Disclosing the Impact of Carcinogenic SF3b Mutations on Pre-mRNA Recognition Via All-Atom Simulations

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Structural Models
2.2. Molecular Dynamics (MD) Simulations
2.3. Principal Component Analysis
2.4. Cross Correlation Matrix and Correlation Scores
2.5. Electrostatic Calculations
2.6. SF3B1 Sequencing and Variant Annotation
3. Results
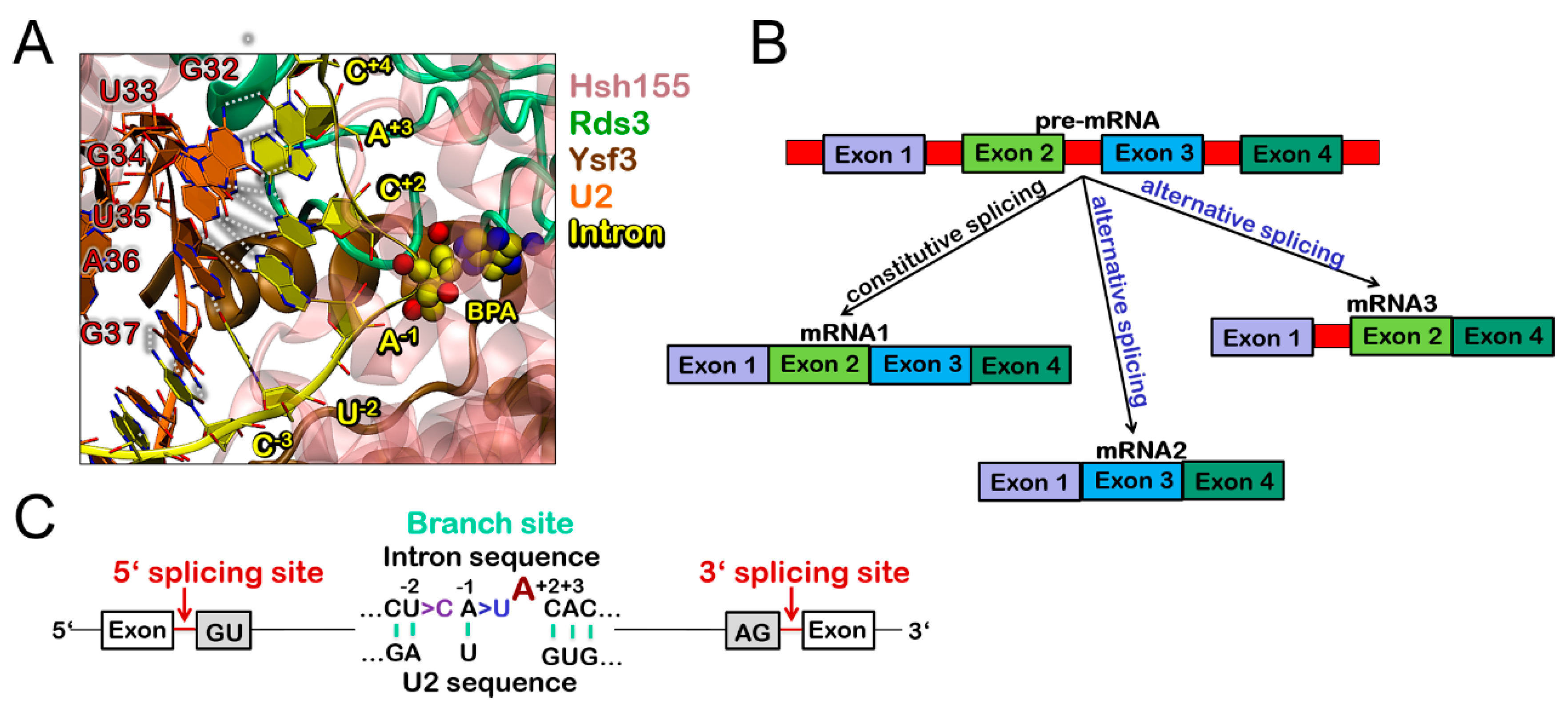
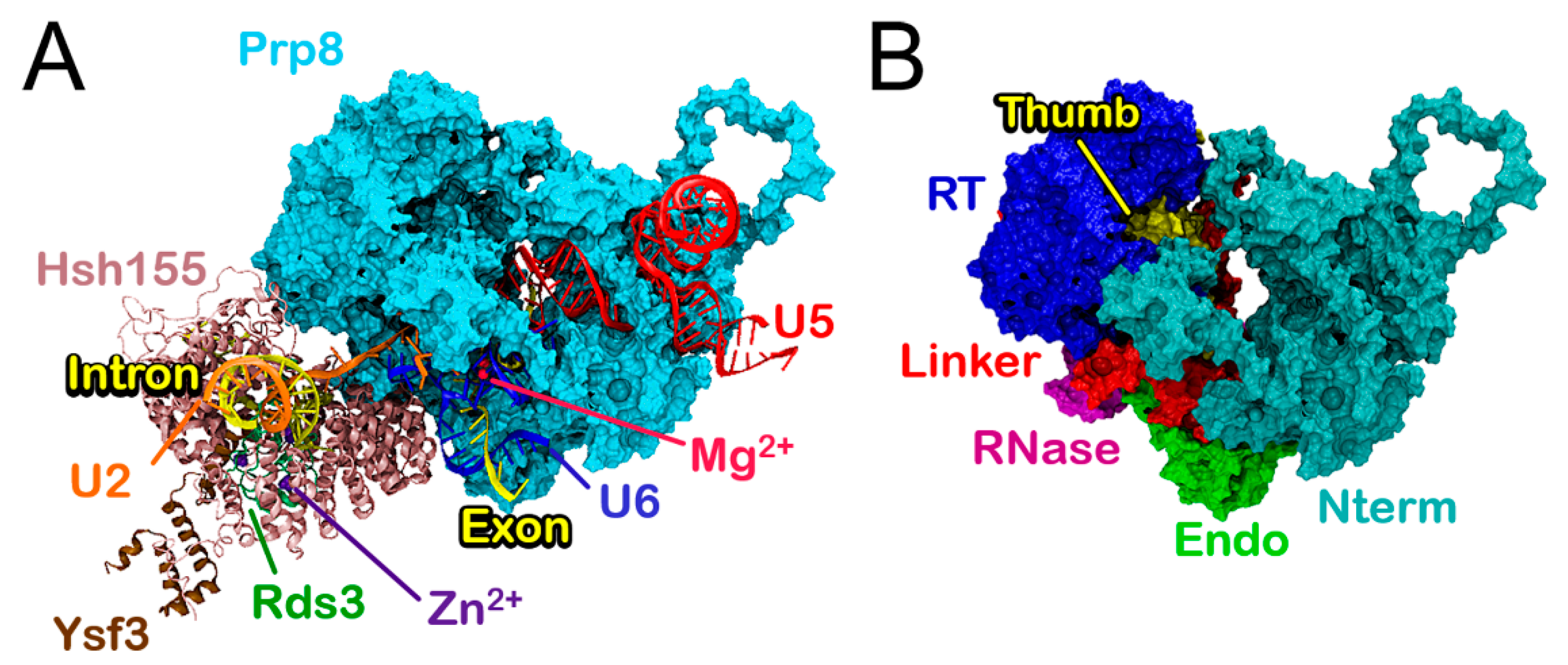
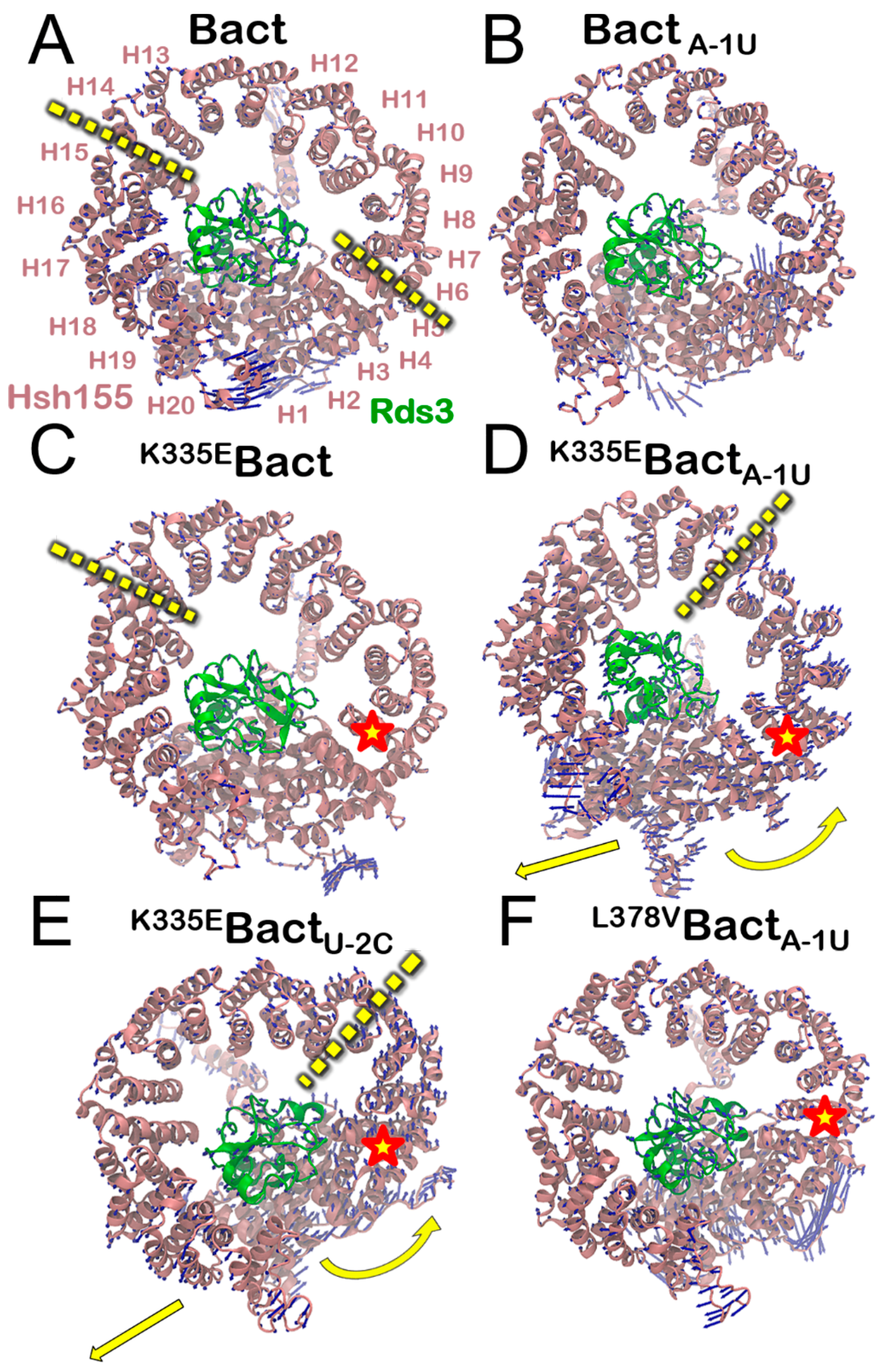
3.1. Molecular Framework of BPS Recognition in the Wild-Type Model
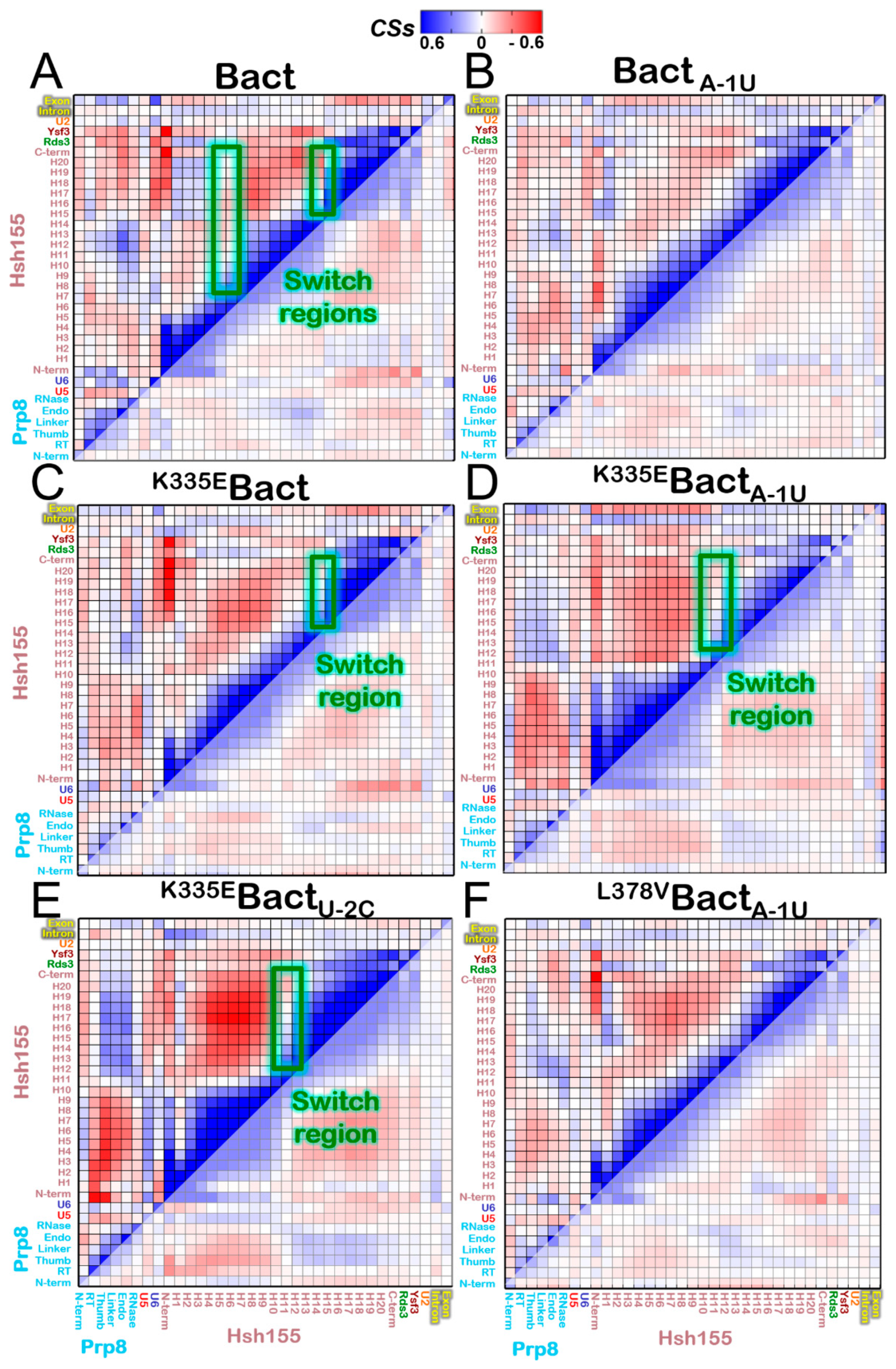
3.2. Functional Dynamics of the Bact Model
3.3. Molecular Framework Underlying Constitutive, Alterative, and Aberrant Splicing
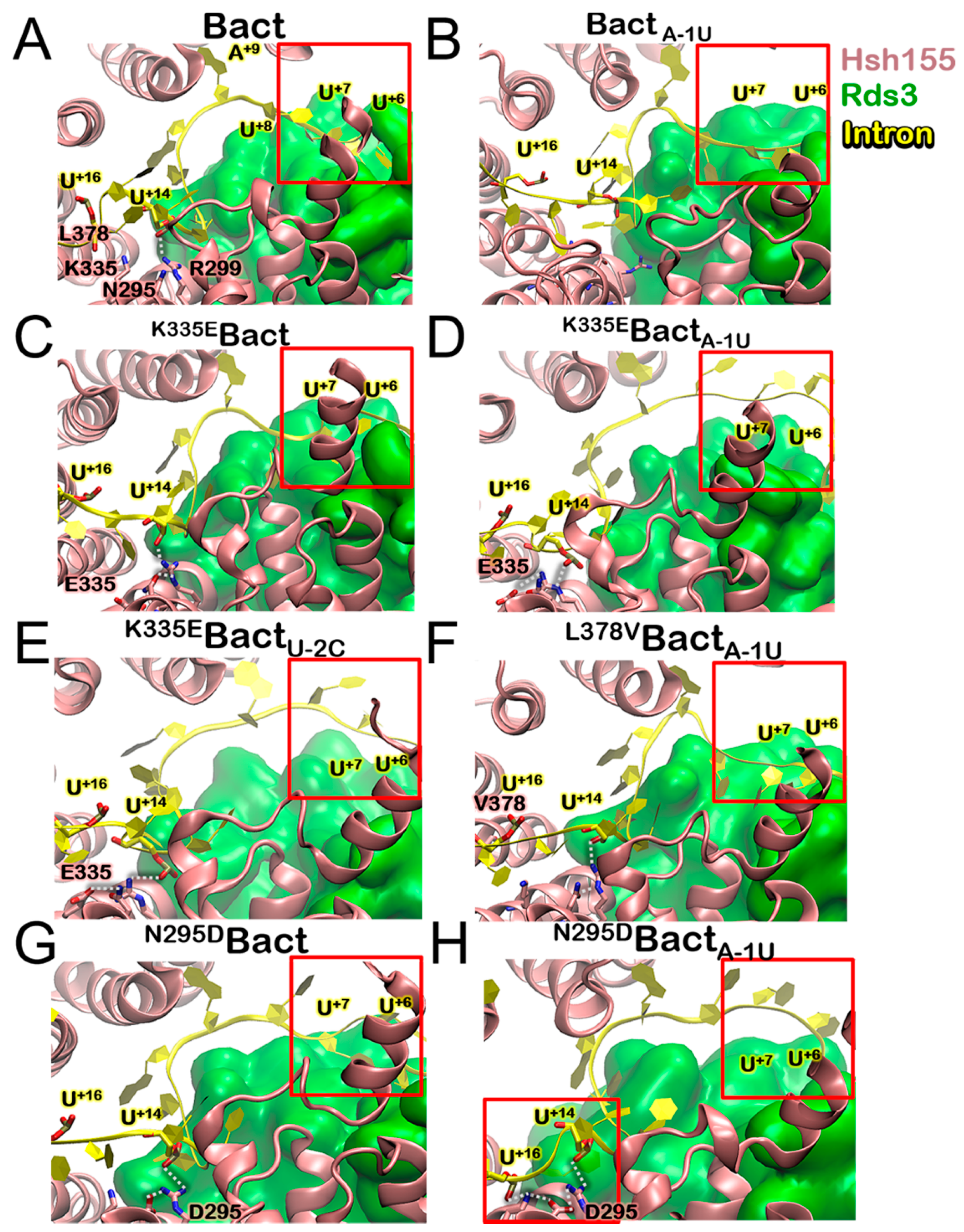
3.4. Impact of the Bact Isoforms on Constitutive/Alternative/Aberrant Splicing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Papasaikas, P.; Valcarcel, J. The Spliceosome: The Ultimate RNA Chaperone and Sculptor. Trends Biochem. Sci. 2016, 41, 386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Yan, C.Y.; Zhan, X.C.; Li, L.J.; Lei, J.L.; Shi, Y.G. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018, 28, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Palermo, G.; Rothlisberger, U.; Magistrato, A. Who Activates the Nucleophile in Ribozyme Catalysis? An Answer from the Splicing Mechanism of Group II Introns. J. Am. Chem. Soc. 2016, 138, 10374–10377. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Palermo, G.; Abdurakhmonova, N.; Rothlisberger, U.; Magistrato, A. Development of Site-Specific Mg2+-RNA Force Field Parameters: A Dream or Reality? Guidelines from Combined Molecular Dynamics and Quantum Mechanics Simulations. J. Chem. Theory. Comput. 2017, 13, 340–352. [Google Scholar] [CrossRef]
- Casalino, L.; Magistrato, A. Structural, dynamical and catalytic interplay between Mg2+ ions and RNA. Vices and virtues of atomistic simulations. Inorg. Chim. Acta 2016, 452, 73–81. [Google Scholar] [CrossRef]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Carrocci, T.J.; Zoerner, D.M.; Paulson, J.C.; Hoskins, A.A. SF3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res. 2017, 45, 4837–4852. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer. 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Lee, S.C.W.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef]
- Buonamici, S.; Yoshimi, A.; Thomas, M.; Seiler, M.; Chan, B.; Caleb, B.; Darman, R.; Fekkes, P.; Karr, C.; Keaney, G.F.; et al. H3B-8800, an Orally Bioavailable Modulator of the SF3b Complex, Shows Efficacy in Spliceosome-Mutant Myeloid Malignancies. Blood 2016, 128, 966. [Google Scholar] [CrossRef]
- Agrawal, A.A.; Yu, L.H.; Smith, P.G.; Buonamici, S. Targeting splicing abnormalities in cancer. Curr. Opin. Genet. Dev. 2018, 48, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.L.; Kielkopf, C.L. Splicing Factor Mutations in Myelodysplasias: Insights from Spliceosome Structures. Trends Genet. 2017, 33, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Hodnefield, J.M.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: Prevalence, clinical correlates, and prognostic relevance. Am. J. Hematol. 2013, 88, 201–206. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.Y.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 3649. [Google Scholar] [CrossRef]
- Yan, C.Y.; Wan, R.X.; Bai, R.; Huang, G.X.Y.; Shi, Y.G. Structure of a yeast activated spliceosome at 3.5 angstrom resolution. Science 2016, 353, 904–911. [Google Scholar] [CrossRef]
- Haselbach, D.; Komarov, I.; Agafonov, D.E.; Hartmuth, K.; Graf, B.; Dybkov, O.; Urlaub, H.; Kastner, B.; Luhrmann, R.; Stark, H. Structure and Conformational Dynamics of the Human Spliceosomal B-act Complex. Cell 2018, 172, 454. [Google Scholar] [CrossRef]
- Finci, L.I.; Zhang, X.F.; Huang, X.L.; Zhou, Q.; Tsai, J.; Teng, T.; Agrawal, A.; Chan, B.; Irwin, S.; Karr, C.; et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Cretu, C.; Schmitzova, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Luhrmann, R.; Pena, V. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cretu, C.; Agrawal, A.A.; Cook, A.; Will, C.L.; Fekkes, P.; Smith, P.G.; Luhrmann, R.; Larsen, N.; Buonamici, S.; Pena, V. Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol. Cell 2018, 70, 265. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Palermo, G.; Spinello, A.; Rothlisberger, U.; Magistrato, A. All-atom simulations disentangle the functional dynamics underlying gene maturation in the intron lariat spliceosome. Proc. Natl. Acad. Sci. USA 2018, 115, 6584–6589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokorna, P.; Kruse, H.; Krepl, M.; Sponer, J. QM/MM Calculations on Protein-RNA Complexes: Understanding Limitations of Classical MD Simulations and Search for Reliable Cost-Effective QM Methods. J. Chem. Theory Comput. 2018, 14, 5419–5433. [Google Scholar] [CrossRef] [PubMed]
- Krepl, M.; Havrila, M.; Stadlbauer, P.; Banas, P.; Otyepka, M.; Pasulka, J.; Stefl, R.; Sponer, J. Can We Execute Stable Microsecond-Scale Atomistic Simulations of Protein-RNA Complexes? J. Chem. Theory Comput. 2015, 11, 1220–1243. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative Protein Modeling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E. Computer program AMBER 2018; University of California, San Francisco: San Francisco, CA, USA, 2018. [Google Scholar]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Perez, A.; Marchan, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinenement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef]
- Sponer, J.; Krepl, M.; Banas, P.; Kuhrova, P.; Zgarbova, M.; Jurecka, P.; Havrila, M.; Otyepka, M. How to understand atomistic molecular dynamics simulations of RNA and protein-RNA complexes? WIREs RNA 2017, 8, e1405. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.G.; Chen, J.S.; Miao, Y.L.; Jinek, M.; Doudna, J.A.; McCammon, J.A.; Palermo, G. Deciphering Off-Target Effects in CRISPR-Cas9 through Accelerated Molecular Dynamics. Acs Cent. Sci. 2019, 5, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, G.; Ricci, C.G.; Fernando, A.; Basak, R.; Jinek, M.; Rivalta, I.; Batista, V.S.; McCammon, J.A. Protospacer Adjacent Motif-Induced Allostery Activates CRISPR-Cas9. J. Am. Chem. Soc. 2017, 139, 16028–16031. [Google Scholar] [CrossRef] [Green Version]
- Krepl, M.; Clery, A.; Blatter, M.; Allain, F.H.T.; Sponer, J. Synergy between NMR measurements and MD simulations of protein/RNA complexes: Application to the RRMs, the most common RNA recognition motifs. Nucleic Acids Res. 2016, 44, 6452–6470. [Google Scholar] [CrossRef]
- Aqvist, J. Ion Water Interaction Potentials Derived from Free-Energy Perturbation Simulations. J. Phys. Chem. 1990, 94, 8021–8024. [Google Scholar] [CrossRef]
- Sgrignani, J.; Magistrato, A. The Structural Role of Mg2+ Ions in a Class I RNA Polymerase Ribozyme: A Molecular Simulation Study. J. Phys. Chem. B 2012, 116, 2259–2268. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008, 112, 9020–9041. [Google Scholar] [CrossRef]
- Pang, Y.P. Novel zinc protein molecular dynamics simulations: Steps toward antiangiogenesis for cancer treatment. J. Mol. Model. 1999, 5, 196–202. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Krepl, M.; Blatter, M.; Clery, A.; Damberger, F.F.; Allain, F.H.T.; Sponer, J. Structural study of the Fox-1 RRM protein hydration reveals a role for key water molecules in RRM-RNA recognition. Nucleic Acids Res. 2017, 45, 8046–8063. [Google Scholar] [CrossRef] [Green Version]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Crystal-Structure and Pair Potentials—A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Bochicchio, A.; Krepl, M.; Yang, F.; Varani, G.; Sponer, J.; Carloni, P. Molecular basis for the increased affinity of an RNA recognition motif with re-engineered specificity: A molecular dynamics and enhanced sampling simulations study. PLoS Comp. Biol. 2018, 14, e1006642. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential Dynamics of Proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [Green Version]
- Palermo, G.; Miao, Y.L.; Walker, R.C.; Jinek, M.; McCammon, J.A. Striking Plasticity of CRISPR-Cas9 and Key Role of Non-target DNA, as Revealed by Molecular Simulations. ACS Cent. Sci. 2016, 2, 756–763. [Google Scholar] [CrossRef]
- Pavlin, M.; Spinello, A.; Pennati, M.; Zaffaroni, N.; Gobbi, S.; Bisi, A.; Colombo, G.; Magistrato, A. A Computational Assay of Estrogen Receptor alpha Antagonists Reveals the Key Common Structural Traits of Drugs Effectively Fighting Refractory Breast Cancers. Sci. Rep. 2018, 8, 649. [Google Scholar] [CrossRef]
- Borišek, J.; Saltalamacchia, A.; Spinello, A.; Magistrato, A. Exploiting Cryo-EM Structural Information and All-Atom Simulations to Decrypt the Molecular Mechanism of Splicing Modulators. J. Chem. Inf. Model. 2019. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Fogolari, F.; Brigo, A.; Molinari, H. The Poisson-Boltzmann equation for biomolecular electrostatics: A tool for structural biology. J. Mol. Recognit. 2002, 15, 377–392. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Palermo, G.; Chen, J.S.; Ricci, C.G.; Rivalta, I.; Jinek, M.; Batista, V.S.; Doudna, J.A.; McCammon, J.A. Key role of the REC lobe during CRISPR-Cas9 activation by ‘sensing’, ‘regulating’, and ‘locking’ the catalytic HNH domain. Q. Rev. Biophys. 2018, 51, e91. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Casalino, L.; Magistrato, A.; Andrew McCammon, J. Understanding the mechanistic basis of non-coding RNA through molecular dynamics simulations. J. Struct. Biol. 2019, 206, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Zachariae, U.; Grubmuller, H. Importin-beta: Structural and dynamic determinants of a molecular spring. Structure 2008, 16, 906–915. [Google Scholar] [CrossRef]
- Yan, C.Y.; Hang, J.; Wan, R.X.; Huang, M.; Wong, C.C.L.; Shi, Y.G. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 2015, 349, 1182–1191. [Google Scholar] [CrossRef]
- Carrocci, T.J.; Paulson, J.C.; Hoskins, A.A. Functional analysis of Hsh155/SF3b1 interactions with the U2 snRNA/branch site duplex. RNA 2018, 24, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Associated | Prediction | Type | Annotation | Frequency |
|---|---|---|---|---|---|
| Q698E (H347) | K666E | 4/6 damaging | Somatic | uncertain | <1% |
| Q670H (Q339) | 9/9 damaging | Germline | uncertain | 1% | |
| I709V (L378) | 4/9 damaging | Germline | benign | <1% | |
| T434P (V103) | 8/9 damaging | Germline | uncertain | <1% | |
| I360V (L29) | 2/9 damaging | Germline | benign | <1% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borišek, J.; Saltalamacchia, A.; Gallì, A.; Palermo, G.; Molteni, E.; Malcovati, L.; Magistrato, A. Disclosing the Impact of Carcinogenic SF3b Mutations on Pre-mRNA Recognition Via All-Atom Simulations. Biomolecules 2019, 9, 633. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100633
Borišek J, Saltalamacchia A, Gallì A, Palermo G, Molteni E, Malcovati L, Magistrato A. Disclosing the Impact of Carcinogenic SF3b Mutations on Pre-mRNA Recognition Via All-Atom Simulations. Biomolecules. 2019; 9(10):633. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100633
Chicago/Turabian StyleBorišek, Jure, Andrea Saltalamacchia, Anna Gallì, Giulia Palermo, Elisabetta Molteni, Luca Malcovati, and Alessandra Magistrato. 2019. "Disclosing the Impact of Carcinogenic SF3b Mutations on Pre-mRNA Recognition Via All-Atom Simulations" Biomolecules 9, no. 10: 633. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100633