; et al. Mitochondrial oxodicarboxylate carrier deficiency is associated with mitochondrial DNA depletion and spinal muscular atrophy-like disease. Genet. Med. 2018, 20, 1224–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripolone, M.; Ronchi, D.; Violano, R.; Vallejo, D.; Fagiolari, G.; Barca, E.; Lucchini, V.; Colombo, I.; Villa, L.; Berardinelli, A.; et al. Impaired Muscle Mitochondrial Biogenesis and Myogenesis in Spinal Muscular Atrophy. JAMA Neurol. 2015, 72, 666–675. [Google Scholar] [CrossRef]
- Cabral-Costa, J.V.; Kowaltowski, A.J. Neurological disorders and mitochondria. Mol. Asp. Med. 2020, 71, 100826. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Motor Neuron Disorders | Pheripheral Nerve Diseases | Neuro-Muscular Junction Diseases | Muscular Dytrophies | Myopathies |
|---|---|---|---|---|
| Amiotrophic Lateral Sclerosis (ALS) | Charcot-Marie-Tooth disease (CMT) | Myastenia Gravis (MG) | Duchenne Muscular Dystrophy (DMD) | Inflammatory Myopathies |
| Spinal muscle atrophy (SMA) | Giant Axonal Neuropathy | Congenital Myasthenic Syndromes (CMS) | Becker Muscular Dystrophy (BMD) | Metabolic Myopathies |
| ALS and SMA Similarities |
|---|
| Neuroinflammation |
| Motor neuron death |
| Common molecular pathways |
| Loss of muscle mass and strenght |
| Weakness, breathing difficulties |
| Muscle atrophy |
| M1 Neuroinflammation Induction | M2 Homeostasis Mainteinance |
|---|---|
| IL-17 | IL-4 |
| IFNγ | IL-10 |
| TNF-α | TGF-β |
| Inflammatory cytochine production | anti-inflammatory molecule production |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salucci, S.; Bartoletti Stella, A.; Battistelli, M.; Burattini, S.; Bavelloni, A.; Cocco, L.I.; Gobbi, P.; Faenza, I. How Inflammation Pathways Contribute to Cell Death in Neuro-Muscular Disorders. Biomolecules 2021, 11, 1109. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081109
Salucci S, Bartoletti Stella A, Battistelli M, Burattini S, Bavelloni A, Cocco LI, Gobbi P, Faenza I. How Inflammation Pathways Contribute to Cell Death in Neuro-Muscular Disorders. Biomolecules. 2021; 11(8):1109. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081109
Chicago/Turabian StyleSalucci, Sara, Anna Bartoletti Stella, Michela Battistelli, Sabrina Burattini, Alberto Bavelloni, Lucio Ildebrando Cocco, Pietro Gobbi, and Irene Faenza. 2021. "How Inflammation Pathways Contribute to Cell Death in Neuro-Muscular Disorders" Biomolecules 11, no. 8: 1109. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081109