
(Pro)Renin Receptor Antagonism Attenuates High-Fat-Diet–Induced Hepatic Steatosis
 , and
, and
Abstract
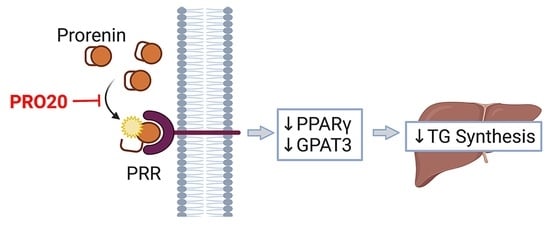
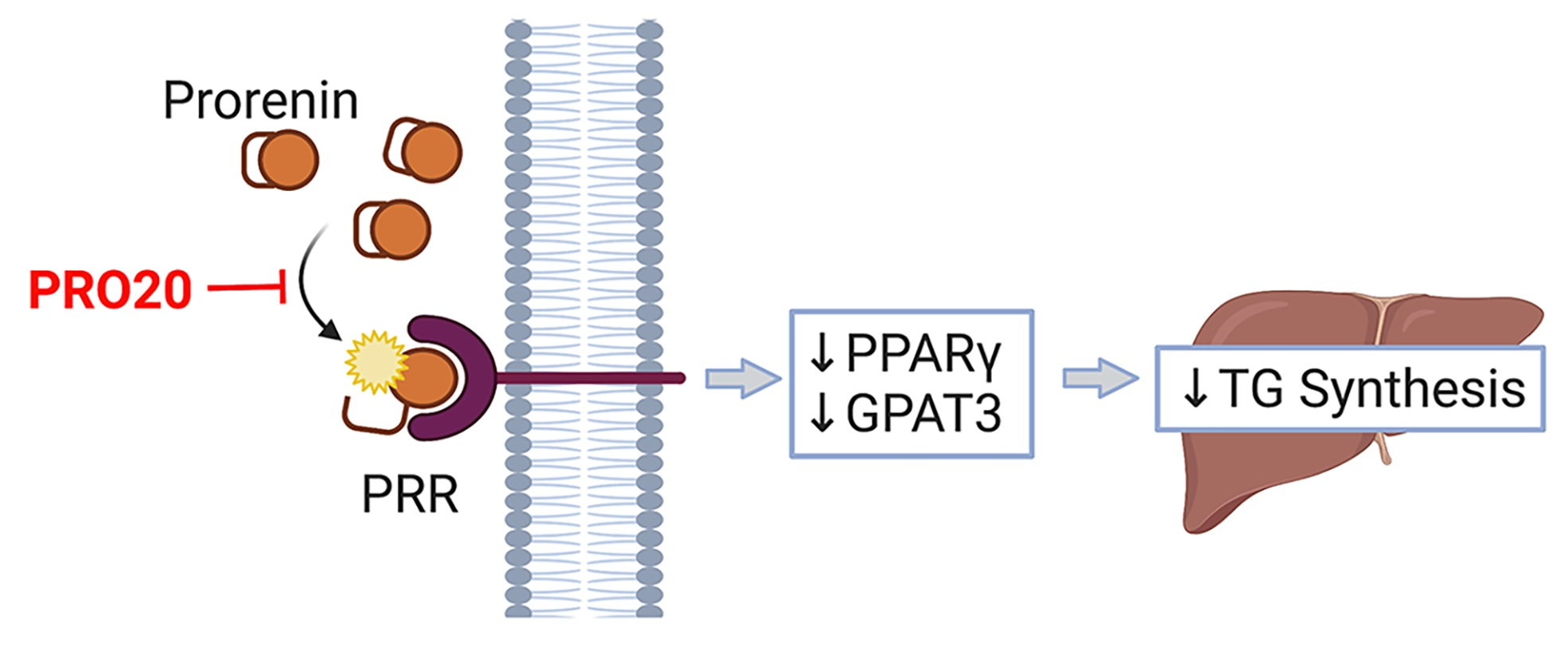
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Subcutaneous Administration of PRO20, Losartan, and Controls Using Osmotic Minipumps
2.3. Fasting Blood Glucose Measurements and Glucose Tolerance Tests
2.4. Oil Red O Staining
2.5. Hematoxylin & Eosin Staining
2.6. Assignment of Histological NAFLD Scores
2.7. Picrosirius Red Staining
2.8. Liver Alanine and Aspartate Aminotransferase Activity Assay
2.9. Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-qPCR)
2.10. Western Blot Analysis
2.11. Statistial Analysis
3. Results
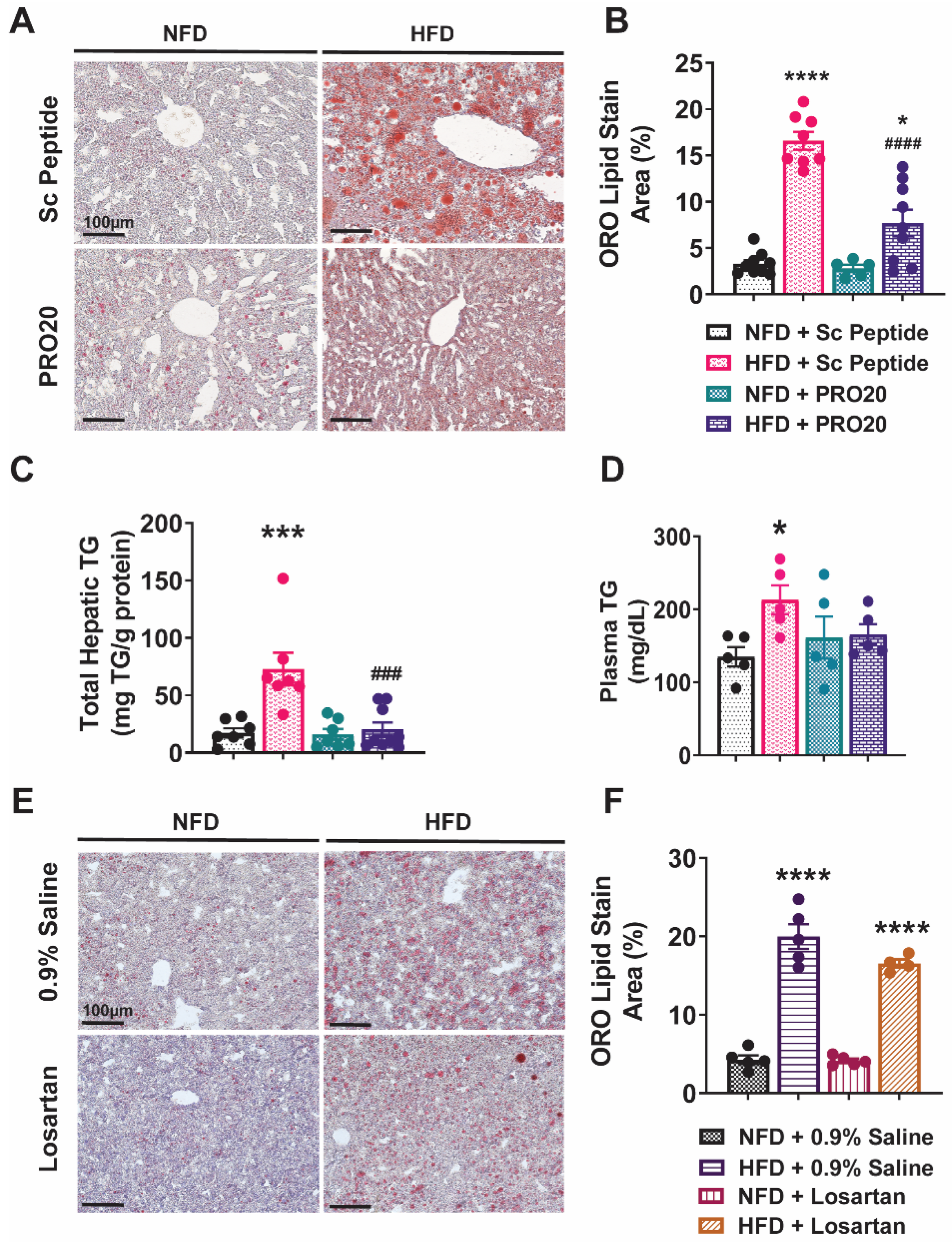
3.1. PRR Antagonism Attenuates HFD-Induced Lipid Accumulation in the Liver
3.2. Subcutaneous Infusion of Losartan Does Not Reduce Hepatic Lipid Accumulation in HFD-Fed Mice
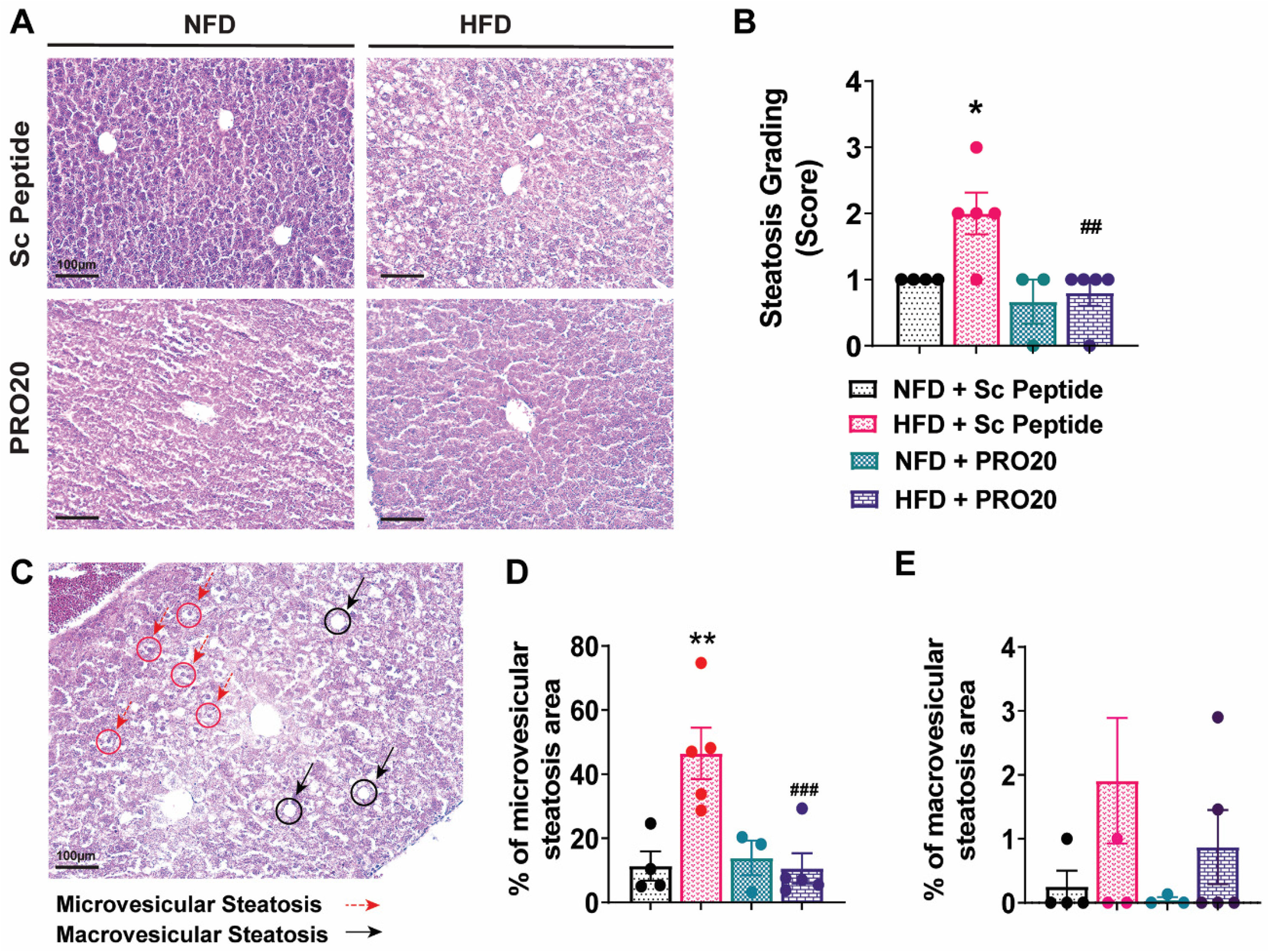
3.3. PRO20 Attenuates HFD-Induced Hepatic Steatosis
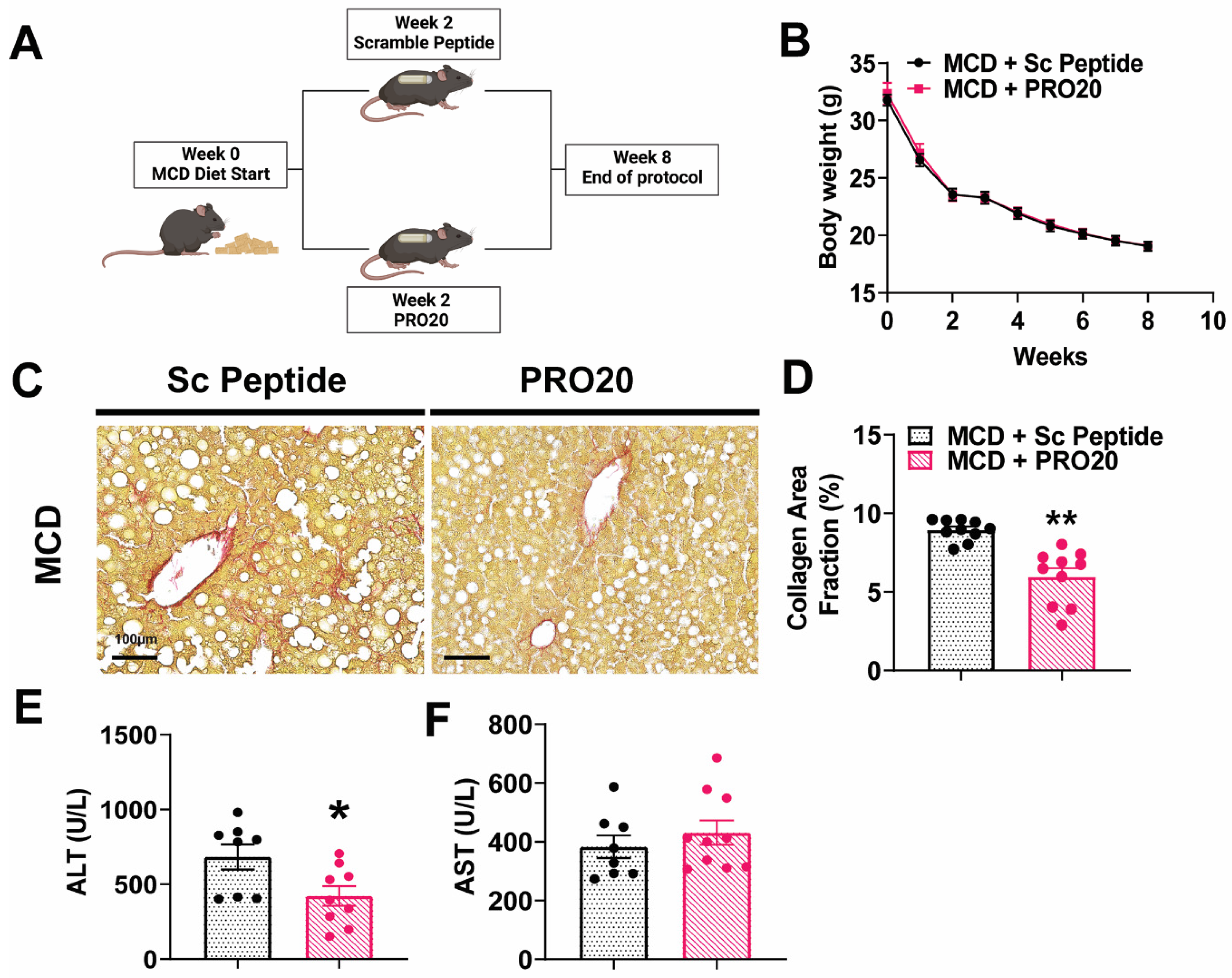
3.4. PRO20 Decreases Hepatic Fibrosis Development and Liver Injury in MCD Diet-Fed Mice
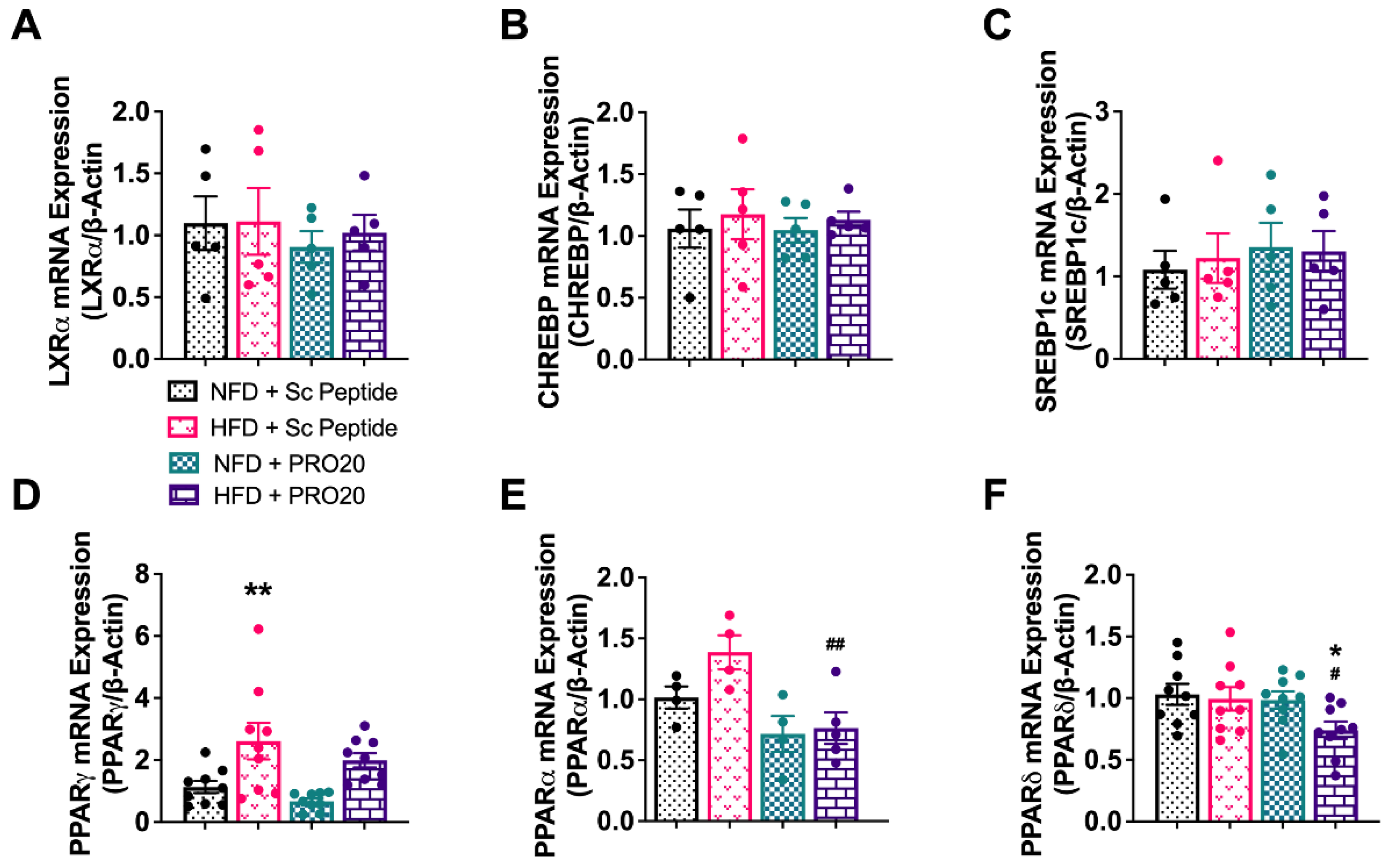
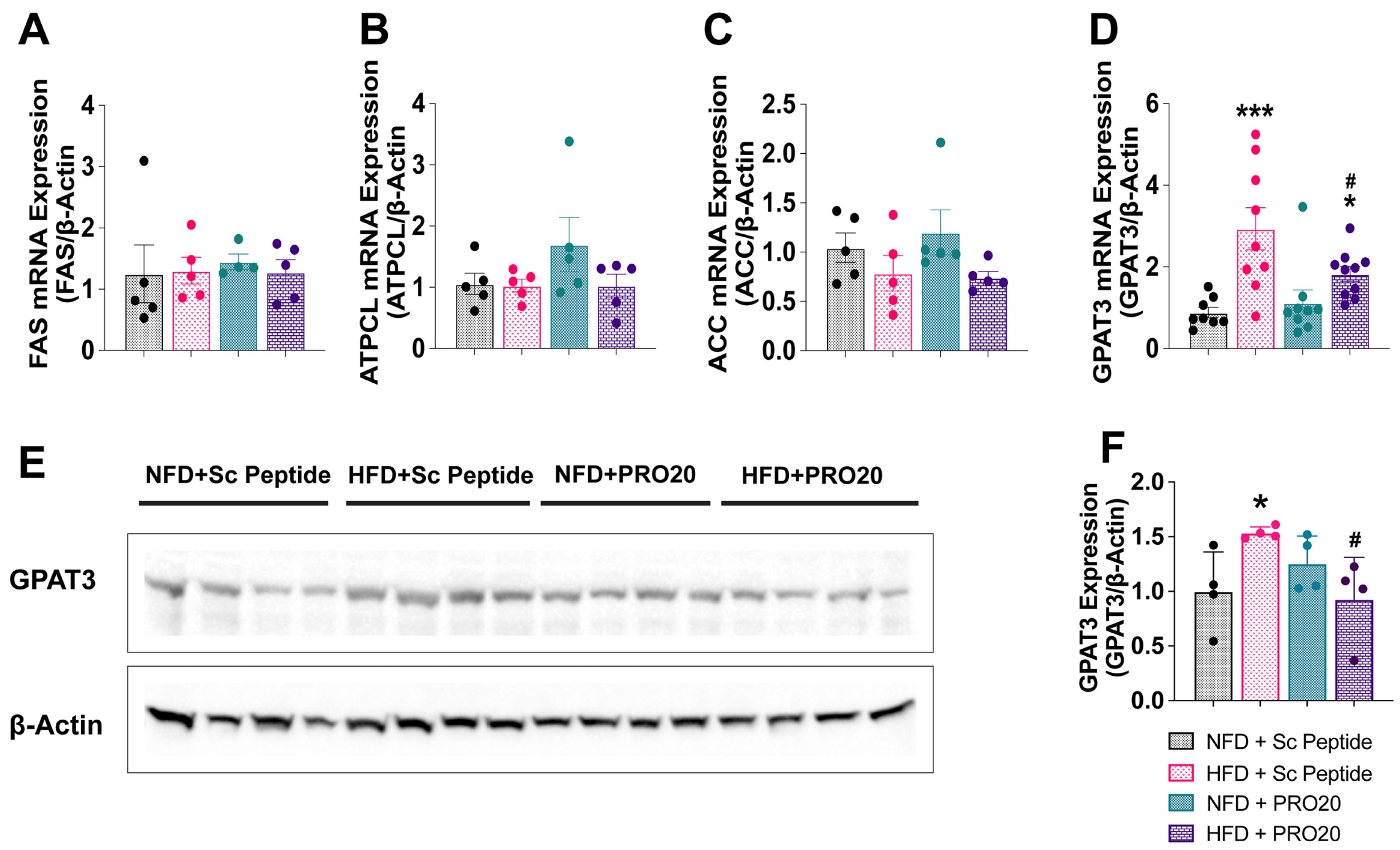
3.5. PRR Antagonism Attenuates HFD-Induced Elevation of PPARγ and GPAT3 in the Liver
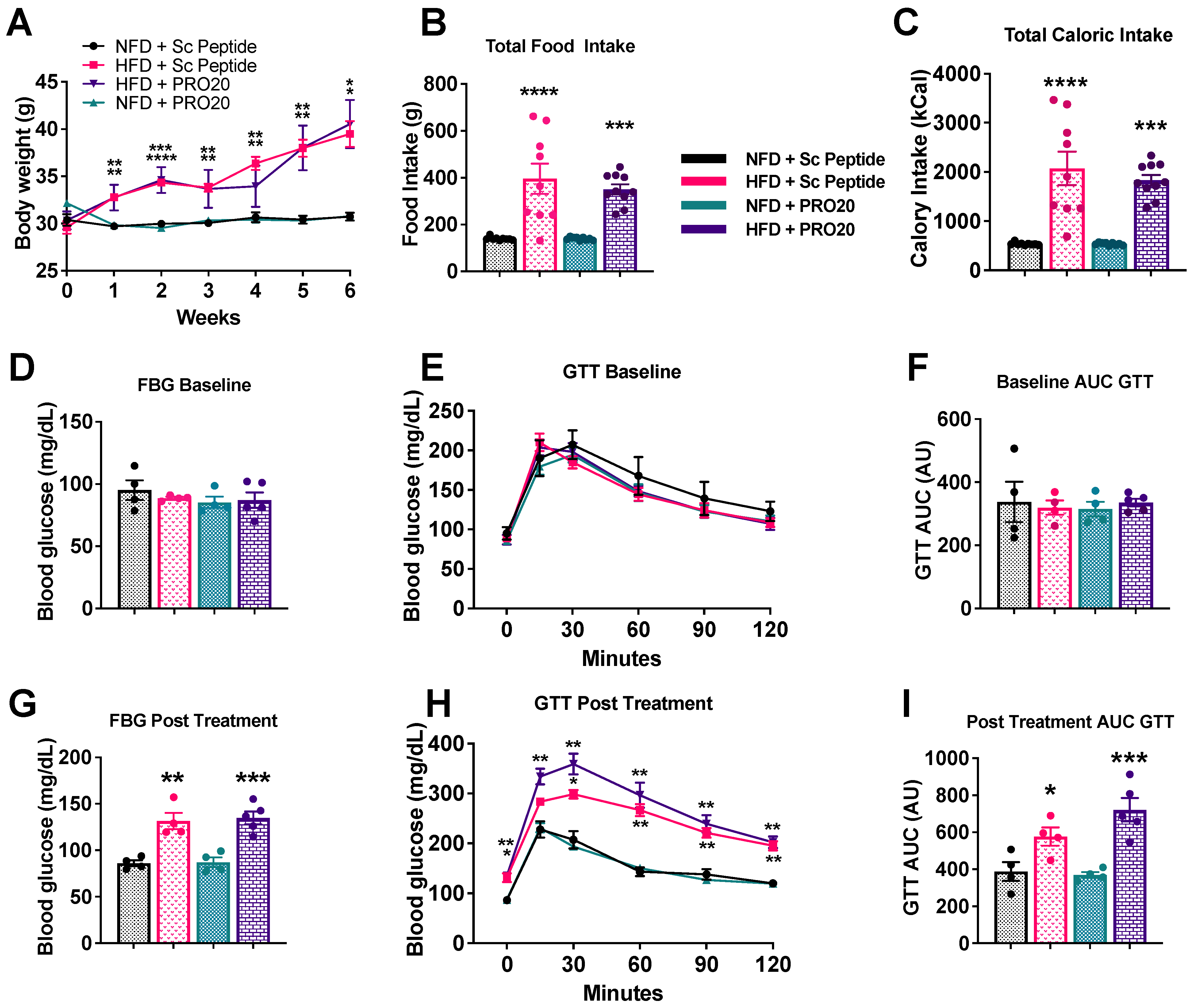
3.6. PRR Antagonism Has No Effect on Body Weight or Glucose Homeostasis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of non-alcoholic fatty liver disease. QJM Int. J. Med. 2010, 103, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Postic, C.; Girard, J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008, 34, 643–648. [Google Scholar] [CrossRef]
- Povsic, M.; Wong, O.Y.; Perry, R.; Bottomley, J. A Structured Literature Review of the Epidemiology and Disease Burden of Non-Alcoholic Steatohepatitis (NASH). Adv. Ther. 2019, 36, 1574–1594. [Google Scholar] [CrossRef] [Green Version]
- Yosypiv, I.V.; Sequeira-Lopez, M.L.S.; Song, R.; De Goes Martini, A. Stromal prorenin receptor is critical for normal kidney development. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R640–R650. [Google Scholar] [CrossRef] [PubMed]
- Satofuka, S.; Ichihara, A.; Nagai, N.; Tsubota, K.; Itoh, H.; Ishida, S. Pathologic roles of prorenin and (pro)renin receptor in the eye. Front. Biosci. 2008, 13, 3884–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Noble, N.A.; Border, W.A.; Owens, R.T.; Huang, Y. Receptor-dependent prorenin activation and induction of PAI-1 expression in vascular smooth muscle cells. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E810–E819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.; Delarue, F.; Burckle, C.; Bouzhir, L.; Giller, T.; Sraer, J.D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Investig. 2002, 109, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Gatineau, E.; Arthur, G.; Poupeau, A.; Nichols, K.; Spear, B.T.; Shelman, N.R.; Graf, G.A.; Temel, R.E.; Yiannikouris, F.B. The prorenin receptor and its soluble form contribute to lipid homeostasis. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E609–E618. [Google Scholar] [CrossRef]
- Xiong, J.; Cao, X.; Qiao, S.; Yu, S.; Li, L.; Yu, Y.; Fu, C.; Jiang, F.; Dong, B.; Su, Q. (Pro)renin Receptor is Involved in Myocardial Damage in Alcoholic Cardiomyopathy. Alcohol. Clin. Exp. Res. 2019, 43, 2344–2353. [Google Scholar] [CrossRef]
- Xu, Q.; Jensen, D.D.; Peng, H.; Feng, Y. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol. Ther. 2016, 164, 126–134. [Google Scholar] [CrossRef]
- Kanda, A.; Ishida, S. (Pro)renin receptor: Involvement in diabetic retinopathy and development of molecular targeted therapy. J. Diabetes Investig. 2019, 10, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Worker, C.J.; Li, W.; Feng, C.Y.; Souza, L.A.C.; Gayban, A.J.B.; Cooper, S.G.; Afrin, S.; Romanick, S.; Ferguson, B.S.; Feng Earley, Y. The neuronal (pro)renin receptor and astrocyte inflammation in the central regulation of blood pressure and blood glucose in mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E765–E778. [Google Scholar] [CrossRef]
- Wu, C.H.; Mohammadmoradi, S.; Thompson, J.; Su, W.; Gong, M.; Nguyen, G.; Yiannikouris, F. Adipocyte (Pro)Renin-Receptor Deficiency Induces Lipodystrophy, Liver Steatosis and Increases Blood Pressure in Male Mice. Hypertension 2016, 68, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Strong, A.; Musunuru, K. (Pro)renin Receptor and LDL Clearance: An Old Player Joins A New Game. Circ. Res. 2016, 118, 187–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sullivan, M.N.; Zhang, S.; Worker, C.J.; Xiong, Z.; Speth, R.C.; Feng, Y. Intracerebroventricular infusion of the (Pro)renin receptor antagonist PRO20 attenuates deoxycorticosterone acetate-salt-induced hypertension. Hypertension 2015, 65, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lu, X.; Liu, M.; Feng, Y.; Zhou, S.F.; Yang, T. Renal medullary (pro)renin receptor contributes to angiotensin II-induced hypertension in rats via activation of the local renin-angiotensin system. BMC Med. 2015, 13, 278. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Yang, K.; Wang, F.; Xu, C.; Yang, T. (Pro)renin receptor decoy peptide PRO20 protects against adriamycin-induced nephropathy by targeting the intrarenal renin-angiotensin system. Am. J. Physiol. Renal. Physiol. 2020, 319, F930–F940. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.C.; Liu, C.H.; Tsai, Y.C.; Li, S.J.; Chen, C.Y.; Chu, C.H.; Chen, M.F. Time-dependent cellular response in the liver and heart in a dietary-induced obese mouse model: The potential role of ER stress and autophagy. Eur. J. Nutr. 2016, 55, 2031–2043. [Google Scholar] [CrossRef]
- Podrini, C.; Cambridge, E.L.; Lelliott, C.J.; Carragher, D.M.; Estabel, J.; Gerdin, A.K.; Karp, N.A.; Scudamore, C.L.; Sanger Mouse Genetics, P.; Ramirez-Solis, R.; et al. High-fat feeding rapidly induces obesity and lipid derangements in C57BL/6N mice. Mamm. Genome 2013, 24, 240–251. [Google Scholar] [CrossRef]
- Gauthier, M.S.; Favier, R.; Lavoie, J.M. Time course of the development of non-alcoholic hepatic steatosis in response to high-fat diet-induced obesity in rats. Br. J. Nutr. 2006, 95, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, L.A.C.; Earley, Y.F. (Pro)renin Receptor and Blood Pressure Regulation: A Focus on the Central Nervous System. Curr. Hypertens. Rev. 2022, 18, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Ko, E.H.; Kim, J.E.; Kim, E.; Lee, H.; Choi, H.; Yu, J.H.; Kim, H.J.; Seong, J.K.; Kim, K.S.; et al. Nuclear receptor PPARgamma-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13656–13661. [Google Scholar] [CrossRef] [Green Version]
- Brocker, C.N.; Patel, D.P.; Velenosi, T.J.; Kim, D.; Yan, T.; Yue, J.; Li, G.; Krausz, K.W.; Gonzalez, F.J. Extrahepatic PPARalpha modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J. Lipid Res. 2018, 59, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Bojic, L.A.; Telford, D.E.; Fullerton, M.D.; Ford, R.J.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Gros, R.; Kemp, B.E.; Steinberg, G.R.; et al. PPARdelta activation attenuates hepatic steatosis in Ldlr-/- mice by enhanced fat oxidation, reduced lipogenesis, and improved insulin sensitivity. J. Lipid Res. 2014, 55, 1254–1266. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Yang, F.; Shi, T.; Yuan, M.; Xin, Z.; Xie, R.; Li, S.; Li, H.; Yang, J.K. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis activates Akt signaling to ameliorate hepatic steatosis. Sci. Rep. 2016, 6, 21592. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Ma, K.L.; Zhang, Y.; Wen, Y.; Wang, G.H.; Hu, Z.B.; Liu, L.; Lu, J.; Chen, P.P.; Ruan, X.Z.; et al. Lipid disorder and intrahepatic renin-angiotensin system activation synergistically contribute to non-alcoholic fatty liver disease. Liver Int. 2016, 36, 1525–1534. [Google Scholar] [CrossRef]
- Santos, S.H.; Braga, J.F.; Mario, E.G.; Porto, L.C.; Rodrigues-Machado Mda, G.; Murari, A.; Botion, L.M.; Alenina, N.; Bader, M.; Santos, R.A. Improved lipid and glucose metabolism in transgenic rats with increased circulating angiotensin-(1-7). Arterioscler. Thromb. Vasc. Biol. 2010, 30, 953–961. [Google Scholar] [CrossRef]
- Ren, L.; Sun, Y.; Lu, H.; Ye, D.; Han, L.; Wang, N.; Daugherty, A.; Li, F.; Wang, M.; Su, F.; et al. (Pro)renin Receptor Inhibition Reprograms Hepatic Lipid Metabolism and Protects Mice From Diet-Induced Obesity and Hepatosteatosis. Circ. Res. 2018, 122, 730–741. [Google Scholar] [CrossRef] [Green Version]
- Rocha Dos Santos, C.; Grigorova, Y.; McDevitt, R.; Zernetkina, V.; Wei, W.; Juhasz, O.; Lakatta, E.G.; Fedorova, O.V. Beneficial effects of AT1 antagonists treatment, losartan, on cardiovascular and cognitive function on aged male Dahl salt sensitive rats. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Castoldi, G.; Carletti, R.; Ippolito, S.; Stella, A.; Zerbini, G.; Pelucchi, S.; Zatti, G.; di Gioia, C.R.T. Angiotensin Type 2 and Mas Receptor Activation Prevents Myocardial Fibrosis and Hypertrophy through the Reduction of Inflammatory Cell Infiltration and Local Sympathetic Activity in Angiotensin II-Dependent Hypertension. Int. J. Mol. Sci. 2021, 22, 13678. [Google Scholar] [CrossRef] [PubMed]
- Al-Majed, A.R.; Assiri, E.; Khalil, N.Y.; Abdel-Aziz, H.A. Losartan: Comprehensive Profile. Profiles Drug Subst. Excip. Relat. Methodol. 2015, 40, 159–194. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.R.; Rong, J.B.; Lu, H.S.; Daugherty, A.; Shi, P.; Ke, C.L.; Zhang, Z.C.; Xu, Y.C.; Wang, J.A. Angiotensinogen in hepatocytes contributes to Western diet-induced liver steatosis. J. Lipid Res. 2019, 60, 1983–1995. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Liu, H.M.; Chang, Z.Y.; Huang, T.H.; Lee, T.Y. Losartan Prevents Hepatic Steatosis and Macrophage Polarization by Inhibiting HIF-1alpha in a Murine Model of NAFLD. Int. J. Mol. Sci. 2021, 22, 7841. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmalski, M.; Ziolkowska, S.; Czarny, P.; Szemraj, J.; Pietras, T. The Coexistence of Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. J. Clin. Med. 2022, 11, 1375. [Google Scholar] [CrossRef]
- Skat-Rordam, J.; Hojland Ipsen, D.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor gamma in non-alcoholic fatty liver disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef]
- Yu, S.; Matsusue, K.; Kashireddy, P.; Cao, W.Q.; Yeldandi, V.; Yeldandi, A.V.; Rao, M.S.; Gonzalez, F.J.; Reddy, J.K. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J. Biol. Chem. 2003, 278, 498–505. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Hernandez-Ono, A.; Siri, P.; Weisberg, S.; Conlon, D.; Graham, M.J.; Crooke, R.M.; Huang, L.S.; Ginsberg, H.N. Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J. Biol. Chem. 2006, 281, 37603–37615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Loh, K.; Song, Z.Y.; Yang, H.Q.; Zhang, Y.; Lin, S. Update on glycerol-3-phosphate acyltransferases: The roles in the development of insulin resistance. Nutr. Diabetes 2018, 8, 34. [Google Scholar] [CrossRef]
- Cao, J.; Li, J.L.; Li, D.; Tobin, J.F.; Gimeno, R.E. Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19695–19700. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.J.; Choi, H.S.; Lee, Y.J.; Hwang, J.H.; Lee, O.H.; Seo, M.J.; Kim, K.J.; Lee, B.Y. Seapolynol Extracted from Ecklonia cava Inhibits Adipocyte Differentiation in Vitro and Decreases Fat Accumulation in Vivo. Molecules 2015, 20, 21715–21731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindhelm, R.K.; Diamant, M.; Dekker, J.M.; Tushuizen, M.E.; Teerlink, T.; Heine, R.J. Alanine aminotransferase as a marker of non-alcoholic fatty liver disease in relation to type 2 diabetes mellitus and cardiovascular disease. Diabetes Metab. Res. Rev. 2006, 22, 437–443. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5’-3’) |
|---|---|
| β-actin | FWD: CCAGCCTTCCTTCTTGGGTA REV: AGAGGTCTTTACGGATGTCAACG |
| Peroxisome proliferator-activated receptor alpha (PPARα) | FWD: GTTCACGCATGTGAAGGCTG REV: GCGAATTGCATTGTGTGACATC |
| Peroxisome proliferator-activated receptor delta (PPARδ) | FWD: GCTCGAGTATGAGAAGTGCGA REV: CGGATAGCGTTGTGCGACAT |
| Peroxisome proliferator-activated receptor gamma (PPARγ) | FWD: GCTTGTGAAGGATGCAAGGGTTT REV: ATCCGCCCAAACCTGATGG |
| Fatty acid synthase (FAS) | FWD: CTGACTCGGCTACTGACACG REV: AATGGGGTGCACAAGGAACA |
| Acetyl-CoA carboxylase alpha (ACC) | FWD: GCCTTTCACATGAGATCCAGC REV: CTGCAATACCATTGTTGGCGA |
| ATP citrate lyase (ATPCL) | FWD: CCCAAGATTCAGTCCCAAGTC REV: TTGTGATCCCCAGTGAAAGG |
| Liver X receptor alpha (LXRa) | FWD: CTCTGCAATCGAGGTCATGCT REV: CAGCTCATTCATGGCTCTGGA |
| Sterol regulatory element-binding protein 2 (SREBP-2) | FWD: CTTCGAAGGCTGGCCCATA REV: AGGTGTCTACCTCTCCATGCTT |
| Sterol regulatory element-binding protein 1c (SREBP-1c) | FWD: GGAGCCATGGATTGCACATT REV: GGCCCGGGAAGTCACTGT |
| Carbohydrate response element binding protein (CHREBP) | FWD: AGTGCTTGAGCCTGGCCTAC REV: TTGTTCAGGCGGATCTTGTC |
| Glycerol-3-phosphate acyltransferase 3 (GPAT3) | FWD: AGCTTTGAAATCGGAGGAACC REV: AACTGCGTCTTCTCCTTCCTCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gayban, A.J.B.; Souza, L.A.C.; Cooper, S.G.; Regalado, E.; Kleemann, R.; Feng Earley, Y. (Pro)Renin Receptor Antagonism Attenuates High-Fat-Diet–Induced Hepatic Steatosis. Biomolecules 2023, 13, 142. https://0-doi-org.brum.beds.ac.uk/10.3390/biom13010142
Gayban AJB, Souza LAC, Cooper SG, Regalado E, Kleemann R, Feng Earley Y. (Pro)Renin Receptor Antagonism Attenuates High-Fat-Diet–Induced Hepatic Steatosis. Biomolecules. 2023; 13(1):142. https://0-doi-org.brum.beds.ac.uk/10.3390/biom13010142
Chicago/Turabian StyleGayban, Ariana Julia B., Lucas A. C. Souza, Silvana G. Cooper, Erick Regalado, Robert Kleemann, and Yumei Feng Earley. 2023. "(Pro)Renin Receptor Antagonism Attenuates High-Fat-Diet–Induced Hepatic Steatosis" Biomolecules 13, no. 1: 142. https://0-doi-org.brum.beds.ac.uk/10.3390/biom13010142