Reliable Generation of Native-Like Decoys Limits Predictive Ability in Fragment-Based Protein Structure Prediction


Abstract
:1. Introduction
- Is it simply the case that fragment set composition (interacting with the sampling algorithms) renders native-like states rarely accessible?
- Is it the case that such structures were in fact sampled during many trajectories, but were not retained because of relatively unfavourable score values?
1.1. Alternative Decoy Acceptance Criteria in Protein Structure Prediction
1.1.1. Multiobjectivisation by Scoring Function Decomposition
1.1.2. Diversity-Based Decoy Acceptance Criteria
1.2. Bilevel and Iterated Local Search (ILS) Protocols
- Each run applies alternating rounds of perturbation and local search operators to the folding candidate structure. The perturbation steps are designed to encourage conformational exploration, and the operator comprises a single fragment insertion operation which is accepted regardless of its impact on energy or score value (which helps to escape from local minima). The local search operator performs fragment insertions and accepts new solutions greedily until 50 fragment insertions have been attempted without acceptance. At the end of each round of local search, the optimisation is said to have reached a local minimum (LMin).
- A newly accessed LMin is compared to the LMin last encountered, and the Metropolis criterion [37] is used to define whether a new LMin should be accepted or rejected. The Metropolis temperature parameter is varied by a simulated annealing scheme.
- An external archive maintains the set of the most favourable LMin structures over the course of each run, under the scoring function in use at any given time. All encountered LMins are considered for addition to this archive. Once stored, the archived solutions are not used to inform the search process.
- Our sampling strategies are active in Stages 2 and 3 of the Rosetta low-resolution protocol. Stage 1 can be seen as a randomisation/initialisation routine, and is used without modification. Following Stage 3, each structure retained in the archive is put through Stage 4 of the standard Rosetta low-resolution protocol.
1.3. Outline and Contributions of This Study
2. Results
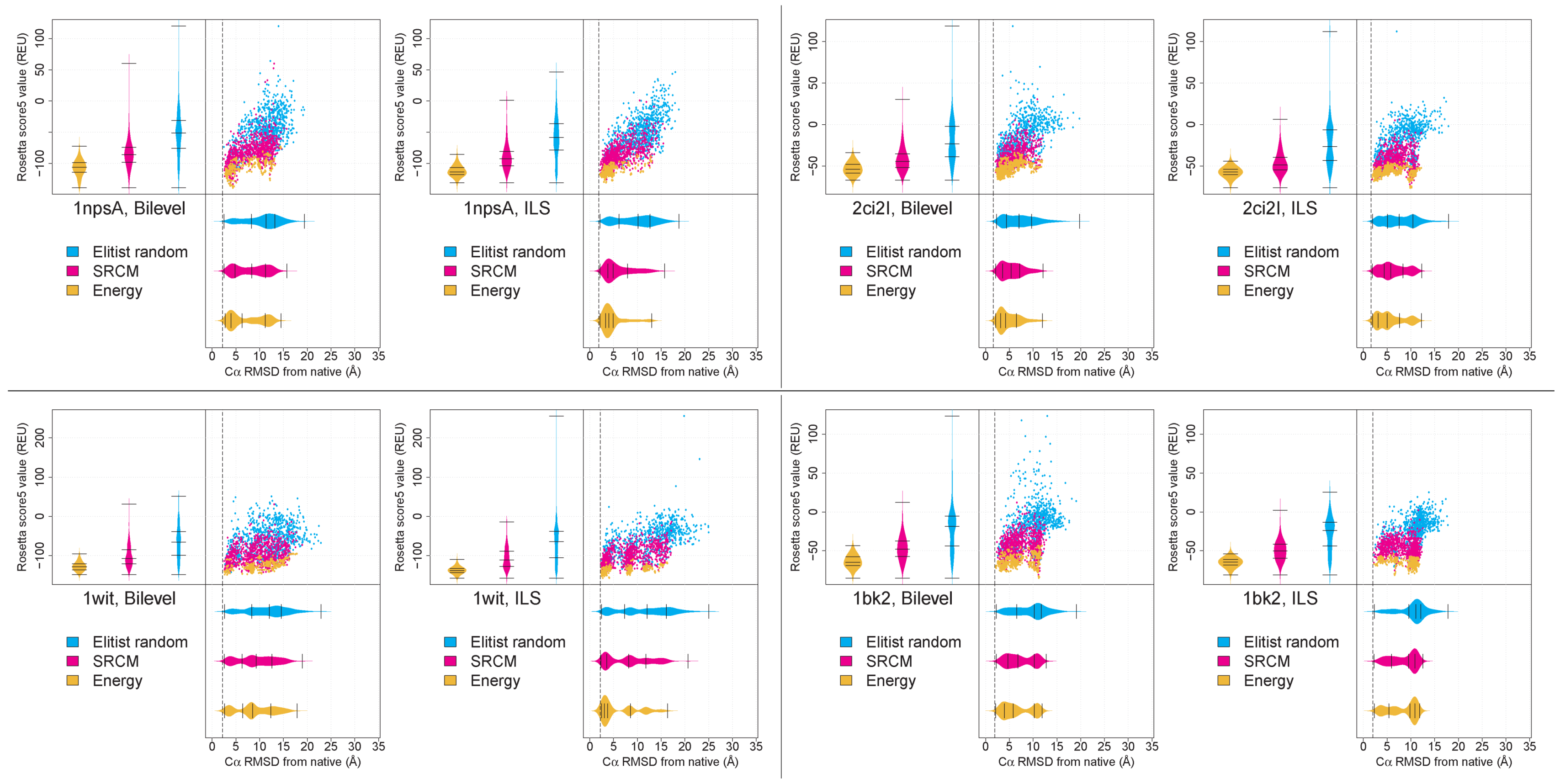
2.1. The SRCM and ER Archivers Succeed in Retaining Structurally Diverse Conformations
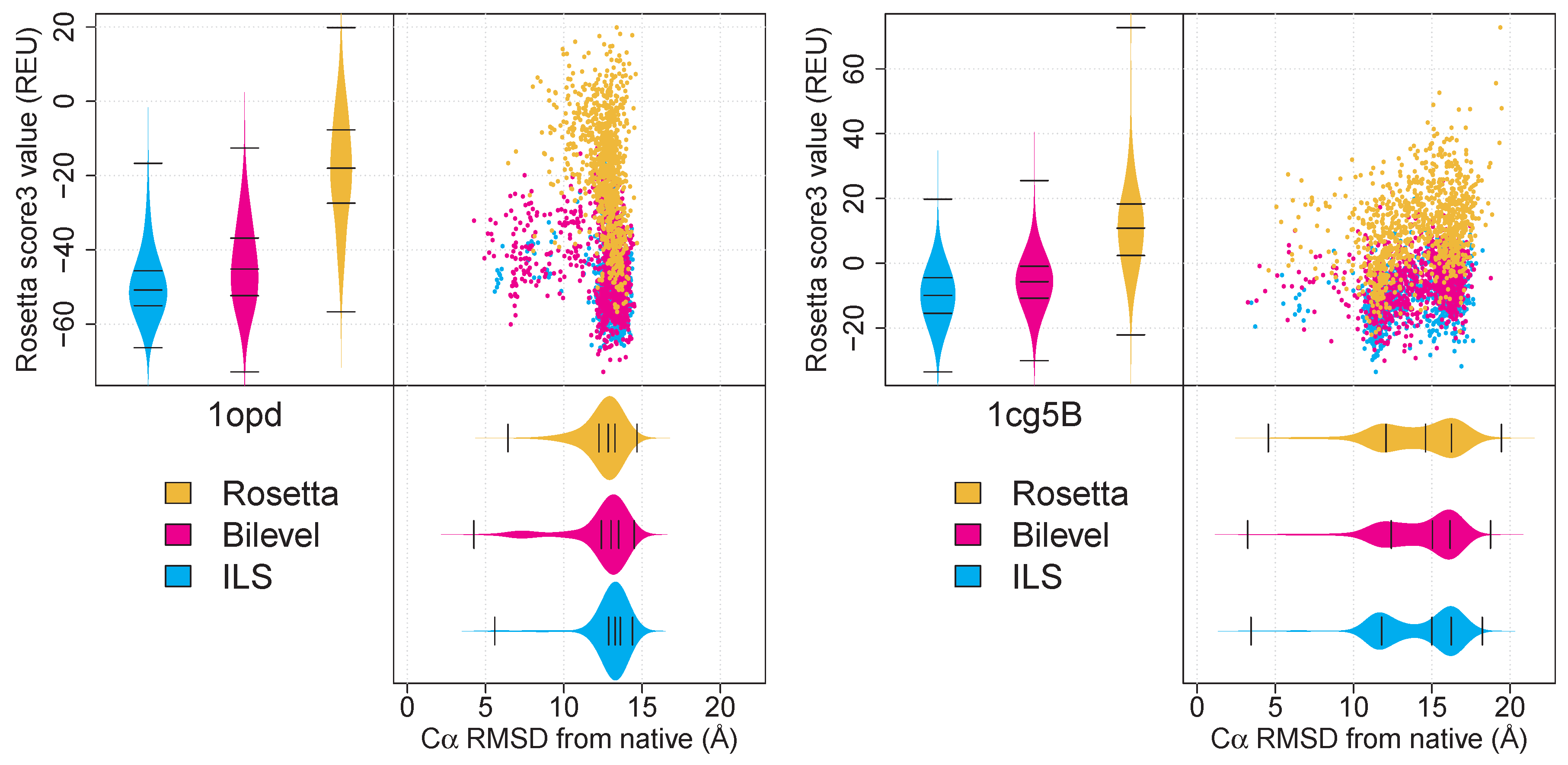
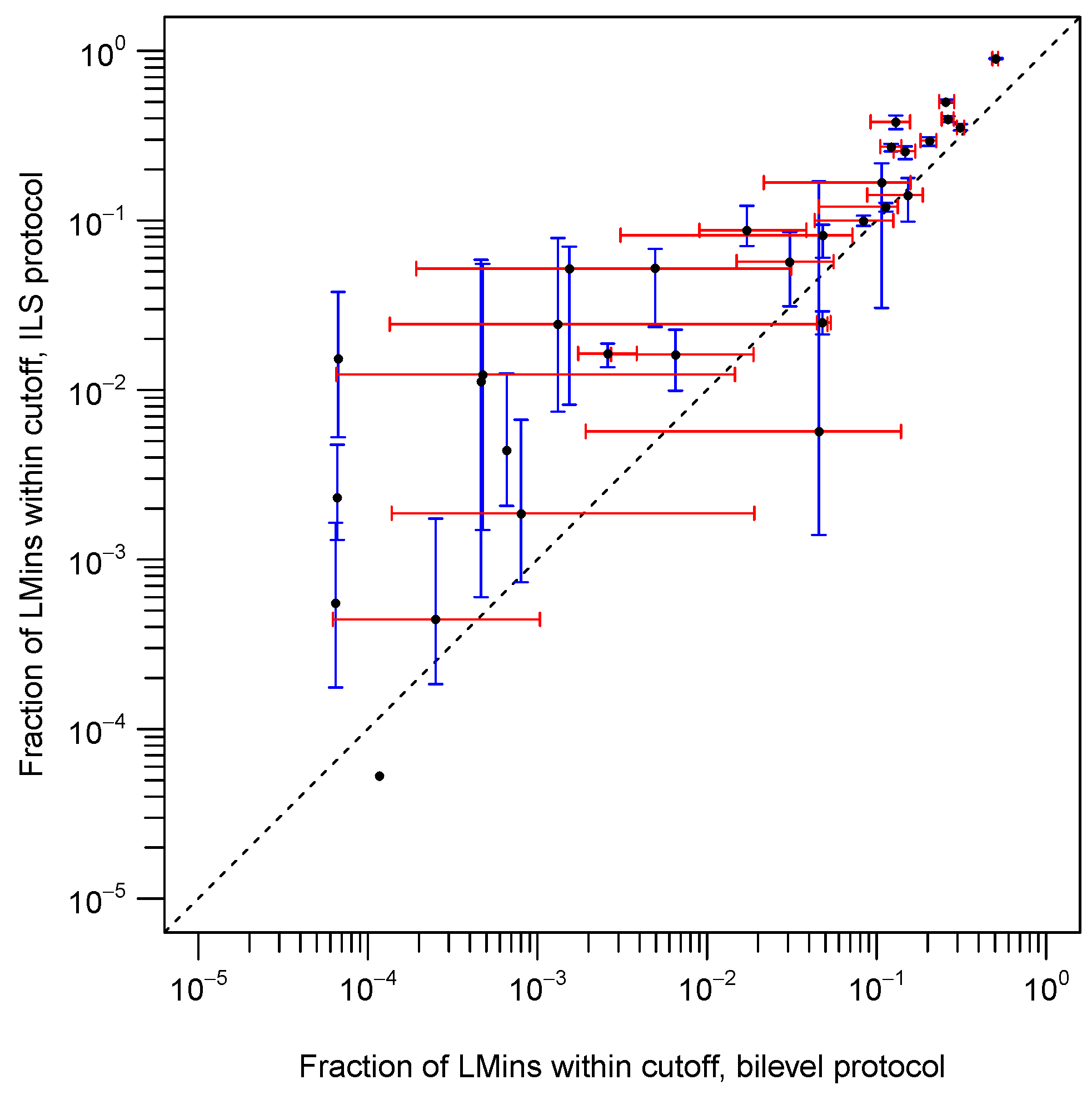
2.2. The Bilevel and ILS Protocols Access Native-Like Local Minima (LMins) with Differing Frequencies
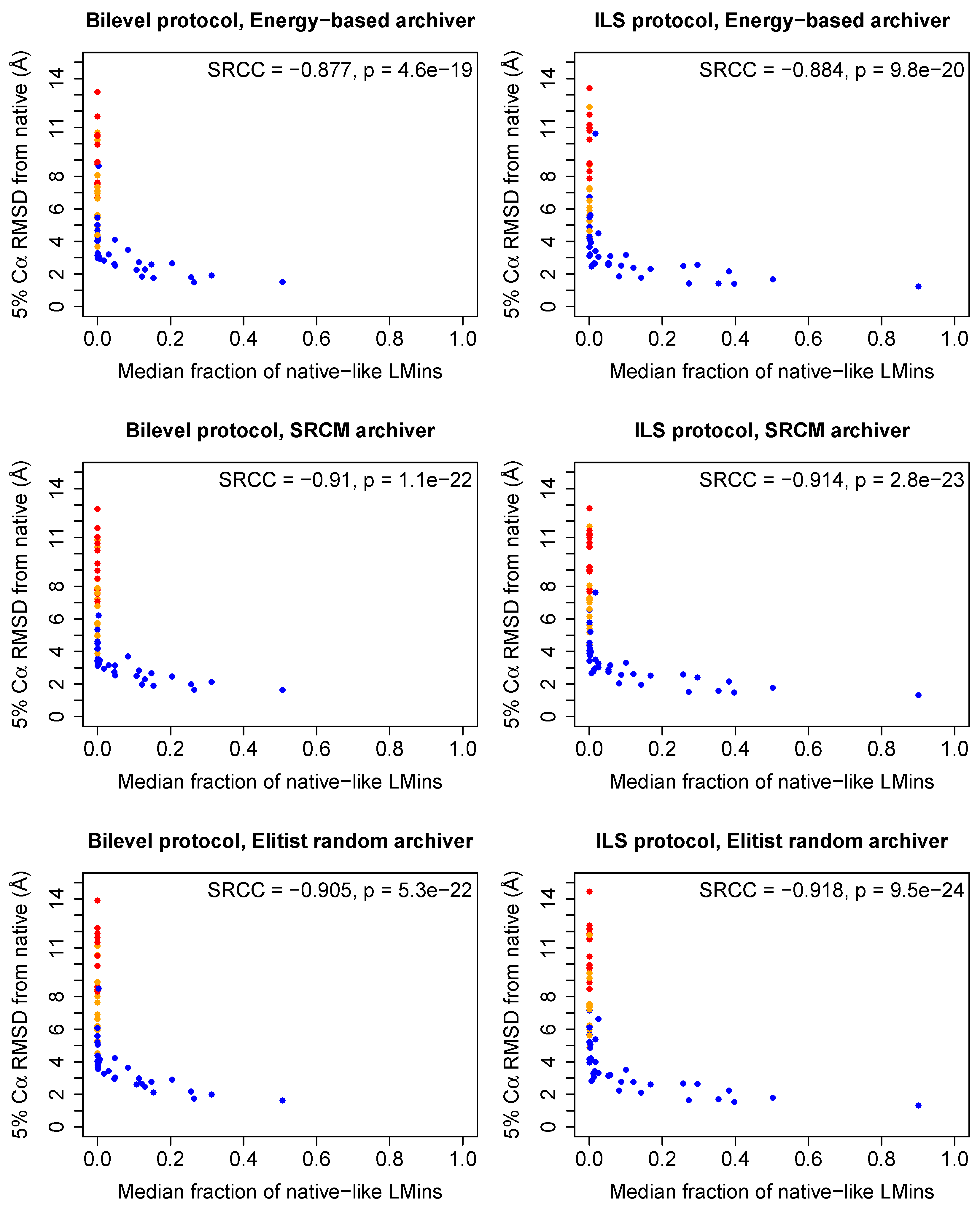
2.3. Effective Conformational Sampling Has a Greater Impact Than Choice of Archiver
2.4. Decoys Retained by Energy-Based Archiving Tend to Be of Higher Quality Following Stage 4 of the Low-Resolution Search
3. Discussion
4. Materials and Methods
4.1. Assessment of LMin Accessibility and Predictive Accuracy
4.2. Stochastic Ranking-Based Archiving Strategy
| Algorithm 1 Generalised stochastic ranking-based procedure. |
| Require: Solution list (), ranking criteria (), bias parameter () Ensure: Ranked list of solutions ()
|
4.3. Elitist Step and Choice of Ranking Criteria
4.4. Implementation within the Bilevel and ILS Protocols
4.5. Elitist Random (ER) Archiver
4.6. Experimental Setup
- A purely energy-based archiver, as in [5];
- a stochastic ranking-based archiver using contact maps (SRCM); and
- an elitist random (ER) archiver.
4.6.1. Intra-Archive Diversity Assessment over the Course of Each Run
4.6.2. Number of Native-Like Solutions Retained
4.7. Code and Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ILS | Iterated local search |
| LMin | Local minimum; a decoy structure at a local minimum in the energy landscape |
| SR | Stochastic ranking |
| SRCM | Stochastic ranking using contact maps |
| ER | Elitist random |
| RMSD | Root mean square deviation |
| ECDF | Empirical cumulative distribution function |
References
- Simons, K.T.; Kooperberg, C.; Huang, E.; Baker, D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 1997, 268, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.T.; Ruczinski, I.; Kooperberg, C.; Fox, B.A.; Bystroff, C.; Baker, D. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins Struct. Funct. Bioinform. 1999, 34, 82–95. [Google Scholar] [CrossRef]
- Bowman, G.R.; Pande, V.S. Simulated tempering yields insight into the low-resolution Rosetta scoring functions. Proteins Struct. Funct. Bioinform. 2009, 74, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Das, R. Four Small Puzzles That Rosetta Doesn’t Solve. PLoS ONE 2011, 6, e20044. [Google Scholar] [CrossRef]
- Kandathil, S.M.; Garza-Fabre, M.; Handl, J.; Lovell, S.C. Improved fragment-based protein structure prediction by redesign of search heuristics. Sci. Rep. 2018, 8, 13694. [Google Scholar] [CrossRef]
- Bradley, P.; Misura, K.M.S.; Baker, D. Toward High-Resolution De Novo Struct. Predict. Small Proteins. Science 2005, 309, 1868–1871. [Google Scholar] [CrossRef]
- Tyka, M.D.; Keedy, D.A.; André, I.; DiMaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.; Baker, D. Protein structure prediction using Rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar]
- Miettinen, K. Nonlinear Multiobjective Optimization; International Series in Operations Research & Management Science; Springer: New York, NY, USA, 1999. [Google Scholar]
- Deb, K.; Pratap, A.; Agarwal, S.; Meyarivan, T. A fast and elitist multiobjective genetic algorithm: NSGA-II. IEEE Trans. Evol. Comput. 2002, 6, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Knowles, J.D.; Corne, D.W. The Pareto archived evolution strategy: A new baseline algorithm for Pareto multiobjective optimisation. In Proceedings of the IEEE 1999 Congress on Evolutionary Computation (CEC 99), Washington, DC, USA, 6–9 July 1999; Volume 1, pp. 98–105. [Google Scholar] [CrossRef]
- Kim, M.; Hiroyasu, T.; Miki, M.; Watanabe, S. SPEA2+: Improving the Performance of the Strength Pareto Evolutionary Algorithm 2. In Parallel Problem Solving from Nature—PPSN VIII; Lecture Notes in Computer Science; Yao, X., Burke, E.K., Lozano, J.A., Smith, J., Merelo-Guervós, J.J., Bullinaria, J.A., Rowe, J.E., Tiňo, P., Kabán, A., Schwefel, H.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; Volume 3242, pp. 742–751. [Google Scholar] [CrossRef]
- Knowles, J.D.; Watson, R.A.; Corne, D.W. Reducing Local Optima in Single-Objective Problems by Multi-objectivization. In Proceedings of the Evolutionary Multi-Criterion Optimization (EMO 2001), Zurich, Switzerland, 7–9 March 2001; Zitzler, E., Thiele, L., Deb, K., Coello Coello, C.A., Corne, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2001; Volume 1993, pp. 269–283. [Google Scholar] [CrossRef]
- Brockhoff, D.; Friedrich, T.; Hebbinghaus, N.; Klein, C.; Neumann, F.; Zitzler, E. Do Additional Objectives Make a Problem Harder? In Proceedings of the 9th Annual Conference on Genetic and Evolutionary Computation, London, UK, 7–11 July 2007; ACM: New York, NY, USA, 2007; pp. 765–772. [Google Scholar] [CrossRef]
- Day, R.; Zydallis, J.; Lamont, G. Solving the protein structure prediction problem through a multiobjective genetic algorithm. In Proceedings of the 2002 International Conference on Computational Nanoscience and Nanotechnology, San Juan, PR, USA, 21–25 April 2002; Volume 2, pp. 32–35. [Google Scholar]
- Cutello, V.; Narzisi, G.; Nicosia, G. A multi-objective evolutionary approach to the protein structure prediction problem. J. R. Soc. Interface 2006, 3, 139–151. [Google Scholar] [CrossRef]
- Cutello, V.; Narzisi, G.; Nicosia, G. A Class of Pareto Archived Evolution Strategy Algorithms Using Immune Inspired Operators for Ab-Initio Protein Structure Prediction. In Proceedings of the Applications of Evolutionary Computing (EvoWorkshops 2005), Lausanne, Switzerland, 30 March–1 April 2005; Rothlauf, F., Branke, J., Cagnoni, S., Corne, D.W., Drechsler, R., Jin, Y., Machado, P., Marchiori, E., Romero, J., Smith, G.D., et al., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3449, pp. 54–63. [Google Scholar] [CrossRef]
- Judy, M.; Ravichandran, K.; Murugesan, K. A multi-objective evolutionary algorithm for protein structure prediction with immune operators. Comput. Methods Biomech. Biomed. Eng. 2009, 12, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Becerra, D.; Sandoval, A.; Restrepo-Montoya, D.; Luis, F.N. A parallel multi-objective ab initio approach for protein structure prediction. In Proceedings of the 2010 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Hong Kong, China, 18–21 December 2010; pp. 137–141. [Google Scholar] [CrossRef]
- Handl, J.; Lovell, S.C.; Knowles, J. Investigations into the Effect of Multiobjectivization in Protein Structure Prediction. In Parallel Problem Solving from Nature—PPSN X; Lecture Notes in Computer Science; Rudolph, G., Jansen, T., Beume, N., Lucas, S., Poloni, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 5199, pp. 702–711. [Google Scholar] [CrossRef]
- Olson, B.; Shehu, A. Multi-Objective Stochastic Search for Sampling Local Minima in the Protein Energy Surface. In Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics, Washington DC, USA, 22–25 September 2013; ACM: New York, NY, USA, 2013; pp. 430:430–430:439. [Google Scholar] [CrossRef]
- Venske, S.M.S.; Gonçalves, R.A.; Benelli, E.M.; Delgado, M.R. A Multiobjective Algorithm for Protein Structure Prediction Using Adaptive Differential Evolution. In Proceedings of the IEEE 2013 Brazilian Conference on Intelligent Systems, Fortaleza, Brazil, 19–24 October 2013; pp. 263–268. [Google Scholar] [CrossRef]
- Rocha, G.K.; Custódio, F.L.; Barbosa, H.J.C.; Dardenne, L.E. A multiobjective approach for protein structure prediction using a steady-state genetic algorithm with phenotypic crowding. In Proceedings of the 2015 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Niagara Falls, ON, Canada, 12–15 August 2015; pp. 1–8. [Google Scholar] [CrossRef]
- Garza-Fabre, M.; Toscano-Pulido, G.; Rodriguez-Tello, E. Multi-objectivization, Fitness Landscape Transformation and Search Performance: A Case of Study on the HP model for Protein Structure Prediction. Eur. J. Oper. Res. 2015, 243, 405–422. [Google Scholar] [CrossRef]
- Venske, S.M.; Gonçalves, R.A.; Benelli, E.M.; Delgado, M.R. ADEMO/D: An adaptive differential evolution for protein structure prediction problem. Expert Syst. Appl. 2016, 56, 209–226. [Google Scholar] [CrossRef]
- Rocha, G.K.; Custódio, F.L.; Barbosa, H.J.; Dardenne, L.E. Using Crowding-Distance in a Multiobjective Genetic Algorithm for Protein Structure Prediction. In Proceedings of the 2016 Genetic and Evolutionary Computation Conference Companion, Denver, CO, USA, 20–24 July 2016; ACM: New York, NY, USA, 2016; pp. 1285–1292. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem. 2004, 25, 865–871. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Shortle, D.; Simons, K.T.; Baker, D. Clustering of low-energy conformations near the native structures of small proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 11158–11162. [Google Scholar] [CrossRef] [Green Version]
- Shehu, A.; Olson, B. Guiding the Search for Native-like Protein Conformations with an Ab Initio Tree-Based Explor. Int. J. Robot. Res. 2010, 29, 1106–1127. [Google Scholar] [CrossRef]
- Ballester, P.J.; Richards, W.G. Ultrafast shape recognition to search compound databases for similar molecular shapes. J. Comput. Chem. 2007, 28, 1711–1723. [Google Scholar] [CrossRef]
- Molloy, K.; Saleh, S.; Shehu, A. Probabilistic Search and Energy Guidance for Biased Decoy Sampling in Ab Initio Protein Struct. Predict. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 1162–1175. [Google Scholar] [CrossRef]
- Črepinšek, M.; Liu, S.H.; Mernik, M. Exploration and Exploitation in Evolutionary Algorithms: A Survey. ACM Comput. Surv. 2013, 45, 35:1–35:33. [Google Scholar] [CrossRef]
- Custódio, F.L.; Barbosa, H.J.; Dardenne, L.E. A multiple minima genetic algorithm for protein structure prediction. Appl. Soft Comput. 2014, 15, 88–99. [Google Scholar] [CrossRef]
- Garza-Fabre, M.; Kandathil, S.M.; Handl, J.; Knowles, J.; Lovell, S.C. Generating, Maintaining, and Exploiting Diversity in a Memetic Algorithm for Protein Structure Prediction. Evol. Comput. 2016, 24, 577–607. [Google Scholar] [CrossRef] [PubMed]
- Runarsson, T.; Yao, X. Stochastic ranking for constrained evolutionary optimization. IEEE Trans. Evol. Comput. 2000, 4, 284–294. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, J.; McGill, J.T. Mathematical Programs with Optimization Problems in the Constraints. Oper. Res. 1973, 21, 37–44. [Google Scholar] [CrossRef]
- Colson, B.; Marcotte, P.; Savard, G. An overview of bilevel optimization. Ann. Oper. Res. 2007, 153, 235–256. [Google Scholar] [CrossRef]
- Kandathil, S.M.; Handl, J.; Lovell, S.C. Toward a detailed understanding of search trajectories in fragment assembly approaches to protein structure prediction. Proteins Struct. Funct. Bioinform. 2016, 84, 411–426. [Google Scholar] [CrossRef] [Green Version]
- Saleh, S.; Olson, B.; Shehu, A. A population-based evolutionary search approach to the multiple minima problem in De Novo Protein Struct. Predict. BMC Struct. Biol. 2013, 13, S4. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 2012, 80, 1715–1735. [Google Scholar] [CrossRef]
- Mack, G.A.; Skillings, J.H. A Friedman-Type Rank Test for Main Effects in a Two-Factor ANOVA. J. Am. Stat. Assoc. 1980, 75, 947–951. [Google Scholar] [CrossRef]
- Hollander, M.; Wolfe, D. Nonparametric Statistical Methods; Wiley Series in Probability and Statistics; Wiley: Hoboken, NJ, USA, 1999; Chapter 7; pp. 270–362. [Google Scholar]
- Kampstra, P. Beanplot: A Boxplot Alternative for Visual Comparison of Distributions. J. Stat. Softw. Code Snippets 2008, 28, 1–9. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Median (ILS) | Median (Bilevel) | ILS:Bilevel Ratio |
|---|---|---|---|
| 1a19A | 0.02445 | 0.00132 | 18.52273 |
| 1bk2 | 0.00188 | 0.00080 | 2.34170 |
| 1bm8 | 0.00056 | 0.00000 | N/A |
| 1bq9A | 0.01620 | 0.00651 | 2.48772 |
| 1c9oA | 0.00005 | 0.00012 | 0.45392 |
| 1ctf | 0.00000 | 0.00007 | 0 |
| 1ew4A | 0.00007 | 0.00000 | N/A |
| 1fna | 0.05202 | 0.00154 | 33.77922 |
| 1gvp | 0.00005 | 0.00000 | N/A |
| 1ig5A | 0.08739 | 0.01719 | 5.08377 |
| 1iibA | 0.01129 | 0.00047 | 24.27957 |
| 1npsA | 0.01531 | 0.00007 | 229.32894 |
| 1pgx | 0.27250 | 0.12170 | 2.23911 |
| 1shfA | 0.01236 | 0.00048 | 26.01558 |
| 1tif | 0.00233 | 0.00007 | 35.28610 |
| 1tig | 0.00012 | 0.00000 | N/A |
| 1urnA | 0.00056 | 0.00006 | 8.60307 |
| 1utg | 0.01643 | 0.00260 | 6.32653 |
| 1vcc | 0.00442 | 0.00066 | 6.71324 |
| 1wit | 0.05264 | 0.00495 | 10.63649 |
| 256bA | 0.38180 | 0.12960 | 2.94599 |
| 2ci2I | 0.00569 | 0.04567 | 0.12463 |
| 4ubpA | 0.00247 | 0.00000 | N/A |
| Archive Size Parameters | |
| desired_size | 10 |
| base_size | 100 |
| max_size | 200 |
| Running Parameters | |
| increase_cycles | 100 |
| Number of runs | 100 |
| Total number of structures (low-resolution) | 1000 |
| (SRCM archiver) | 0.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandathil, S.M.; Garza-Fabre, M.; Handl, J.; Lovell, S.C. Reliable Generation of Native-Like Decoys Limits Predictive Ability in Fragment-Based Protein Structure Prediction. Biomolecules 2019, 9, 612. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100612
Kandathil SM, Garza-Fabre M, Handl J, Lovell SC. Reliable Generation of Native-Like Decoys Limits Predictive Ability in Fragment-Based Protein Structure Prediction. Biomolecules. 2019; 9(10):612. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100612
Chicago/Turabian StyleKandathil, Shaun M., Mario Garza-Fabre, Julia Handl, and Simon C. Lovell. 2019. "Reliable Generation of Native-Like Decoys Limits Predictive Ability in Fragment-Based Protein Structure Prediction" Biomolecules 9, no. 10: 612. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100612