
Commonly Used Therapeutics Associated with Changes in Arousal Inhibit GABAAR Activation
 , ,
, ,
Abstract
:1. Introduction
2. Compounds Tested and Rationale
3. Methods
3.1. Molecular Biology and Cell Culture
3.2. Conventional Patch Clamp
3.3. Microfluidic Patch Clamp
3.4. In Silico Modeling Methods
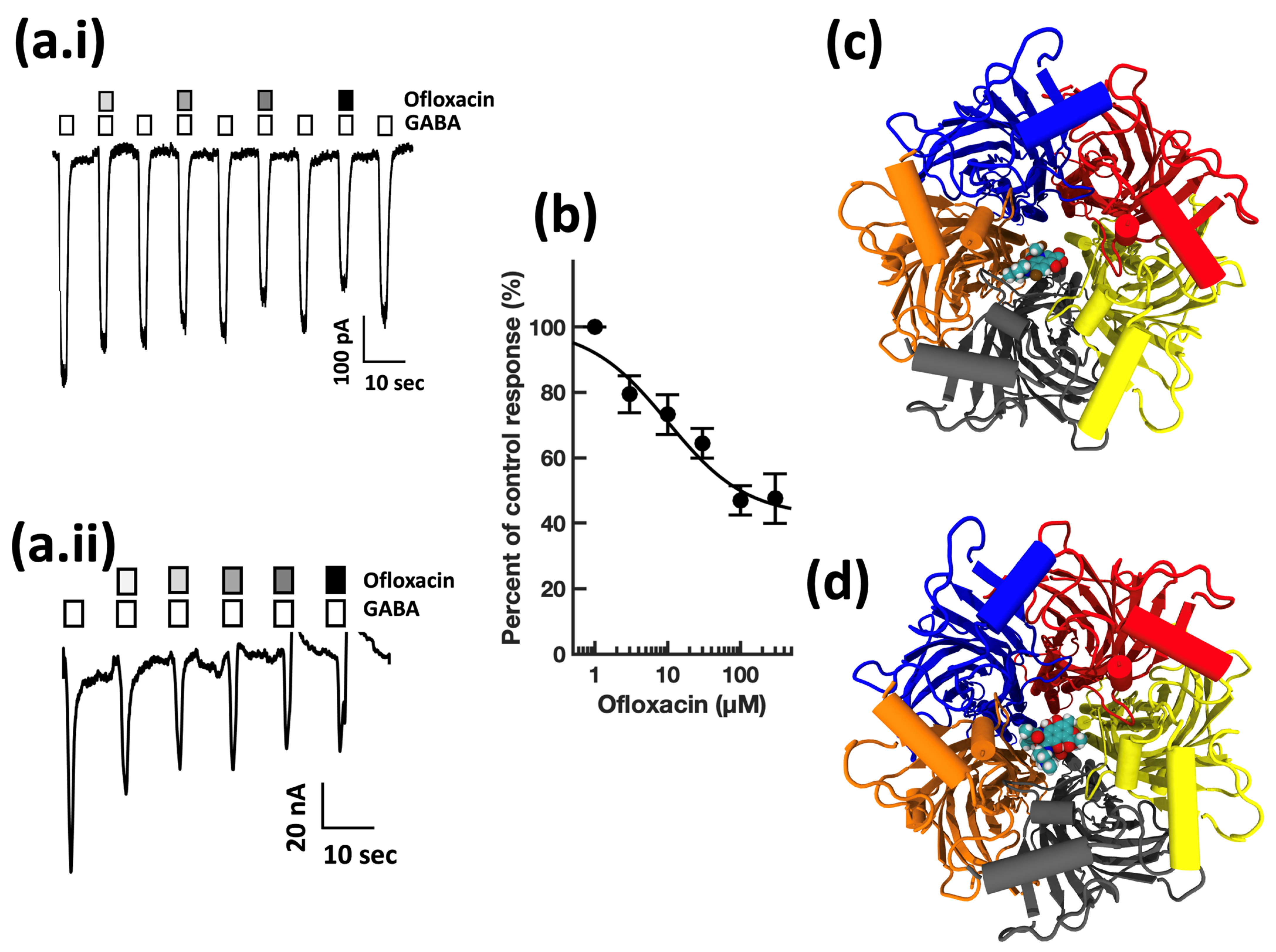
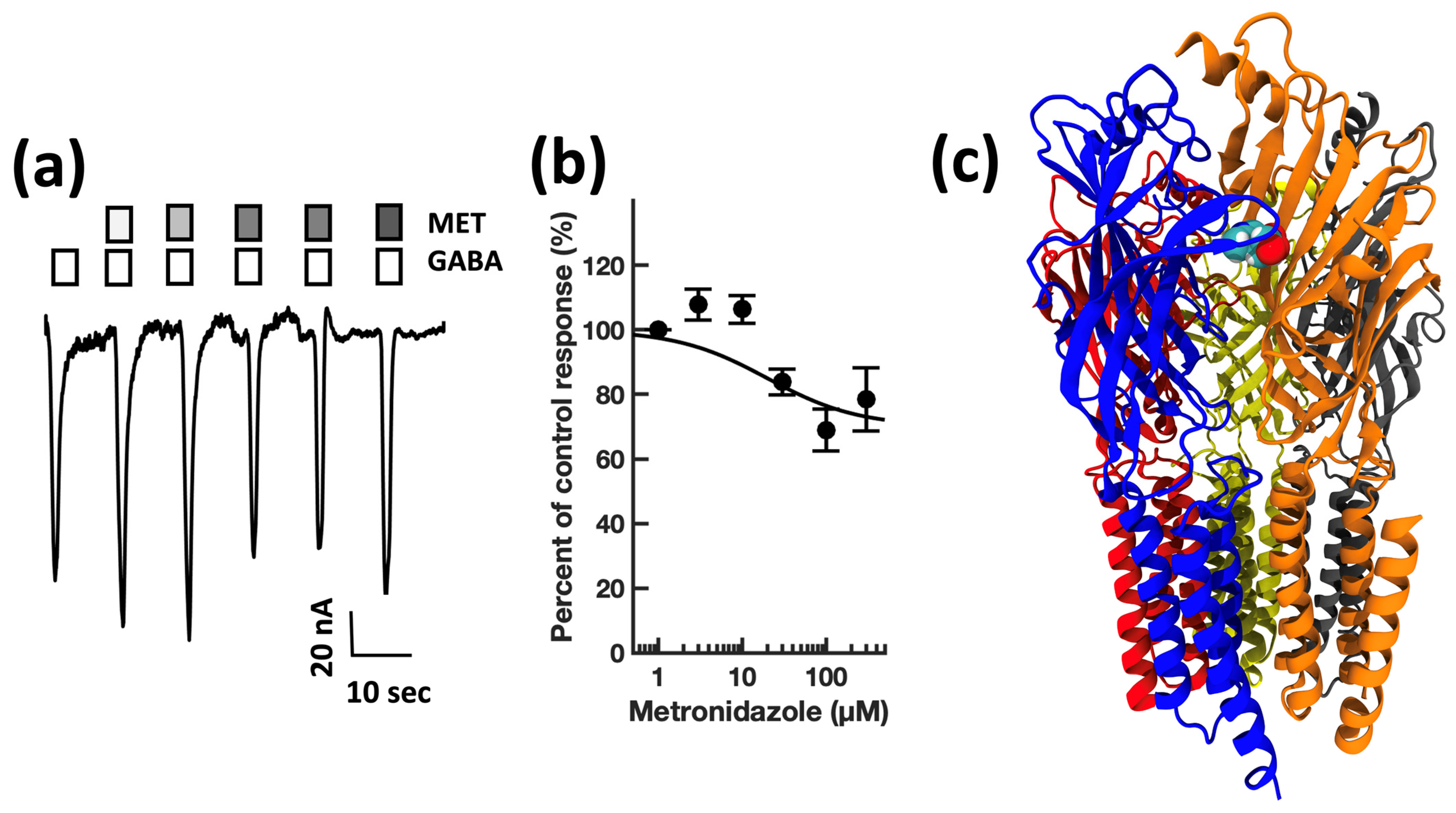
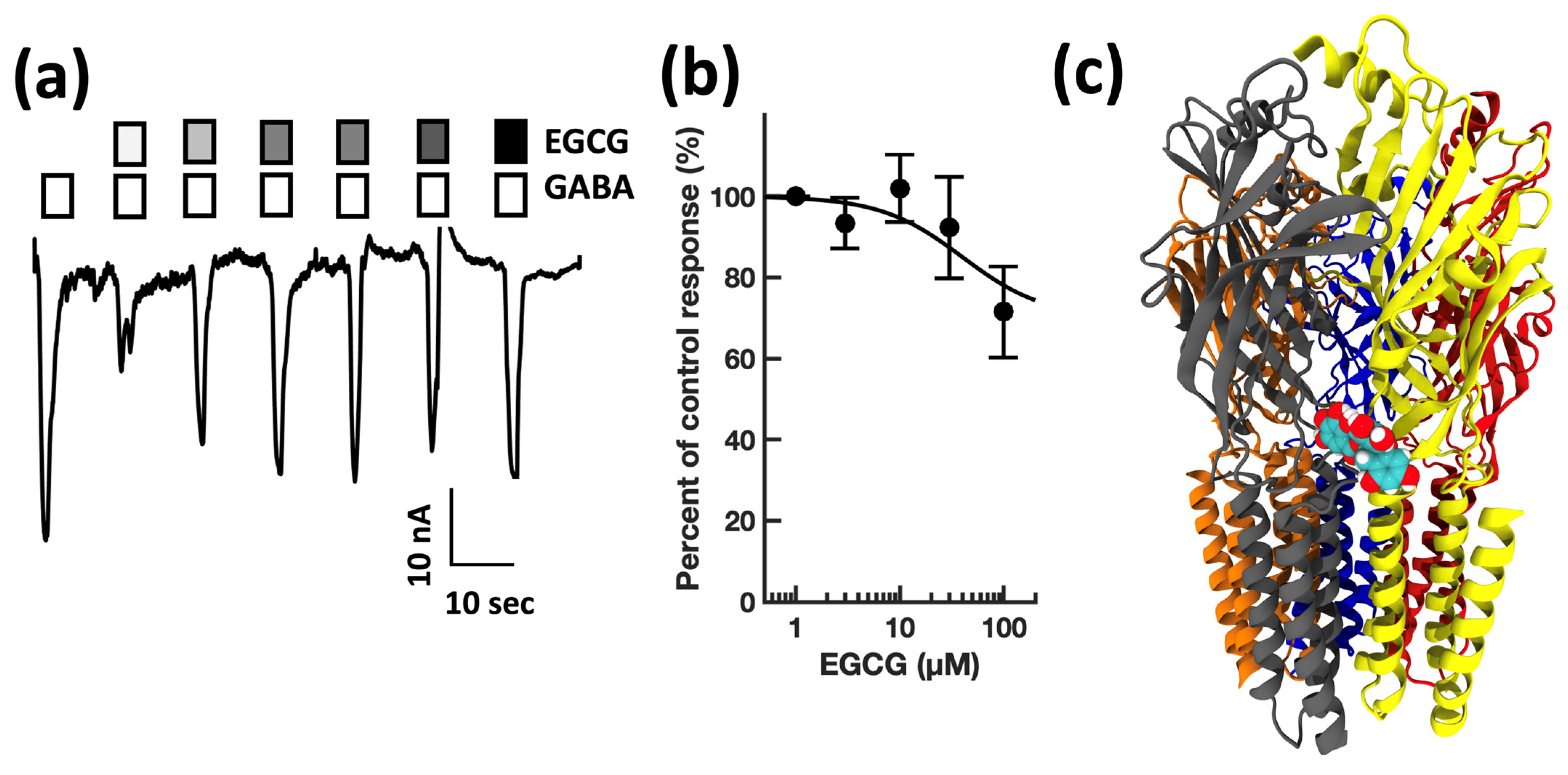
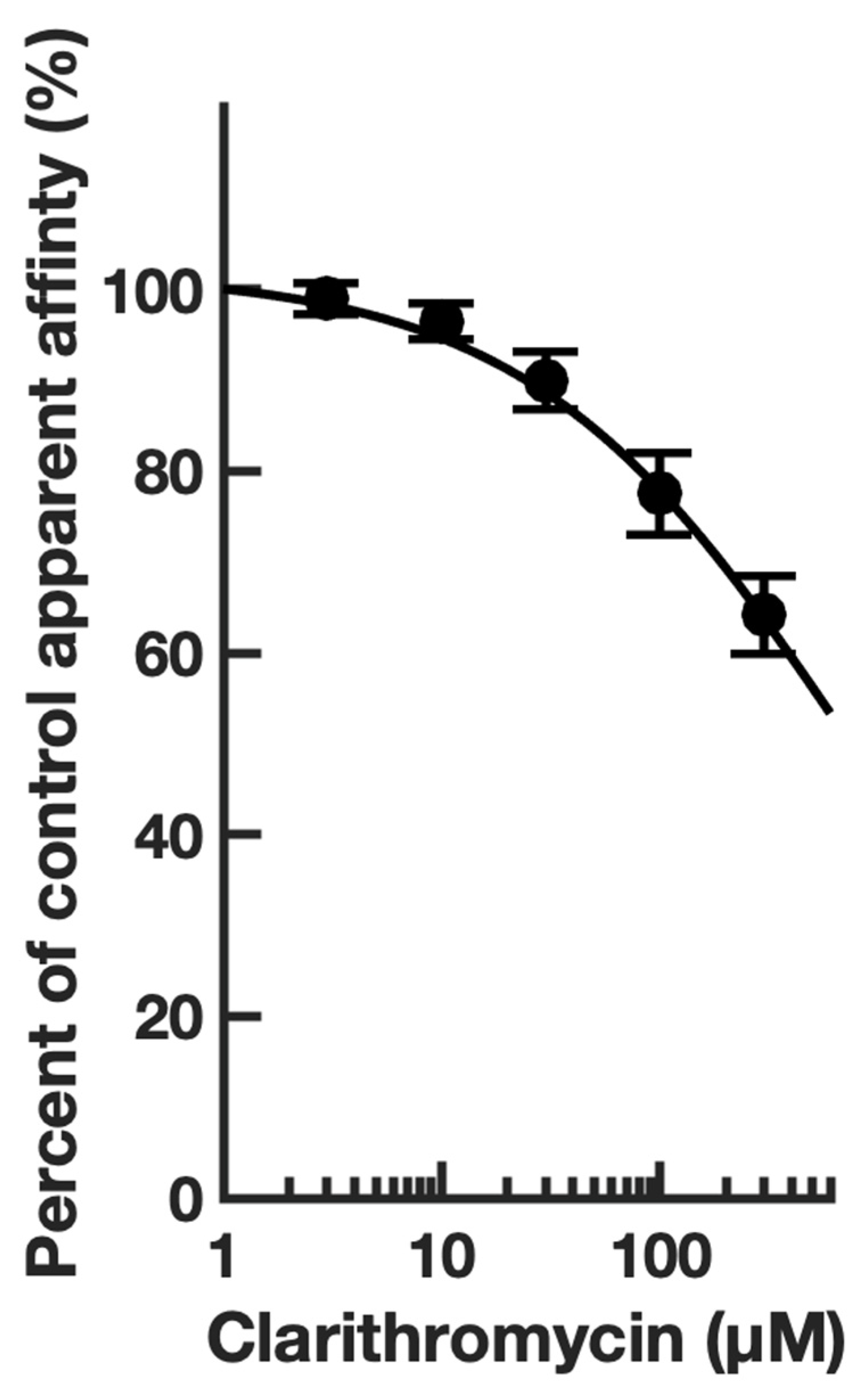
4. Results
4.1. Electrophysiology
4.2. Modeling
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fritschy, J.-M.; Mohler, H. GABAA-receptor heterogeneity in the adult rat brain: Differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 1995, 359, 154–194. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, D.E.; Ryan, S.J.; Hazra, R.; Guo, J.D.; Rainnie, D.G. Postnatal maturation of GABAergic transmission in the rat basolateral amygdala. J. Neurophysiol. 2013, 110, 926–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Low, C.-M.; Moody, O.A.; Jenkins, A.; Traynelis, S.F. Ionotropic GABA and Glutamate Receptor Mutations and Human Neurologic Diseases. Mol. Pharmacol. 2015, 88, 203–217. [Google Scholar] [CrossRef]
- Gulinello, M.; Smith, S.S. Anxiogenic effects of neurosteroid exposure: Sex differences and altered GABAA receptor pharmacology in adult rats. J. Pharmacol. Exp. Ther. 2003, 305, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewska, M.D.; Harrison, N.L.; Schwartz, R.D.; Barker, J.L.; Paul, S.M. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 1986, 232, 1004–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, N.P. General anaesthesia: From molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 2008, 9, 370–386. [Google Scholar] [CrossRef] [PubMed]
- Rye, D.B.; Bliwise, D.L.; Parker, K.; Trotti, L.M.; Saini, P.; Fairley, J.; Freeman, A.; Garcia, P.S.; Owens, M.J.; Ritchie, J.C.; et al. Modulation of vigilance in the primary hypersomnias by endogenous enhancement of GABAA receptors. Sci. Transl. Med. 2012, 4, 161ra51. [Google Scholar] [CrossRef] [PubMed]
- Safavynia, M.S.A.; Keating, G.; Speigel, I.; Fidler, B.J.A.; Kreuzer, M.; Rye, M.D.B.; Jenkins, A.; García, M.P.S. Effects of gamma-Aminobutyric Acid Type A Receptor Modulation by Flumazenil on Emergence from General Anesthesia. Anesthesiology 2016, 125, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Trotti, L.M.; Saini, P.; Bliwise, D.L.; Freeman, A.A.; Jenkins, A.; Rye, D.B. Clarithromycin in gamma-aminobutyric acid-Related hypersomnolence: A randomized, crossover trial. Ann. Neurol. 2015, 78, 454–465. [Google Scholar] [CrossRef] [Green Version]
- Trotti, L.M.; Saini, P.; Freeman, A.A.; Bliwise, D.L.; García, P.S.; Jenkins, A.; Rye, D.B. Improvement in daytime sleepiness with clarithromycin in patients with GABA-related hypersomnia: Clinical experience. J. Psychopharmacol. 2014, 28, 697–702. [Google Scholar] [CrossRef]
- Lamp, K.C.; Freeman, C.D.; Klutman, N.E.; Lacy, M.K. Pharmacokinetics and Pharmacodynamics of the Nitroimidazole Antimicrobials. Clin. Pharmacokinet. 1999, 36, 353–373. [Google Scholar] [CrossRef]
- Brown. R.; Price, R.J.; King, M.G.; Husband, A.J. Are antibiotic effects on sleep behavior in the rat due to modulation of gut bacteria? Physiol. Behav. 1990, 48, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Misrani, A.; Tabassum, S.; Yang, L.; Long, C. Minocycline inhibits sleep deprivation-induced aberrant microglial activation and Keap1-Nrf2 expression in mouse hippocampus. Brain Res. Bull. 2021, 174, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, K.; Nakazawa, Y.; Kotorii, T. Effects of antibiotics, minocycline and ampicillin, on human sleep. Brain Res. 1983, 288, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.L.; Butt, H.; Ball, M.; Lewis, D.P.; Bruck, D. Sleep quality and the treatment of intestinal microbiota imbalance in Chronic Fatigue Syndrome: A pilot study. Sleep Sci. 2015, 8, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Khandelwal, S.K. Ofloxacin induced hypomania. J. Clin. Diagn. Res. 2013, 7, 1469–1470. [Google Scholar] [CrossRef]
- Enginar, N.; Eroglu, L.; Ulak, G. The effect of ofloxacin on pentobarbital-induced sleep in mice. Pharmacol. Biochem. Behav. 1991, 40, 65–67. [Google Scholar] [CrossRef]
- Akaike, N.; Shirasaki, T.; Yakushiji, T. Quinolones and fenbufen interact with GABAA receptor in dissociated hippocampal cells of rat. J. Neurophysiol. 1991, 66, 497–504. [Google Scholar] [CrossRef]
- Thompson, A.J.; Lummis, S.C.R. Antimalarial drugs inhibit human 5-HT(3) and GABA(A) but not GABA(C) receptors. Br. J. Pharmacol. 2008, 153, 1686–1696. [Google Scholar] [CrossRef] [Green Version]
- Steere, A.C.; Angelis, S.M. Therapy for Lyme arthritis: Strategies for the treatment of antibiotic-refractory arthritis. Arthritis Rheum. 2006, 54, 3079–3086. [Google Scholar] [CrossRef]
- Guan, P.; Sun, C.; Chen, Z.; Chen, J.; Ran, R. Long-term hydroxychloroquine therapy improves the quality of sleep in patients with primary Sjogren’s syndrome: A real-world study. Ann. Palliat. Med. 2020, 9, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Trotti, L.M.; Saini, P.; Koola, C.; LaBarbera, V.; Bliwise, D.L.; Rye, D.B. Flumazenil for the Treatment of Refractory Hypersomnolence: Clinical Experience with 153 Patients. J. Clin. Sleep Med. 2016, 12, 1389–1394. [Google Scholar] [CrossRef]
- De Deyn, P.P.; Marescau, B.; MacDonald, R.L. Epilepsy and the GABA-hypothesis a brief review and some examples. Acta Neurol. Belg. 1990, 90, 65–81. [Google Scholar] [PubMed]
- Möhler, H. Cognitive enhancement by pharmacological and behavioral interventions: The murine Down syndrome model. Biochem. Pharmacol. 2012, 84, 994–999. [Google Scholar] [CrossRef]
- Möhler, H. The legacy of the benzodiazepine receptor: From flumazenil to enhancing cognition in Down syndrome and social interaction in autism. Adv. Pharmacol. 2015, 72, 1–36. [Google Scholar] [CrossRef]
- Rudolph, U.; Mohler, H. GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu. Rev. Pharmacol Toxicol. 2014, 54, 483–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giménez, S.; Altuna, M.; Blessing, E.; Osorio, R.; Fortea, J. Sleep Disorders in Adults with Down Syndrome. J. Clin. Med. 2021, 10, 3012. [Google Scholar] [CrossRef]
- Campbell, E.L.; Chebib, M.; Johnston, G.A. The dietary flavonoids apigenin and (-)-epigallocatechin gallate enhance the positive modulation by diazepam of the activation by GABA of recombinant GABA(A) receptors. Biochem. Pharmacol. 2004, 68, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Unno, K.; Pervin, M.; Nakagawa, A.; Iguchi, K.; Hara, A.; Takagaki, A.; Nanjo, F.; Minami, A.; Nakamura, Y. Blood–Brain Barrier Permeability of Green Tea Catechin Metabolites and their Neuritogenic Activity in Human Neuroblastoma SH-SY5Y Cells. Mol. Nutr. Food Res. 2017, 61, 1700294. [Google Scholar] [CrossRef]
- Perszyk, R.E.; Yip, M.C.; McConnell, O.L.; Wang, E.T.; Jenkins, A.; Traynelis, S.F.; Forest, C.R. Automated Intracellular Pharmacological Electrophysiology for Ligand-Gated Ionotropic Receptor and Pharmacology Screening. Mol. Pharmacol. 2021, 100, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Dixon, C.; Sah, P.; Lynch, J.W.; Keramidas, A. GABAA receptor alpha and gamma subunits shape synaptic currents via different mechanisms. J. Biol. Chem. 2014, 289, 5399–5411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallay, L.; Keskin, H.; Ross, A.; Rupji, M.; Moody, O.A.; Wang, X.; Li, G.; Ahmed, T.; Rashid, F.; Stephen, M.R.; et al. Modulating native GABAA receptors in medulloblastoma with positive allosteric benzodiazepine-derivatives induces cell death. J. Neurooncol. 2019, 142, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krummel, D.A.P.; Nasti, T.H.; Kaluzova, M.; Kallay, L.; Bhattacharya, D.; Melms, J.C.; Izar, B.; Xu, M.; Burnham, A.; Ahmed, T.; et al. Melanoma Cell Intrinsic GABAA Receptor Enhancement Potentiates Radiation and Immune Checkpoint Inhibitor Response by Promoting Direct and T Cell-Mediated Antitumor Activity. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 1040–1053. [Google Scholar] [CrossRef]
- Phulera, S.; Zhu, H.; Yu, J.; Claxton, D.P.; Yoder, N.; Yoshioka, C.; Gouaux, E. Cryo-EM structure of the benzodiazepine-sensitive alpha1beta1gamma2S tri-heteromeric GABA(A) receptor in complex with GABA. Elife 2018, 7, e39383. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of Green Tea Catechins in the Brain: Epigallocatechin Gallate and its Metabolites. Int. J. Mol. Sci. 2019, 20, 3630. [Google Scholar] [CrossRef] [Green Version]
- Osifo, N.G. The regional uptake of chloroquine in the rat brain. Toxicol. Appl. Pharmacol. 1979, 50, 109–114. [Google Scholar] [CrossRef]
- Maniu, C.V.; Hellinger, W.C.; Chu, S.; Palmer, R.; Alvarez-Elcoro, S. Failure of Treatment for ChronicMycobacterium abscessusMeningitis Despite Adequate Clarithromycin Levels in Cerebrospinal Fluid. Clin. Infect. Dis. 2001, 33, 745–748. [Google Scholar] [CrossRef]
- Imshenetskaia, V.F. Erythromycin penetration into the cerebrospinal fluid of patients. Antibiotiki 1976, 21, 1002–1004. [Google Scholar]
- Manzo, C.; Gareri, P.; Castagna, A. Psychomotor Agitation Following Treatment with Hydroxychloroquine. Drug Saf. Case Rep. 2017, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Jokipii, A.M.M.; Myllylä, V.V.; Hokkanen, E.; Jokipii, L. Penetration of the blood brain barrier by metronidazole and tinidazole. J. Antimicrob. Chemother. 1977, 3, 239–245. [Google Scholar] [CrossRef]
- Saivin, S.; Houin, G. Clinical Pharmacokinetics of Doxycycline and Minocycline. Clin. Pharmacokinet. 1988, 15, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Nau, R.; Kinzig, M.; Dreyhaupt, T.; Kolenda, H.; Sörgel, F.; Prange, H.W. Kinetics of ofloxacin and its metabolites in cerebrospinal fluid after a single intravenous infusion of 400 milligrams of ofloxacin. Antimicrob. Agents Chemother. 1994, 38, 1849–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramzan, I.M.; Levy, G. Kinetics of drug action in disease states. XIV. Effect of infusion rate on pentylenetetrazol concentrations in serum, brain and cerebrospinal fluid of rats at onset of convulsions. J. Pharmacol. Exp. Ther. 1985, 234, 624–628. [Google Scholar] [PubMed]
- Ruiz-Bedoya, C.A.; Mota, F.; Tucker, E.W.; Mahmud, F.J.; Reyes-Mantilla, M.I.; Erice, C.; Bahr, M.; Flavahan, K.; de Jesus, P.; Kim, J.; et al. High-dose rifampin improves bactericidal activity without increased intracerebral inflammation in animal models of tuberculous meningitis. J. Clin. Investig. 2022, 132, e155851. [Google Scholar] [CrossRef]
- Masiulis, S.; Desai, R.; Uchanski, T.; Martin, I.S.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABA(A) receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Barker, J.L. Pentylenetetrazol and penicillin are selective antagonists of GABA-mediated post-synaptic inhibition in cultured mammalian neurones. Nature 1977, 267, 720–721. [Google Scholar] [CrossRef]
- Rehavi, M.; Skolnick, P.; Paul, S.M. Effects of tetrazole derivatives on [3H]diazepam binding in vitro: Correlation with convulsant potency. Eur. J. Pharmacol. 1982, 78, 353–356. [Google Scholar] [CrossRef]
- Squires, R.F.; Saederup, E.; Crawley, J.N.; Skolnick, P.; Paul, S.M. Convulsant potencies of tetrazoles are highly correlated with actions on GABA/benzodiazepine/picrotoxin receptor complexes in brain. Life Sci. 1984, 35, 1439–1444. [Google Scholar] [CrossRef]
- Ticku, M.K.; Maksay, G. Convulsant/depressant site of action at the allosteric benzodiazepine-gaba receptor-ionophore complex. Life Sci. 1983, 33, 2363–2375. [Google Scholar] [CrossRef]
- Huang, R.Q.; Bell-Horner, C.L.; Dibas, M.I.; Covey, D.F.; Drewe, J.A.; Dillon, G.H. Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: Mechanism and site of action. J. Pharmacol. Exp. Ther. 2001, 298, 986–995. [Google Scholar]
- Bichler, E.K.; Elder, C.C.; García, P.S. Clarithromycin increases neuronal excitability in CA3 pyramidal neurons through a reduction in GABAergic signaling. J. Neurophysiol. 2017, 117, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maski, M.K.; Trotti, L.M.; Kotagal, S.; Auger, R.R.; Swick, T.J.; Rowley, J.A.; Hashmi, M.S.D.; Watson, N.F. Treatment of central disorders of hypersomnolence: An American Academy of Sleep Medicine systematic review, meta-analysis, and GRADE assessment. J. Clin. Sleep Med. 2021, 17, 1895–1945. [Google Scholar] [CrossRef] [PubMed]
- Grahl, J.J.; Stollings, J.L.; Rakhit, S.; Person, A.K.; Wang, L.; Thompson, J.L.; Pandharipande, P.P.; Ely, E.W.; Patel, M.B. Antimicrobial exposure and the risk of delirium in critically ill patients. Crit. Care 2018, 22, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pejčić, A.V. Delirium associated with the use of macrolide antibiotics: A review. Int. J. Psychiatry Clin. Pract. 2022, 26, 29–42. [Google Scholar] [CrossRef]
- Twyman, R.E.; Green, R.M.; MacDonald, R.L. Kinetics of open channel block by penicillin of single GABAA receptor channels from mouse spinal cord neurones in culture. J. Physiol. 1992, 445, 97–127. [Google Scholar] [CrossRef]
- Yakushiji, T.; Shirasaki, T.; Akaike, N. Non-competitive inhibition of GABAA responses by a new class of quinolones and non-steroidal anti-inflammatories in dissociated frog sensory neurones. Br. J. Pharmacol. 1992, 105, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Koniuszewski, F.; Vogel, F.D.; Bampali, K.; Fabjan, J.; Seidel, T.; Scholze, P.; Schmiedhofer, P.B.; Langer, T.; Ernst, M. Molecular Mingling: Multimodal Predictions of Ligand Promiscuity in Pentameric Ligand-Gated Ion Channels. Front. Mol. Biosci. 2022, 9, 860246. [Google Scholar] [CrossRef]
- Jenkins, A.; Andreasen, A.; Trudell, J.R.; Harrison, N.L. Tryptophan scanning mutagenesis in TM4 of the GABA(A) receptor alpha1 subunit: Implications for modulation by inhaled anesthetics and ion channel structure. Neuropharmacology 2002, 43, 669–678. [Google Scholar] [CrossRef]
- Jenkins, A.; Greenblatt, E.P.; Faulkner, H.J.; Bertaccini, E.; Light, A.; Lin, A.; Andreasen, A.; Viner, A.; Trudell, J.R.; Harrison, N.L. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J. Neurosci. 2001, 21, RC136. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Class | Use | [CNS] | GABA EC50 | Imax (%) | nH |
|---|---|---|---|---|---|---|
| EGCG | polyphenol | dietary supplement | 0.18 [35] | 31 ± 4 | 22 ± 8 | 1.1 |
| Chloroquine | aminoquinolone | antimalarial | 1.60 [36] | |||
| Clarithromycin | macrolide | antibiotic | 10 [37] | 20 ± 1 | 85 ± 8 | 1 |
| Erythromycin | macrolide | antibiotic | 0.68 [38] | |||
| Hydroxychloroquine | aminoquinolone | antimalarial | 50 [39] | 40 ± 8 | 29 ± 4 | 1.4 |
| Metronidazole | nitroimidazole | Antibiotic/protozoal | 7010 [40] | 50 ± 11 | 24 ± 9 | 0.8 |
| Minocycline | tetracycline | antibiotic | 1.1 [41] | |||
| Ofloxacin | quinolone | antibiotic | 5.6 [42] | 15 ± 4 | 61 ± 7 | 1 |
| pentylenetetrazole | azepine | stimulant/convulsant | 330 [43] | 10 ± 3 | 68 ± 5 | 1.3 |
| Rifampin | ansamycin | antibiotic | 3.60 [44] |
| Compound | Glide Score | MMGBSA | QPlogPo/w | QPlogkhsa | CNS |
|---|---|---|---|---|---|
| Chloroquine | −9.528 | −69.04 | 4.653 | 0.629 | 1 |
| Erythromycin | N/A | N/A | 2.558 | −0.060 | −1 |
| Metronidazole | −6.505 | −38.49 | −0.019 | −0.688 | −2 |
| Minocycline | −6.690 | −47.42 | 0.323 | 0.178 | −2 |
| Ofloxacin | −4.224 | −30.44 | −0.388 | −0.431 | 0 |
| (-)-Epigallocatechin gallate | −10.319 | −65.68 | −0.275 | −0.447 | −2 |
| Flumazenil | −9.324 | −44.59 | 1.956 | −0.442 | 0 |
| Hydroxychloroquine | −10.956 | −65.90 | 3.564 | 0.207 | 1 |
| Pentylenetetrazol | −6.954 | −33.85 | 0.555 | −0.658 | 0 |
| Clarithromycin | −3.271 | −34.09 | 3.090 | 0.058 | 0 |
| Rifampicin | −3.052 | −41.18 | 3.302 | −0.181 | −2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaplan, A.; Nash, A.I.; Freeman, A.A.H.; Lewicki, L.G.; Rye, D.B.; Trotti, L.M.; Brandt, A.L.; Jenkins, A. Commonly Used Therapeutics Associated with Changes in Arousal Inhibit GABAAR Activation. Biomolecules 2023, 13, 365. https://0-doi-org.brum.beds.ac.uk/10.3390/biom13020365
Kaplan A, Nash AI, Freeman AAH, Lewicki LG, Rye DB, Trotti LM, Brandt AL, Jenkins A. Commonly Used Therapeutics Associated with Changes in Arousal Inhibit GABAAR Activation. Biomolecules. 2023; 13(2):365. https://0-doi-org.brum.beds.ac.uk/10.3390/biom13020365
Chicago/Turabian StyleKaplan, Anling, Abigail I. Nash, Amanda A. H. Freeman, Lauren G. Lewicki, David B. Rye, Lynn Marie Trotti, Asher L. Brandt, and Andrew Jenkins. 2023. "Commonly Used Therapeutics Associated with Changes in Arousal Inhibit GABAAR Activation" Biomolecules 13, no. 2: 365. https://0-doi-org.brum.beds.ac.uk/10.3390/biom13020365