
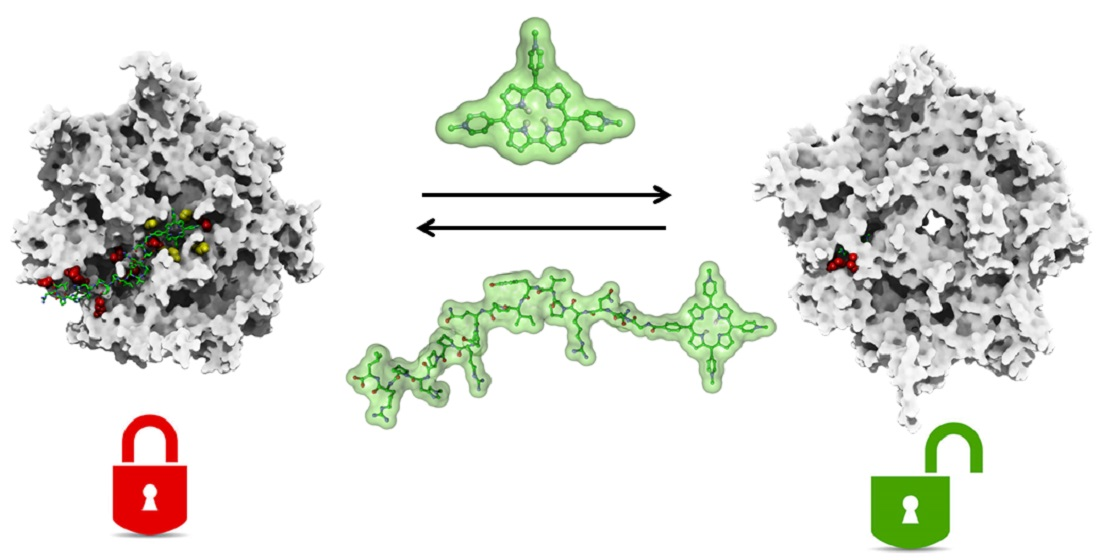
Modulation of the 20S Proteasome Activity by Porphyrin Derivatives Is Steered through Their Charge Distribution

 , ,
, ,  , , , , ,
, , , , ,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
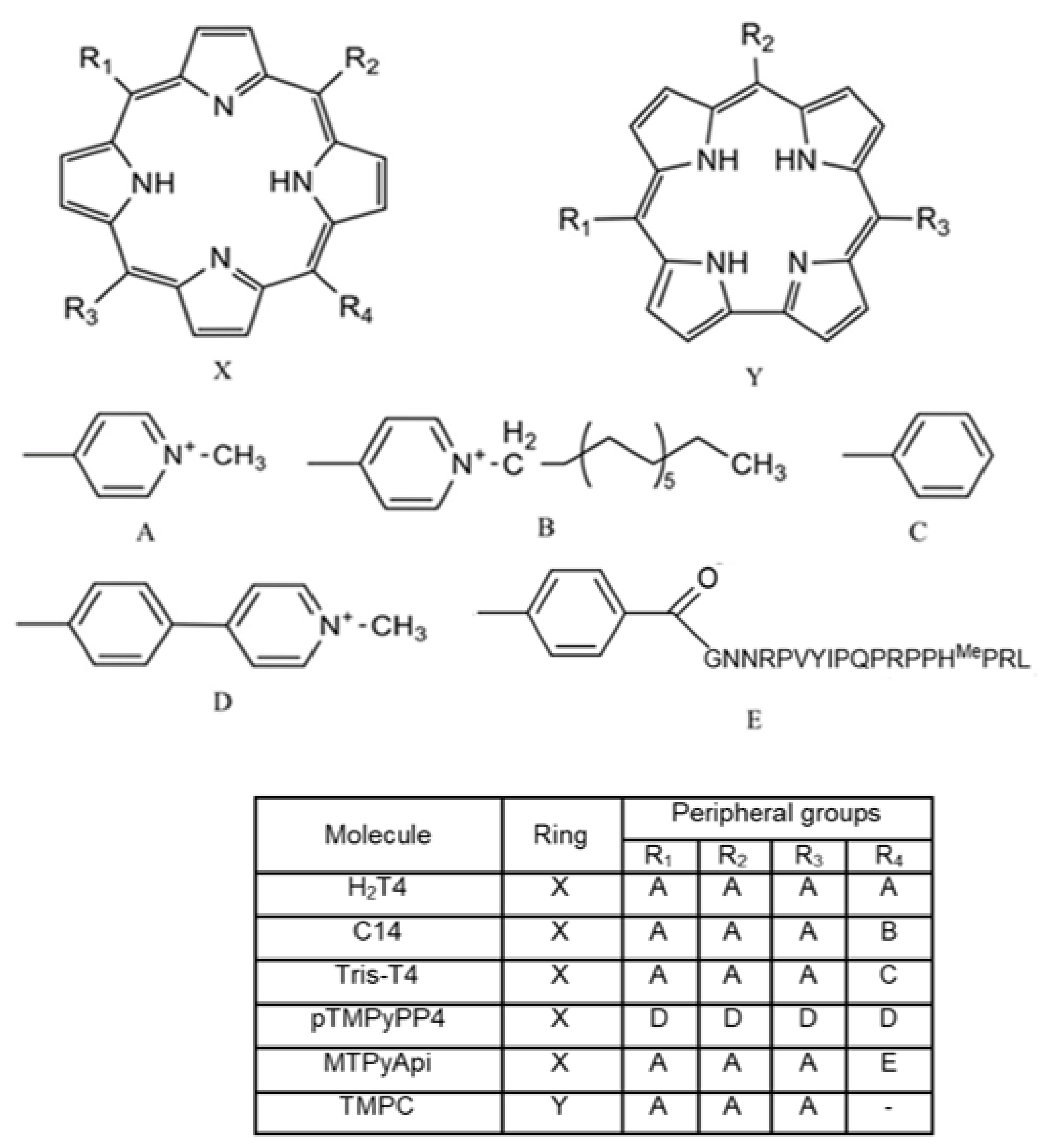
2.1. Chemicals
2.2. Cell Culture
2.3. 20S Proteasomal Chymotryptic-like Activity Assays and Analysis of Kinetic Data
2.4. NMR Spectroscopy
2.5. Molecular Modeling
2.5.1. Calculation of the Chemical–Physical Properties of New Porphyrins
2.5.2. Docking Studies on Human 20S Proteasome
2.6. Cell Viability Assay and Proteasome-GloTM Cell-Based Assay
3. Results and Discussion
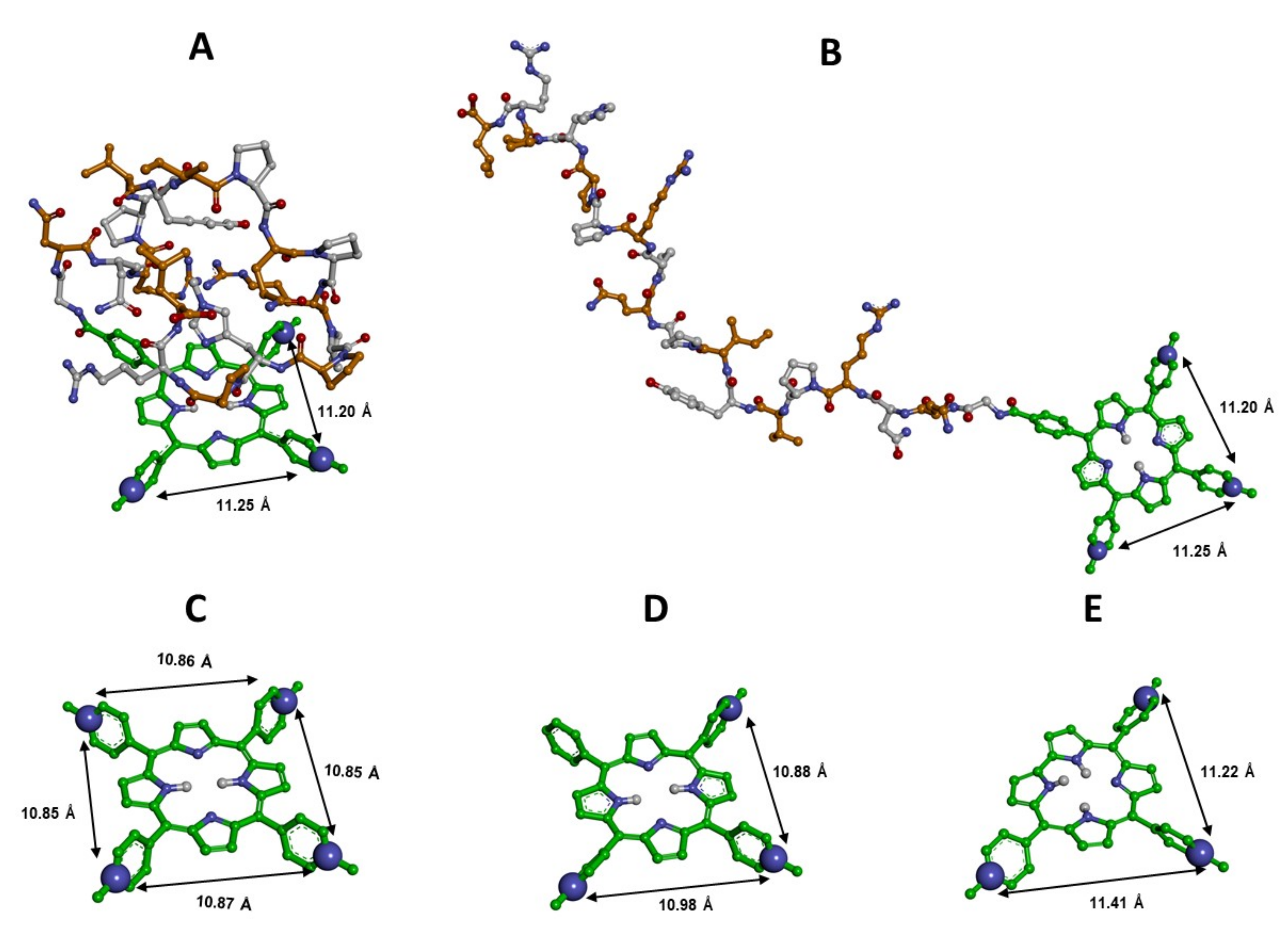
3.1. Structural Features of the New Ligands
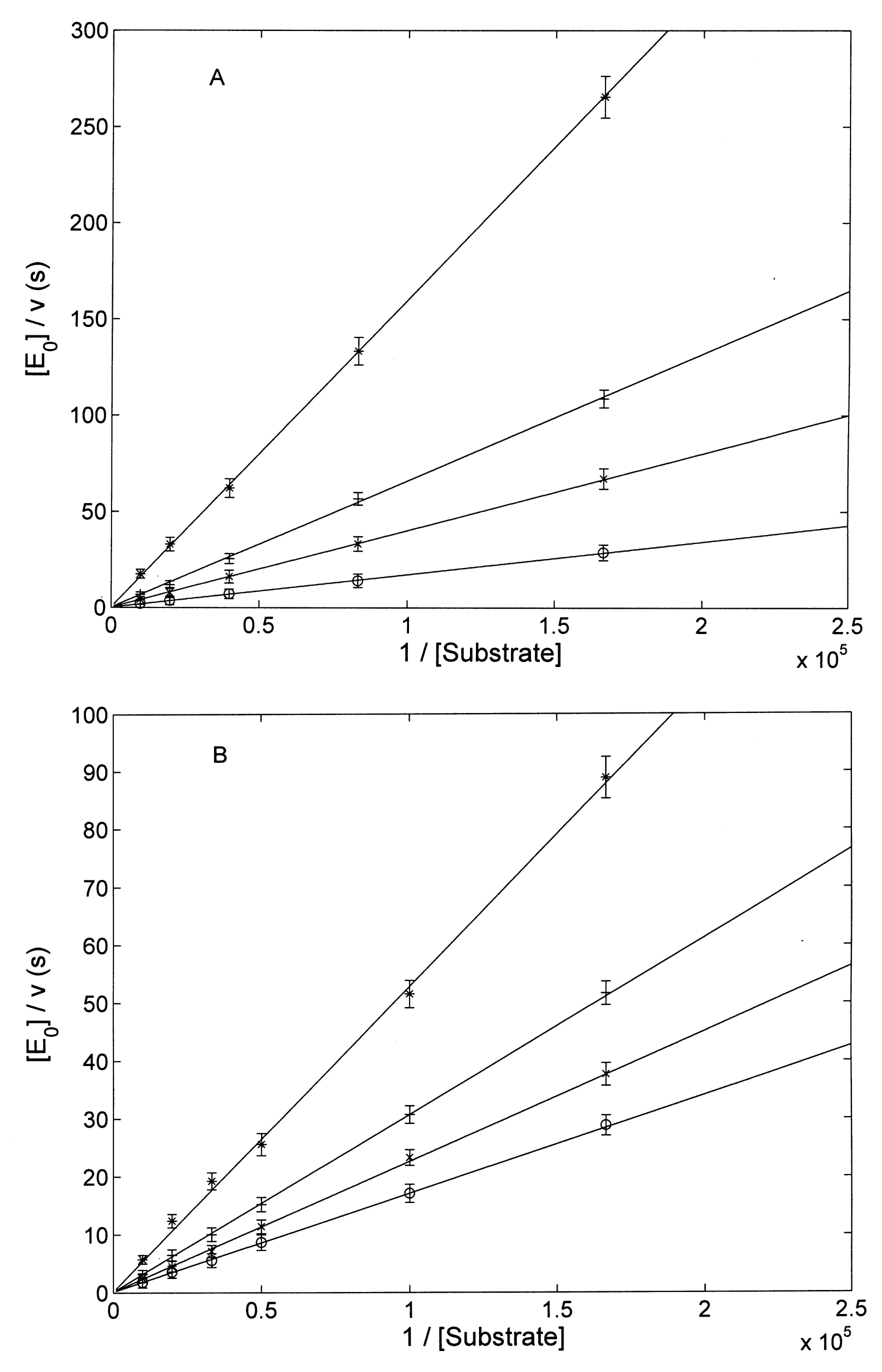
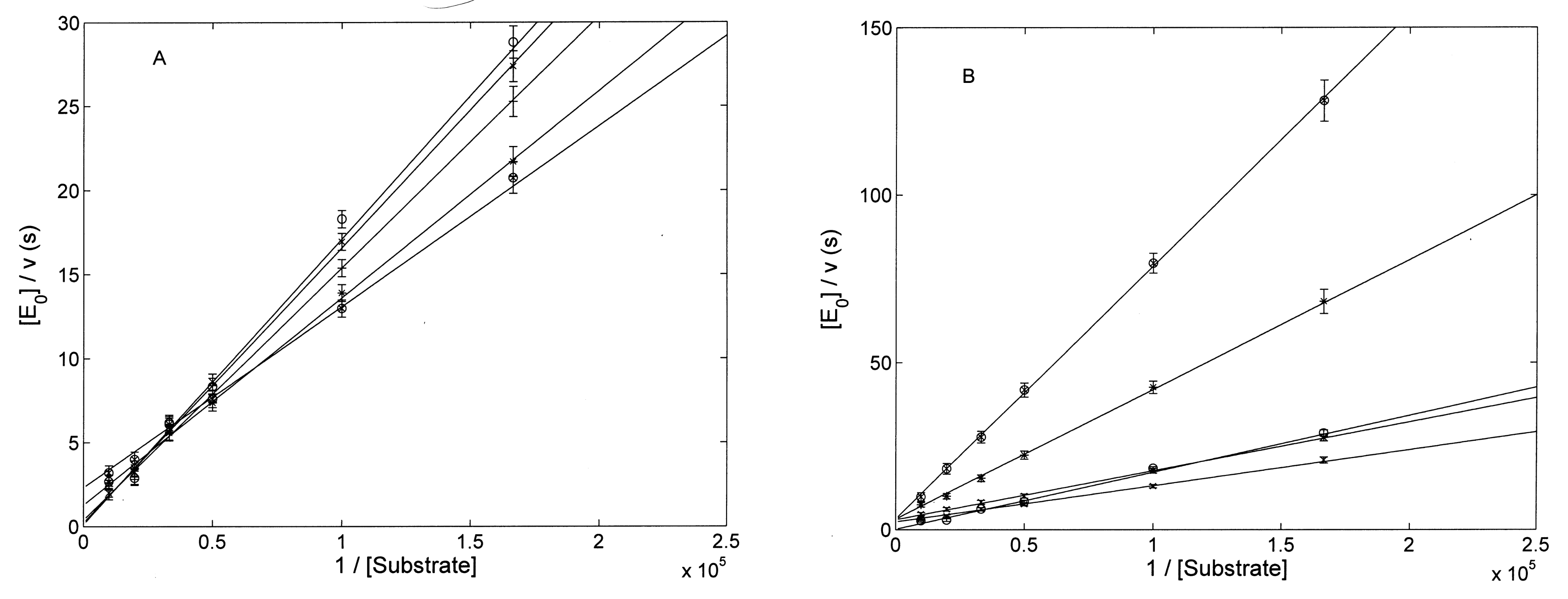
3.2. Proteasome Assays on the Isolated 20S CP
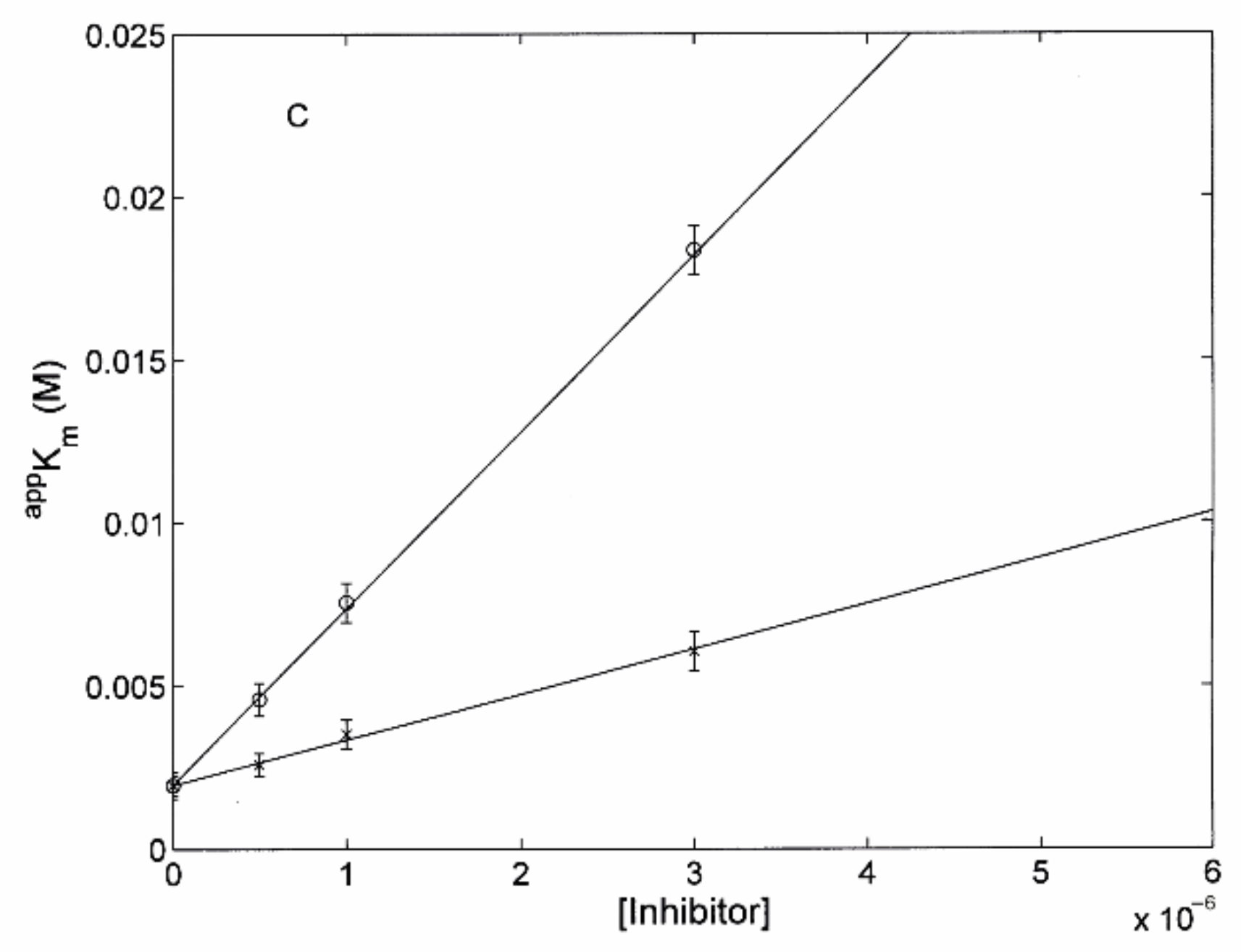
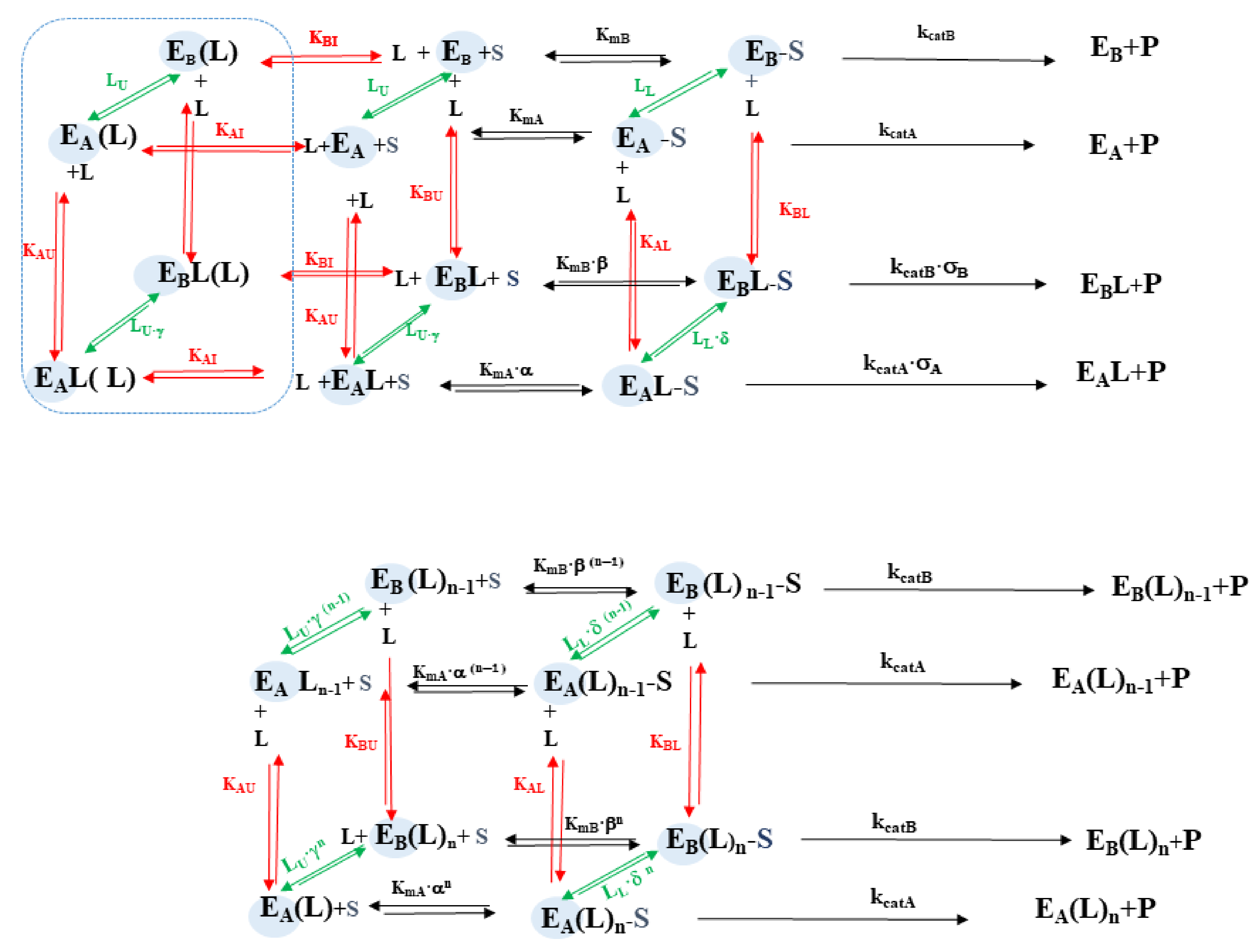
3.2.1. Comparison of H2T4 and MTPyApi
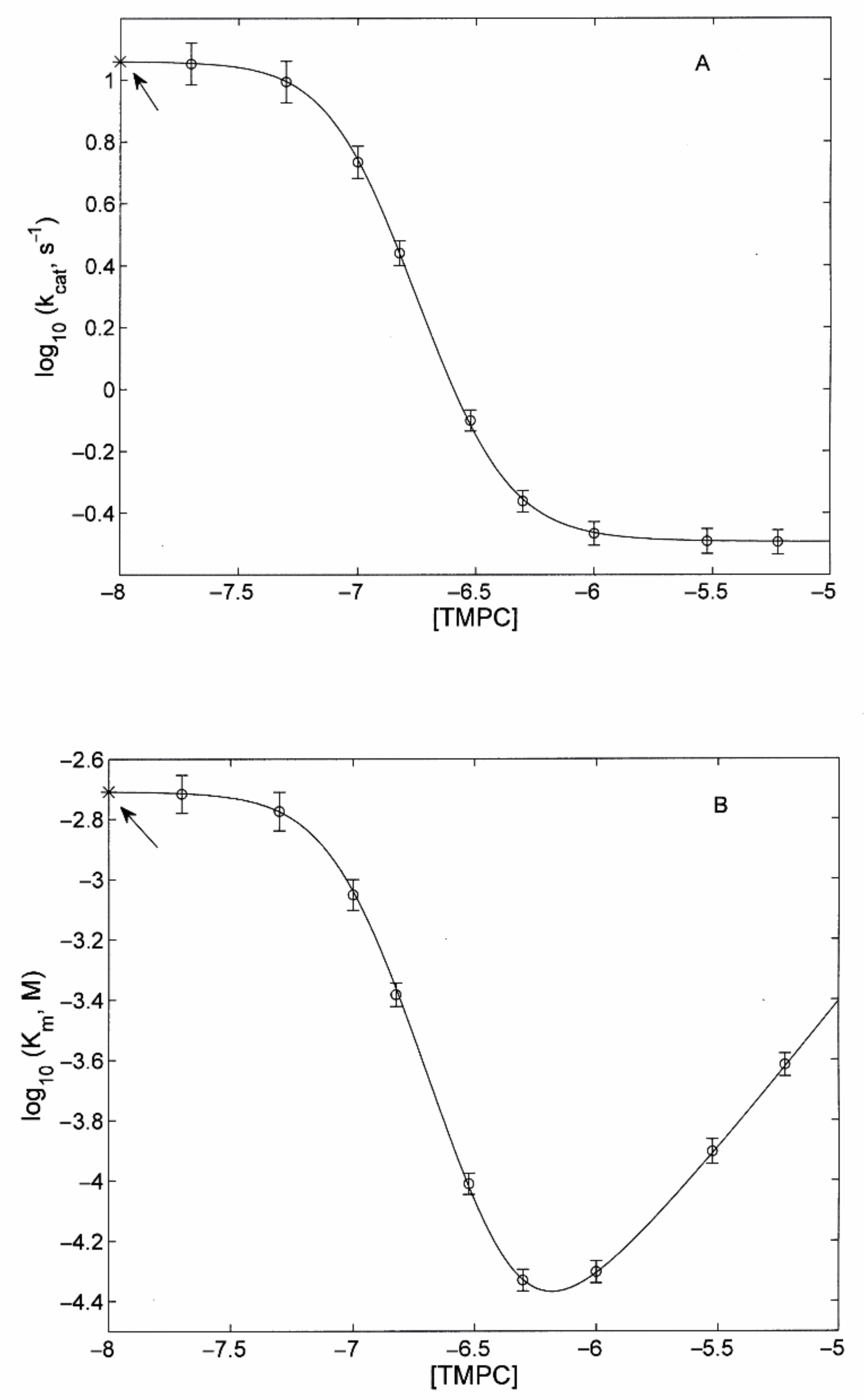
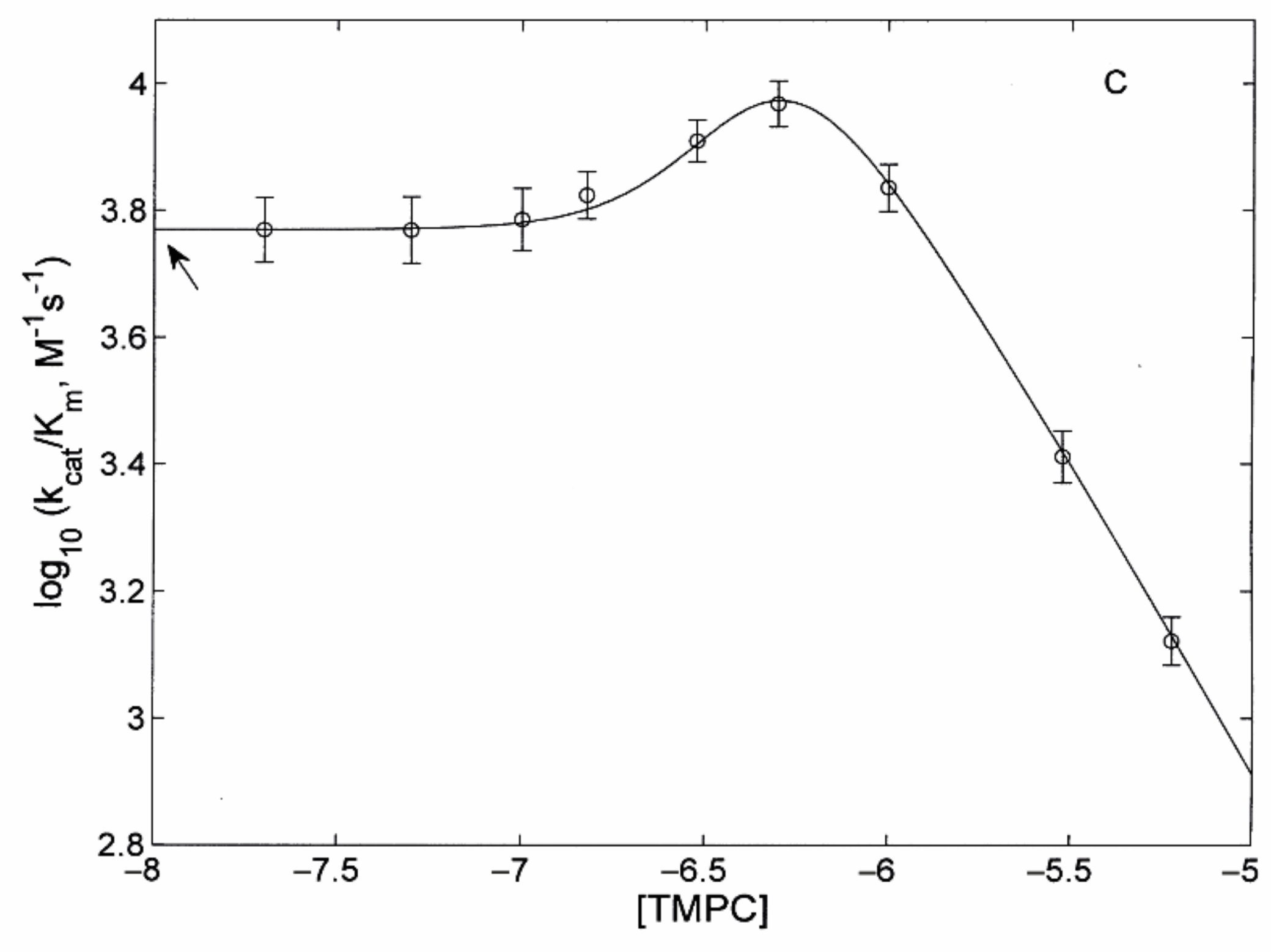
3.2.2. Modulatory Properties of TMPC and Its Comparison with Tris-T4
- (a)
- both porphyrins appear to activate the h20S by inducing a conformational transition from the A to the B functional state through a cooperative action, involving, in both cases, a clustering of (at least) three porphyrin molecules;
- (b)
- the main determinant of the higher affinity of TMPC as a proteasome activator is the binding of the porphyrin to the substrate-bound A state (with an almost thirty-fold larger binding constant KAL with respect to Tris-T4; see Table 1);
- (c)
- in the B state, binding constants for TMPC are only moderately (two- to three-fold) higher than for Tris-T4 (see Table 1);
- (d)
- (e)
- although an inhibitory site for Tris-T4 had been detected only in y20S, its affinity was much lower than that observed here for TMPC in h20S (see Table 1 and Ref. [30]); in y20S the value of KAI for Tris-T4 was very low (i.e., KAI ≤ 104 M−1), so it could be ignored. In the case of TMPC, KAI (= 106 M−1; see Table 1) is instead large enough to play a significant role, and we had to take it into account.
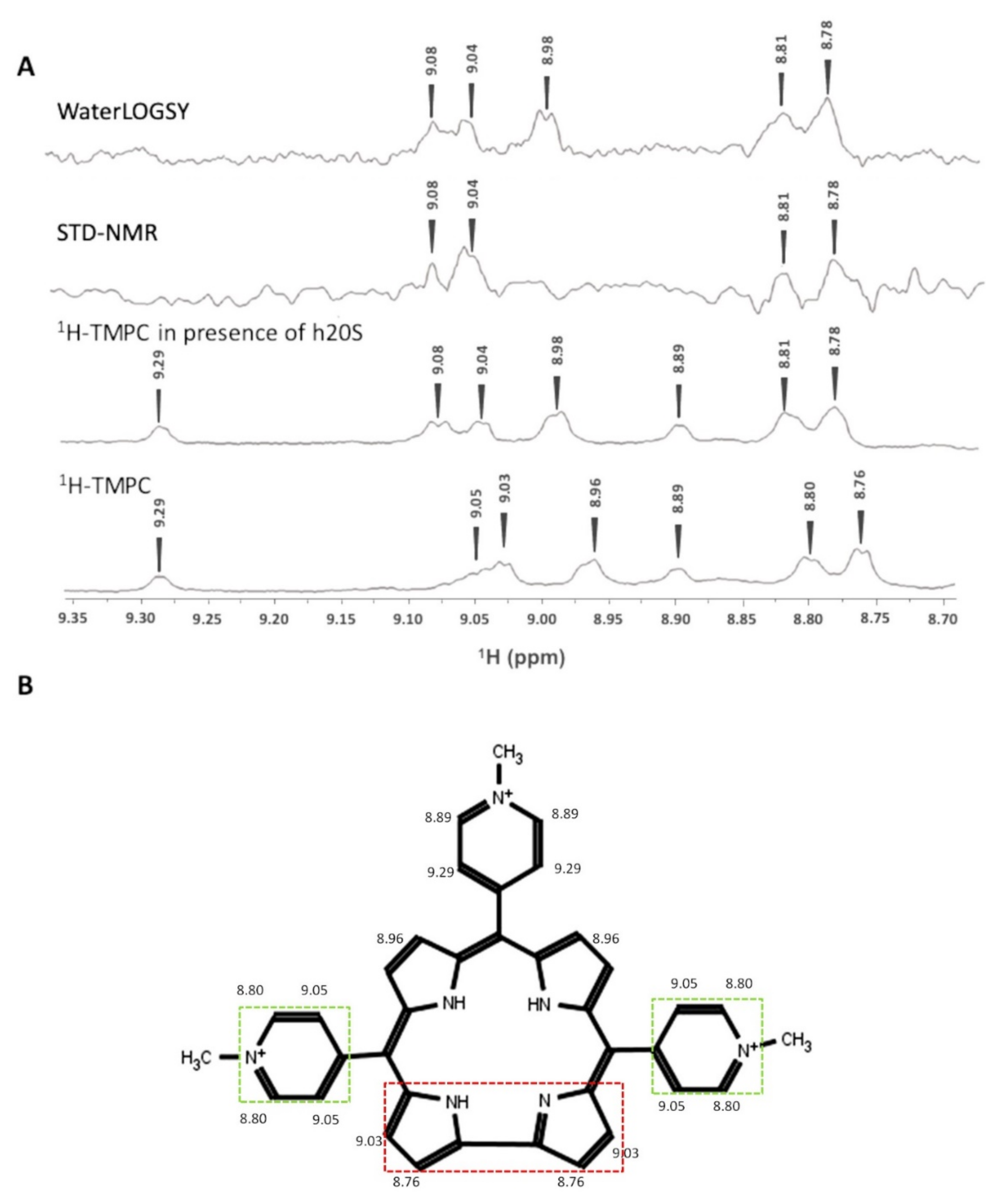
3.3. Interaction of TMPC by NMR Spectroscopy
3.4. Docking Studies
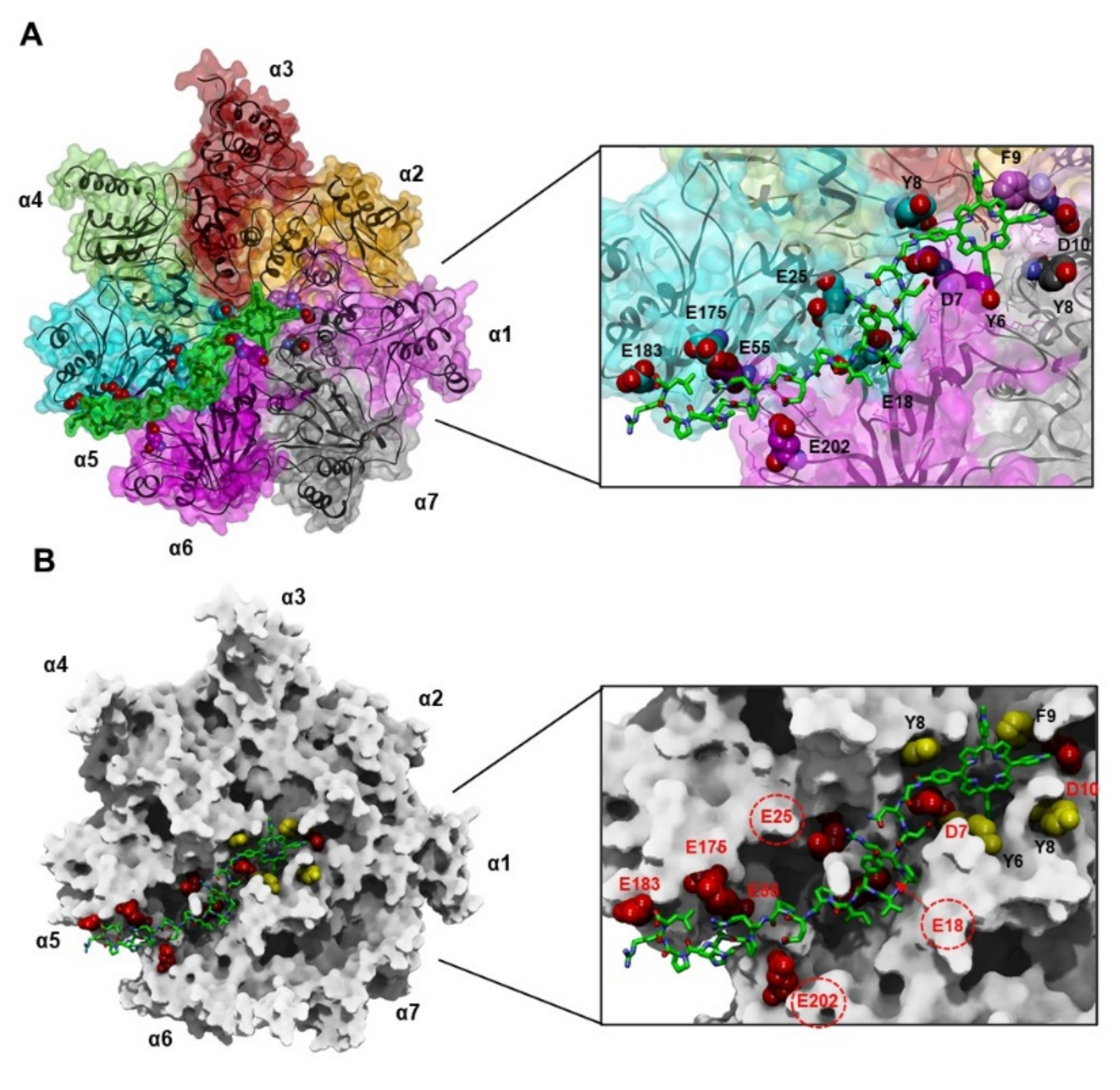
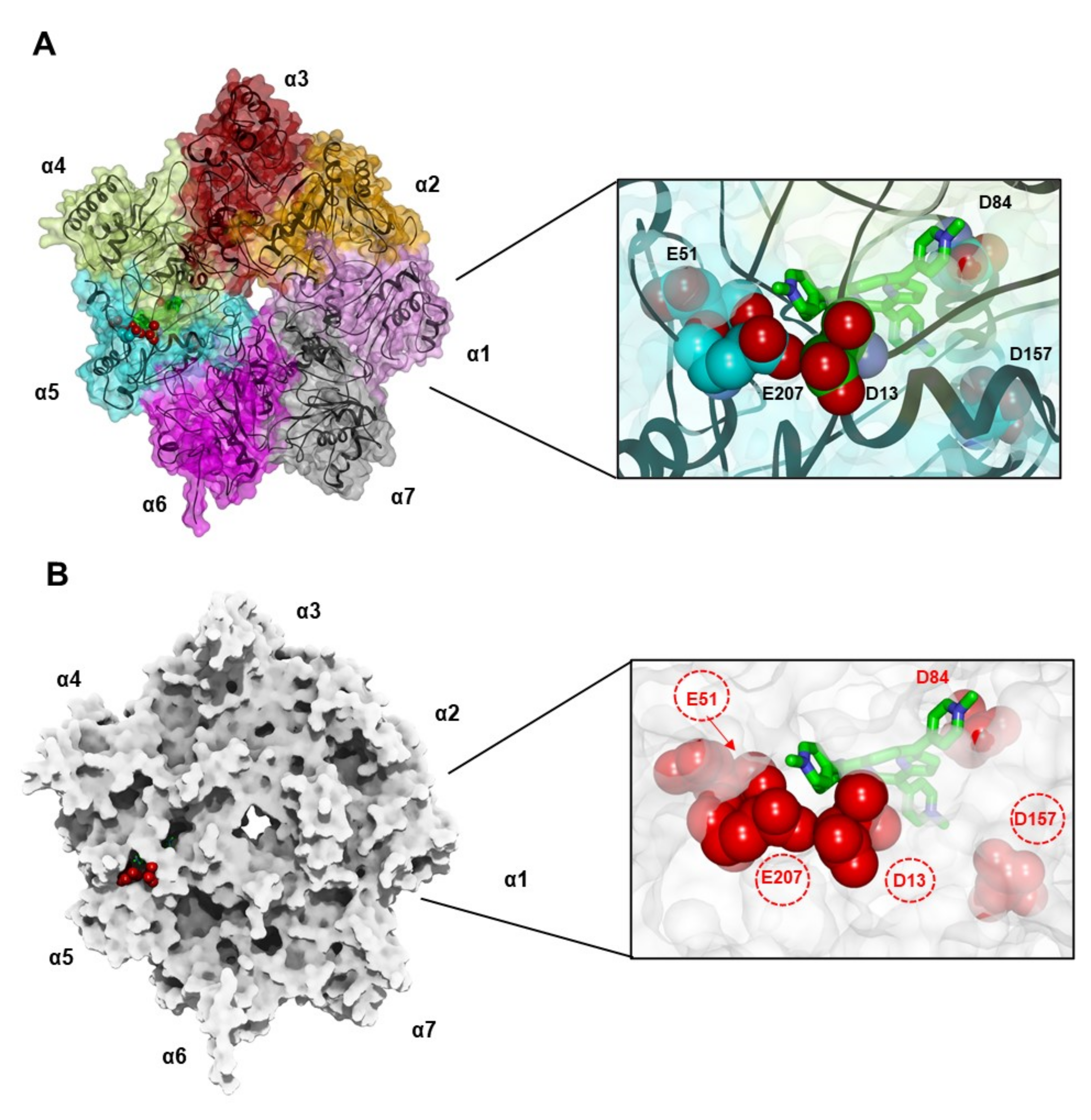
3.4.1. Docking of MTPyApi
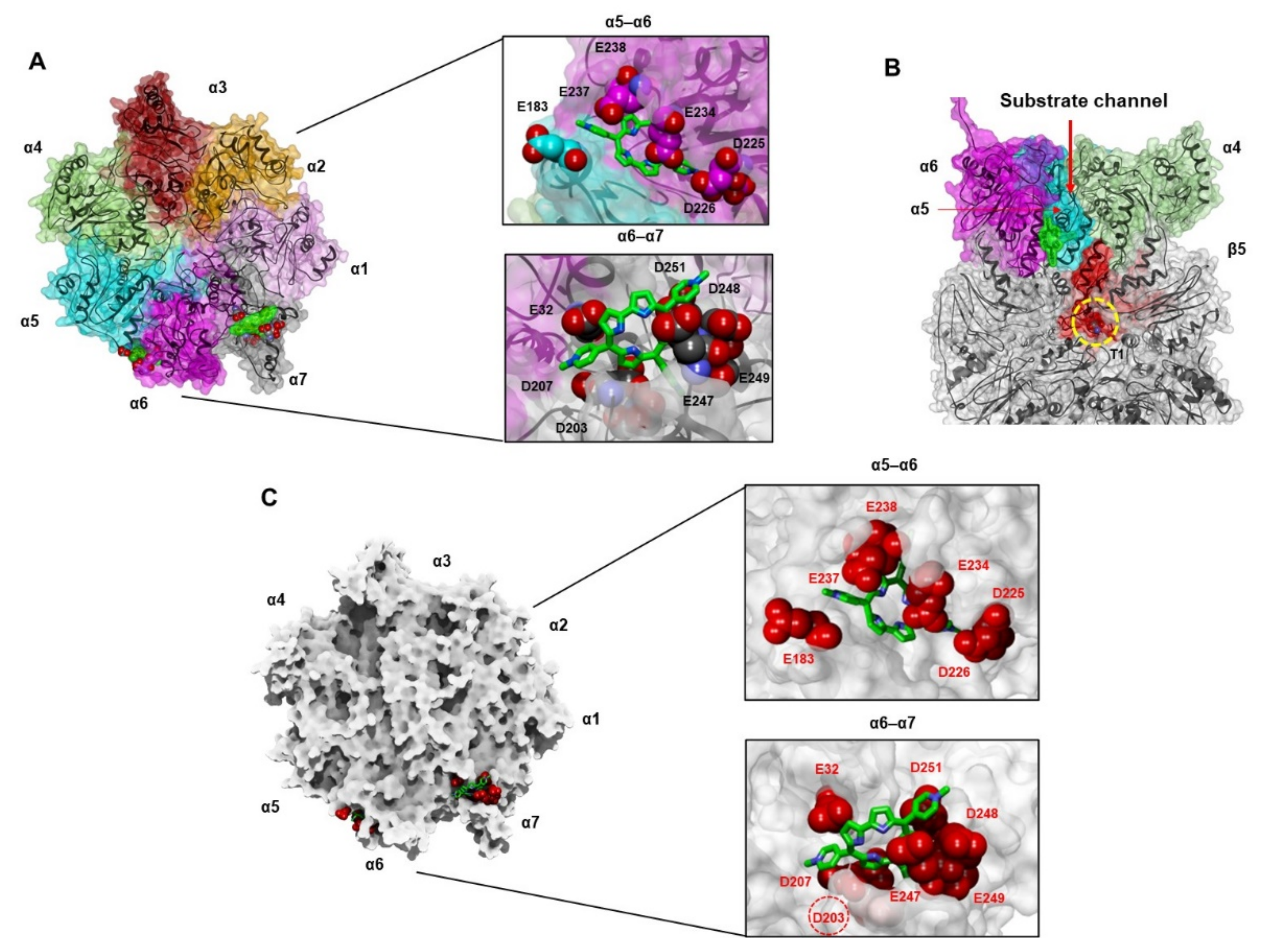
3.4.2. Docking of TMPC
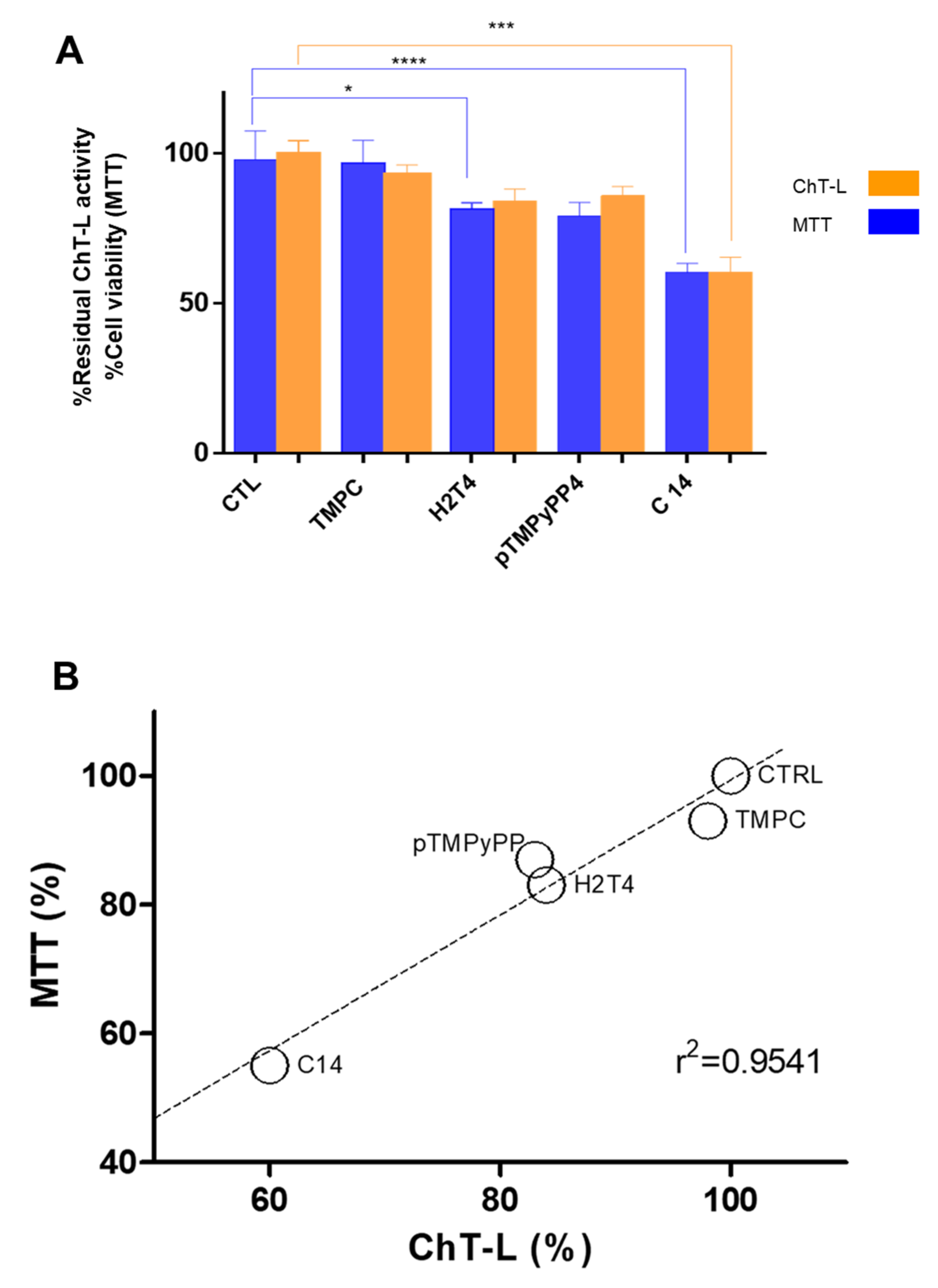
3.5. Porphyrins Inhibit h20S Proteasome in MCF7 Cells and Affect Cell Viability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [Green Version]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The proteasome as a druggable target with multiple therapeutic potentialities: Cutting and non-cutting edges. Pharmacol. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef]
- Oddone, F.; Rossetti, L.; Parravano, M.; Sbardella, D.; Coletta, M.; Ziccardi, L.; Roberti, G.; Carnevale, C.; Romano, D.; Manni, G.; et al. Citicoline in Ophthalmological Neurodegenerative Disease: A Comprehensive Review. Pharmaceuticals 2021, 14, 281. [Google Scholar] [CrossRef] [PubMed]
- Tundo, G.R.; Sbardella, D.; Oddone, F.; Kudriaeva, A.A.; Lacal, P.M.; Belogurov, A.A.; Graziani, G.; Marini, S. At the Cutting Edge against Cancer: A Perspective on Immunoproteasome and Immune Checkpoints Modulation as a Potential Therapeutic Intervention. Cancers 2021, 13, 4852. [Google Scholar] [CrossRef] [PubMed]
- Tundo, G.R.; Sbardella, D.; Coletta, M. Insights into Proteasome Conformation Dynamics and Intersubunit Communication. Trends Biochem. Sci. 2018, 43, 852–853. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Santoro, A.M.; Lanza, V.; Sbardella, D.; Tundo, G.R.; Ciaccio, C.; Marini, S.; Coletta, M.; Milardi, D. The double faced role of copper in Aβ homeostasis: A survey on the interrelationship between metal dyshomeostasis, UPS functioning and autophagy in neurodegeneration. Coord. Chem. Rev. 2017, 347, 1–22. [Google Scholar] [CrossRef]
- Mani, A.; Gelmann, E.P. The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. 2005, 23, 4776–4789. [Google Scholar] [CrossRef]
- Tomko, R.J.; Hochstrasser, M. Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem. 2013, 82, 415–445. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Marshall, R.S.; Vierstra, R.D. Dynamic regulation of the 26S proteasome: From synthesis to degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef]
- Whitby, F.G.; Masters, E.I.; Kramer, L.; Knowlton, J.R.; Yao, Y.; Wang, C.C.; Hill, C.P. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 2000, 408, 115–120. [Google Scholar] [CrossRef]
- Ustrell, V.; Hoffman, L.; Pratt, G.; Rechsteiner, M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002, 21, 3516–3525. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gao, X.; Ortega, J.; Nazif, T.; Joss, L.; Bogyo, M.; Steven, A.C.; Rechsteiner, M. Lysine 188 substitutions convert the pattern of proteasome activation by REGgamma to that of REGs alpha and beta. EMBO J. 2001, 20, 3359–3369. [Google Scholar] [CrossRef] [Green Version]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Bianculli, A.; Orlandi, A.; Desimio, M.G.; Arcuri, G.; Coletta, M.; Marini, S. Insulin-degrading enzyme (IDE): A novel heat shock-like protein. J. Biol. Chem. 2013, 288, 2281–2289. [Google Scholar] [CrossRef] [Green Version]
- Gaczynska, M.; Osmulski, P.A. Targeting Protein–Protein Interactions in the Ubiquitin–Proteasome Pathway. Adv. Protein Chem. Struct. Biol. 2018, 110, 123–165. [Google Scholar]
- Bajorek, M.; Glickman, M.H. Keepers at the final gates: Regulatory complexes and gating of the proteasome channel. Cell. Mol. Life Sci. 2004, 61, 1579–1588. [Google Scholar]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Ruschak, A.M.; Kay, L.E. Proteasome allostery as a population shift between interchanging conformers. Proc. Natl. Acad. Sci. USA 2012, 109, E3454–E3462. [Google Scholar] [CrossRef] [Green Version]
- Akopian, T.N.; Kisselev, A.F.; Goldberg, A.L. Processive degradation of proteins and other catalytic properties of the proteasome from Thermoplasma acidophilum. J. Biol. Chem. 1997, 272, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Köhler, A.; Bajorek, M.; Groll, M.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. The substrate translocation channel of the proteasome. Biochimie 2001, 83, 325–332. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Hochstrasser, M.; Gaczynska, M. A tetrahedral transition state at the active sites of the 20S proteasome is coupled to opening of the alpha-ring channel. Structure 2009, 17, 1137–1147. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, F.K.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The contribution of the 20s proteasome to proteostasis. Biomolecules 2019, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Demasi, M.; da Cunha, F.M. The physiological role of the free 20S proteasome in protein degradation: A critical review. Biochim. Biophys. Acta-Gen. Subj. 2018, 1862, 2948–2954. [Google Scholar] [CrossRef]
- Paoluzzi, L.; O’Connor, O.A. Mechanistic rationale and clinical evidence for the efficacy of proteasome inhibitors against indolent and mantle cell lymphomas. BioDrugs 2006, 20, 13–23. [Google Scholar] [CrossRef]
- Kale, A.J.; Moore, B.S. Molecular mechanisms of acquired proteasome inhibitor resistance. J. Med. Chem. 2012, 55, 10317–10327. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Oddone, F.; Grasso, G.; Marini, S.; Atzori, M.G.; Santoro, A.M.; Milardi, D.; Bellia, F.; Macari, G.; et al. Insulin-Degrading Enzyme Is a Non Proteasomal Target of Carfilzomib and Affects the 20S Proteasome Inhibition by the Drug. Biomolecules 2022, 12, 315. [Google Scholar] [CrossRef]
- Santoro, A.M.; Lo Giudice, M.C.; D’Urso, A.; Lauceri, R.; Purrello, R.; Milardi, D. Cationic porphyrins are reversible proteasome inhibitors. J. Am. Chem. Soc. 2012, 134, 10451–10457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.M.; Cunsolo, A.; D’Urso, A.; Sbardella, D.; Tundo, G.R.; Ciaccio, C.; Coletta, M.; Diana, D.; Fattorusso, R.; Persico, M.; et al. Cationic porphyrins are tunable gatekeepers of the 20S proteasome. Chem. Sci. 2016, 7, 1286–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dato, A.D.; Cunsolo, A.; Persico, M.; Santoro, A.M.; D’Urso, A.; Milardi, D.; Purrello, R.; Stefanelli, M.; Paolesse, R.; Tundo, G.R.; et al. Electrostatic Map Of Proteasome α-Rings Encodes The Design of Allosteric Porphyrin-Based Inhibitors Able To Affect 20S Conformation By Cooperative Binding. Sci. Rep. 2017, 7, 17098. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.M.; D’urso, A.; Cunsolo, A.; Milardi, D.; Purrello, R.; Sbardella, D.; Tundo, G.R.; Diana, D.; Fattorusso, R.; Di Dato, A.; et al. Cooperative binding of the cationic porphyrin tris-t4 enhances catalytic activity of 20s proteasome unveiling a complex distribution of functional states. Int. J. Mol. Sci. 2020, 21, 7190. [Google Scholar] [CrossRef] [PubMed]
- Sugisaki, K.; Usui, T.; Nishiyama, N.; Jang, W.D.; Yanagi, Y.; Yamagami, S.; Amano, S.; Kataoka, K. Photodynamic Therapy for Corneal Neovascularization Using Polymeric Micelles Encapsulating Dendrimer Porphyrins. Investig. Ophthalmol. Vis. Sci. 2008, 49, 894–899. [Google Scholar] [CrossRef]
- Awan, M.A.; Tarin, S.A. Review of photodynamic therapy. Surgeon 2006, 4, 231–236. [Google Scholar] [CrossRef]
- Rishi, E.; Rishi, P.; Sharma, V.; Koundanya, V.; Athanikar, R. Long-term outcomes of combination photodynamic therapy with ranibizumab or bevacizumab for treatment of wet age-related macular degeneration. Oman J. Ophthalmol. 2016, 9, 87–92. [Google Scholar] [CrossRef]
- Dosselli, R.; Tampieri, C.; Ruiz-González, R.; De Munari, S.; Ragàs, X.; Sánchez-García, D.; Agut, M.; Nonell, S.; Reddi, E.; Gobbo, M. Synthesis, characterization, and photoinduced antibacterial activity of porphyrin-type photosensitizers conjugated to the antimicrobial peptide apidaecin 1b. J. Med. Chem. 2013, 56, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Giżyńska, M.; Witkowska, J.; Karpowicz, P.; Rostankowski, R.; Chocron, E.S.; Pickering, A.M.; Osmulski, P.; Gaczynska, M.; Jankowska, E. Proline- and Arginine-Rich Peptides as Flexible Allosteric Modulators of Human Proteasome Activity. J. Med. Chem. 2019, 62, 359–370. [Google Scholar] [CrossRef]
- D’Urso, A.; Nardis, S.; Pomarico, G.; Fragalà, M.E.; Paolesse, R.; Purrello, R. Interaction of tricationic corroles with single/double helix of homopolymeric nucleic acids and DNA. J. Am. Chem. Soc. 2013, 135, 8632–8638. [Google Scholar] [CrossRef] [PubMed]
- Teo, R.D.; Hwang, J.Y.; Termini, J.; Gross, Z.; Gray, H.B. Fighting Cancer with Corroles. Chem. Rev. 2017, 117, 2711–2729. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Grossmann, C. Fletcher, R., Unconstrained Optimization. Practical Methods of Optimization 1. Chichester-New York-Brisbane-Toronto, John Wiley&Sons 1980. VIII, 120 S., £ 8.80. ISBN 0–471–27711–8. ZAMM-Zeitschrift Angew. Math. Mech. 1981, 61, 408. [Google Scholar] [CrossRef]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An antimicrobial peptide that inhibits translation by trapping release factors on the ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef]
- Senderowitz, H.; Guarnieri, F.; Still, W.C. A Smart Monte Carlo Technique for Free Energy Simulations of Multiconformational Molecules. Direct Calculations of the Conformational Populations of Organic Molecules. J. Am. Chem. Soc. 1995, 117, 8211–8219. [Google Scholar] [CrossRef]
- Ding, H.Q.; Karasawa, N.; Goddard, W.A. Atomic level simulations on a million particles: The cell multipole method for Coulomb and London nonbond interactions. J. Chem. Phys. 1992, 97, 4309–4315. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B.; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.C.; Nagpal, S.; Salunke, D.M. Functional mapping of apidaecin through secondary structure correlation. Int. J. Biochem. Cell Biol. 2008, 40, 1005–1015. [Google Scholar] [CrossRef]
- Zahn, M.; Berthold, N.; Kieslich, B.; Knappe, D.; Hoffmann, R.; Sträter, N. Structural studies on the forward and reverse binding modes of peptides to the chaperone DnaK. J. Mol. Biol. 2013, 425, 2463–2479. [Google Scholar] [CrossRef]
- Czihal, P.; Knappe, D.; Fritsche, S.; Zahn, M.; Berthold, N.; Piantavigna, S.; Müller, U.; Van Dorpe, S.; Herth, N.; Binas, A.; et al. Api88 is a novel antibacterial designer peptide to treat systemic infections with multidrug-resistant gram-negative pathogens. ACS Chem. Biol. 2012, 7, 1281–1291. [Google Scholar] [CrossRef]
- Brancaccio, D.; Diana, D.; Di Maro, S.; Di Leva, F.S.; Tomassi, S.; Fattorusso, R.; Russo, L.; Scala, S.; Trotta, A.M.; Portella, L.; et al. Ligand-Based NMR Study of C-X-C Chemokine Receptor Type 4 (CXCR4)-Ligand Interactions on Living Cancer Cells. J. Med. Chem. 2018, 61, 2910–2923. [Google Scholar] [CrossRef]
- Di Stasi, R.; Diana, D.; Capasso, D.; Di Gaetano, S.; De Rosa, L.; Celentano, V.; Isernia, C.; Fattorusso, R.; D’andrea, L.D. VEGFR recognition interface of a proangiogenic VEGF-mimetic peptide determined in vitro and in the presence of endothelial cells by NMR spectroscopy. Chem.-A Eur. J. 2018, 24, 11461–11466. [Google Scholar] [CrossRef]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| h20S | h20S + Tris-T4 | h20S + TMPC | ||
|---|---|---|---|---|
| kcatA (s−1) | 11.5 ± 1.3 | KAU (M−1) | 2.0 (±0.3) × 104 | 1.35 (±0.31) × 104 |
| kcatB (s−1) | 0.32 ± 0.05 | KBU (M−1) | 8.2 (±0.3) × 107 | 2.55 (±0.47) × 108 |
| KmA (M) | 1.95 (±0.37) × 10−3 | KAL (M−1) | 1.2 (±0.3) × 104 | 3.5 (±0.6) × 105 |
| KmB (M) | 1.5 (±0.3) × 10−4 | KBL (M−1) | 1.35 (±0.28) × 108 | 3.1 (±0.6) × 108 |
| LU | 3.0 (±0.5) × 10−6 (3) | KAI (M−1) | - | 1.0 (±0.2) × 106 |
| LL | 3.9 (±0.6) × 10−5 (3) | KBI (M−1) | - | 2.05 (±0.47) × 109 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persico, M.; Santoro, A.M.; D’Urso, A.; Milardi, D.; Purrello, R.; Cunsolo, A.; Gobbo, M.; Fattorusso, R.; Diana, D.; Stefanelli, M.; et al. Modulation of the 20S Proteasome Activity by Porphyrin Derivatives Is Steered through Their Charge Distribution. Biomolecules 2022, 12, 741. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12060741
Persico M, Santoro AM, D’Urso A, Milardi D, Purrello R, Cunsolo A, Gobbo M, Fattorusso R, Diana D, Stefanelli M, et al. Modulation of the 20S Proteasome Activity by Porphyrin Derivatives Is Steered through Their Charge Distribution. Biomolecules. 2022; 12(6):741. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12060741
Chicago/Turabian StylePersico, Marco, Anna Maria Santoro, Alessandro D’Urso, Danilo Milardi, Roberto Purrello, Alessandra Cunsolo, Marina Gobbo, Roberto Fattorusso, Donatella Diana, Manuela Stefanelli, and et al. 2022. "Modulation of the 20S Proteasome Activity by Porphyrin Derivatives Is Steered through Their Charge Distribution" Biomolecules 12, no. 6: 741. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12060741