The Loop of the TPR1 Subdomain of Phi29 DNA Polymerase Plays a Pivotal Role in Primer-Terminus Stabilization at the Polymerization Active Site


Abstract
:1. Introduction
2. Materials and Methods
2.1. Nucleotides and DNAs
2.2. Site-Directed Mutagenesis of Phi29 DNA Polymerase and Terminal Protein
2.3. Polymerase/3’–5’ Exonuclease (Pol/Exo) Coupled Assay
2.4. Hydrolysis of p-Nitrophenol-Thymidine Monophosphate
2.5. 3’-5’ Exonuclease Assay
2.6. DNA Gel Retardation Assay
2.7. Replication of Primed M13 DNA
2.8. Processivity Assay
2.9. Polymerization Activity under Single Binding Conditions
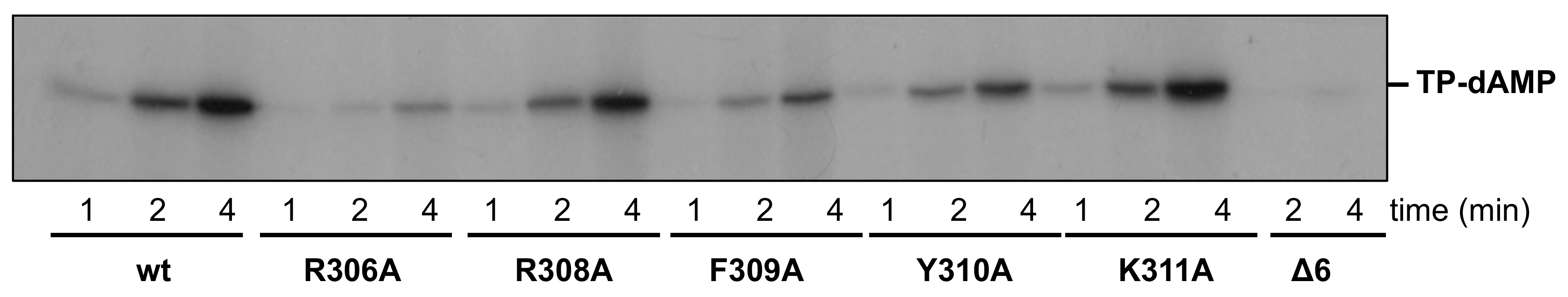
2.10. Protein-Primed Initiation Assay (Terminal Protein-Deoxyadenosine Monophosphate Formation)
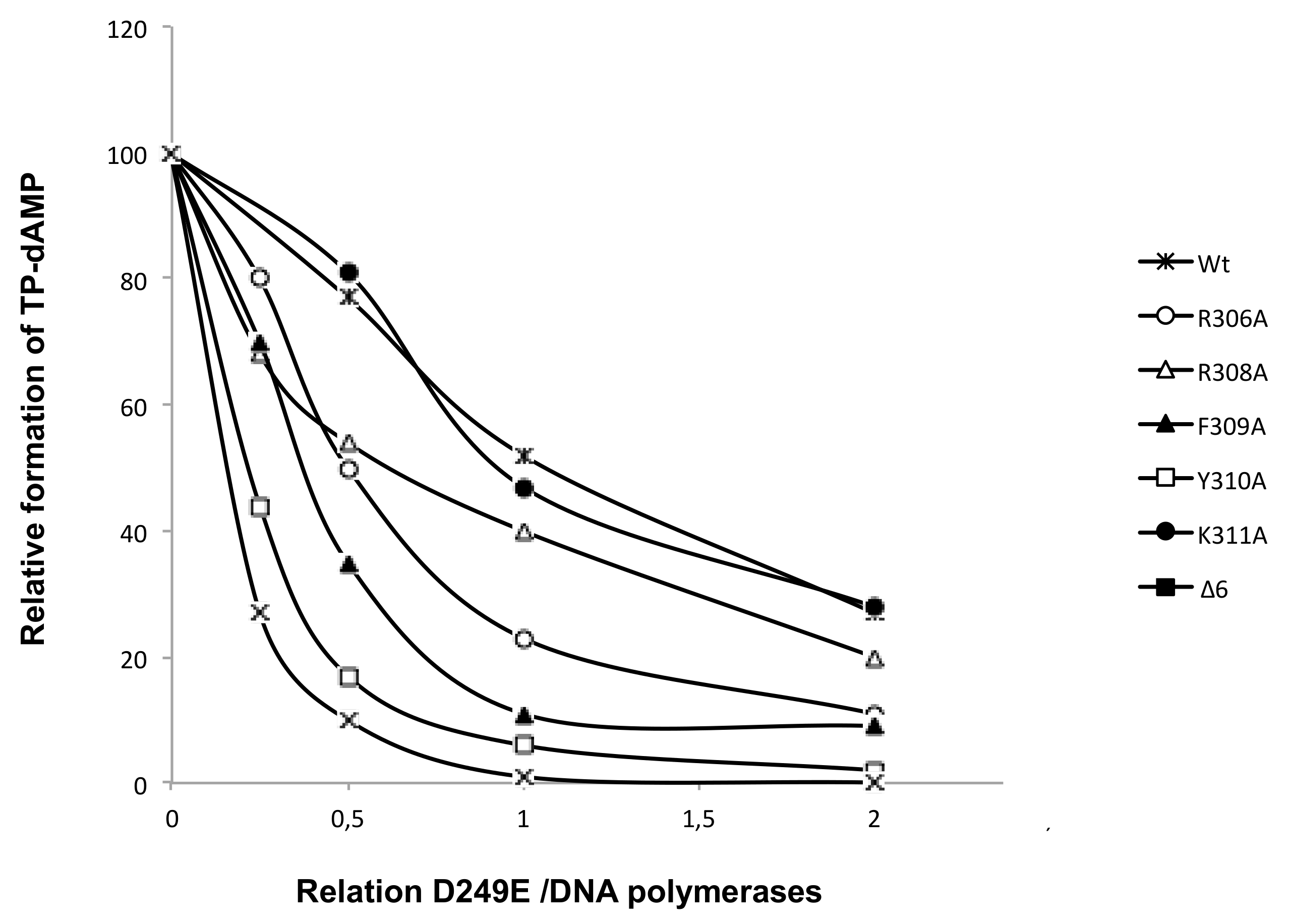
2.11. Interference Assay for Terminal Protein Binding
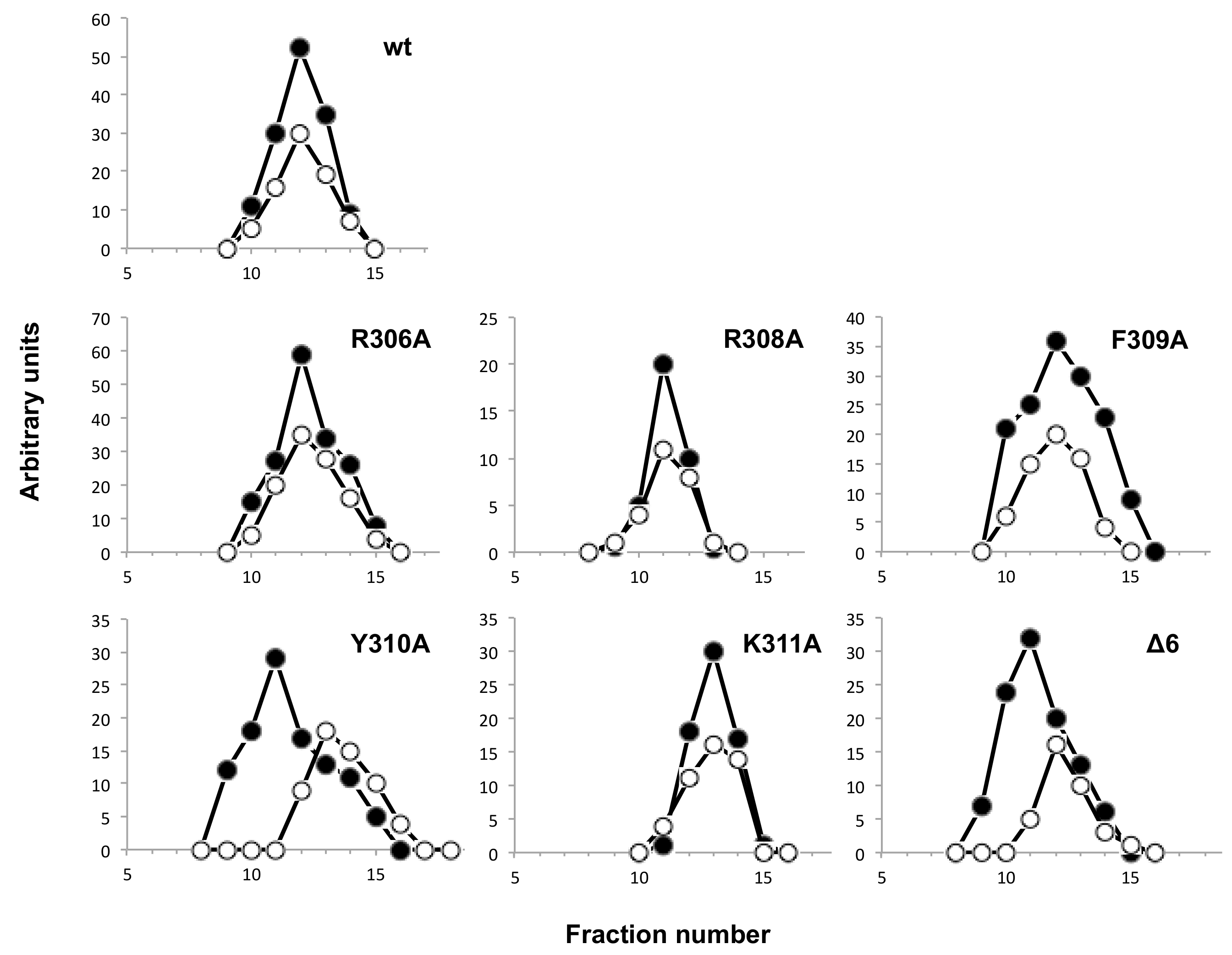
2.12. Analysis of the Interaction between Terminal Protein and DNA Polymerase Mutants by Glycerol-Gradient Ultracentrifugation
2.13. Terminal Protein-DNA Replication Assay
3. Results
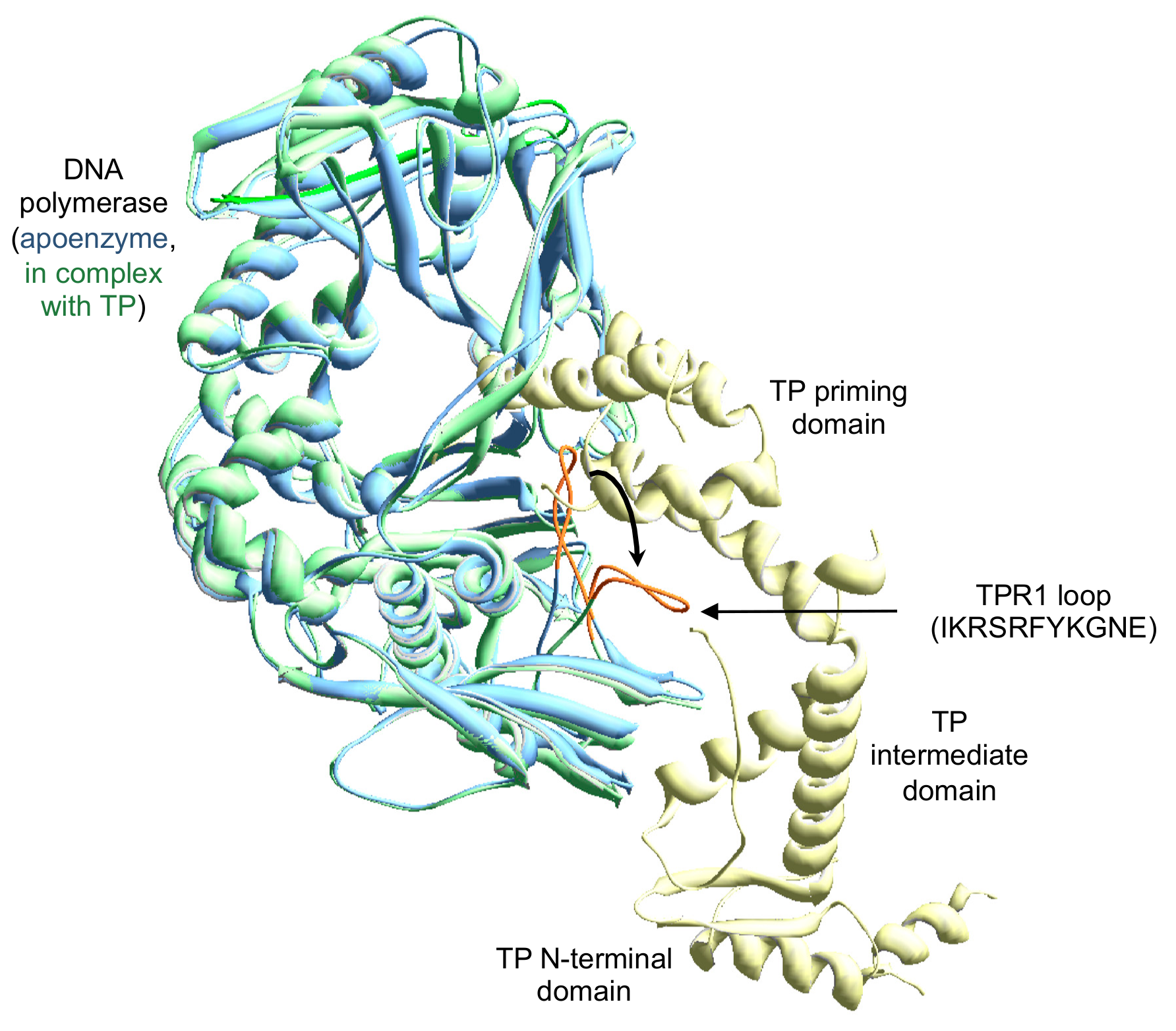
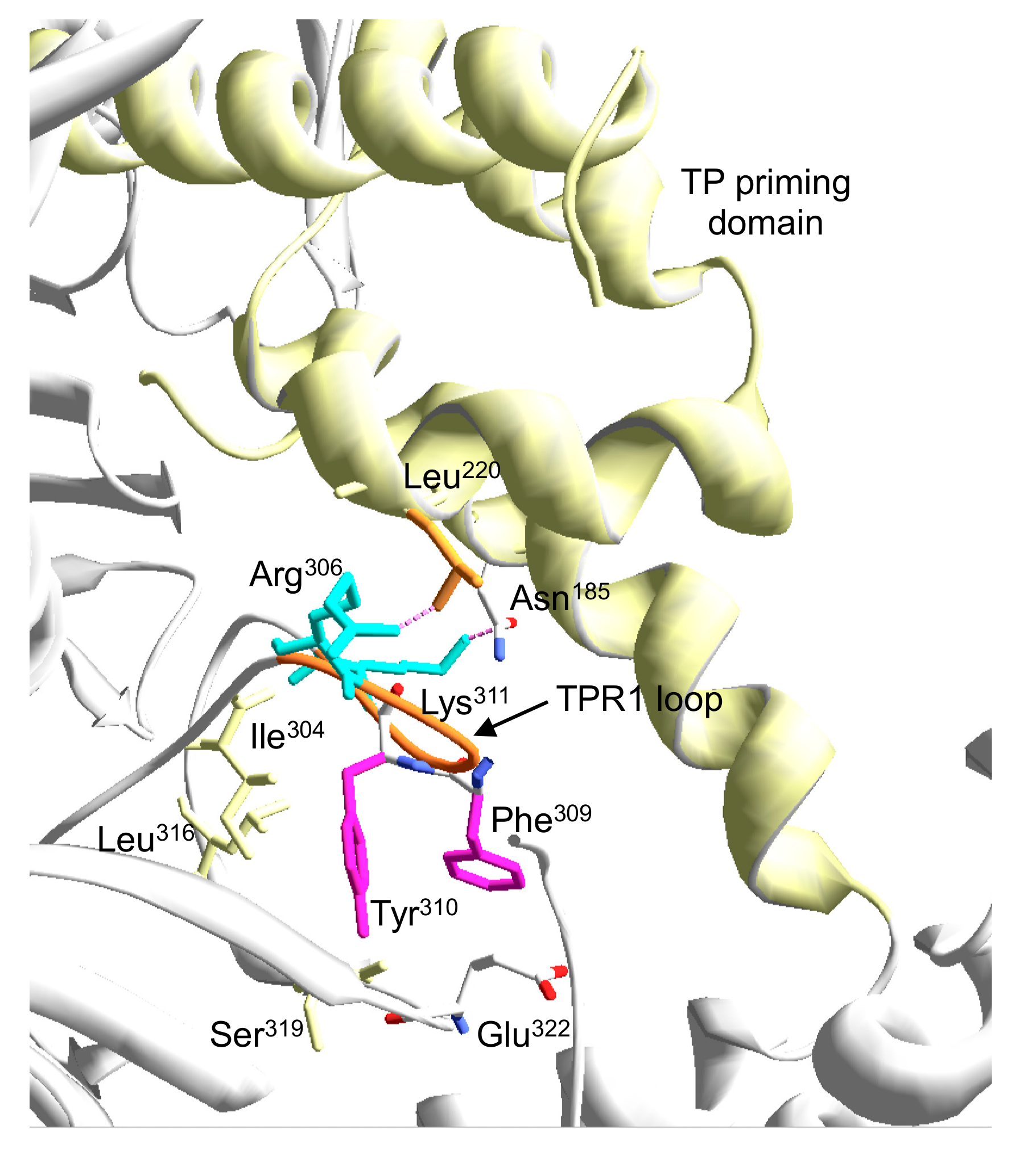
3.1. Site-Directed Mutagenesis at Terminal Protein Region 1 Phi29 DNA Polymerase Residues
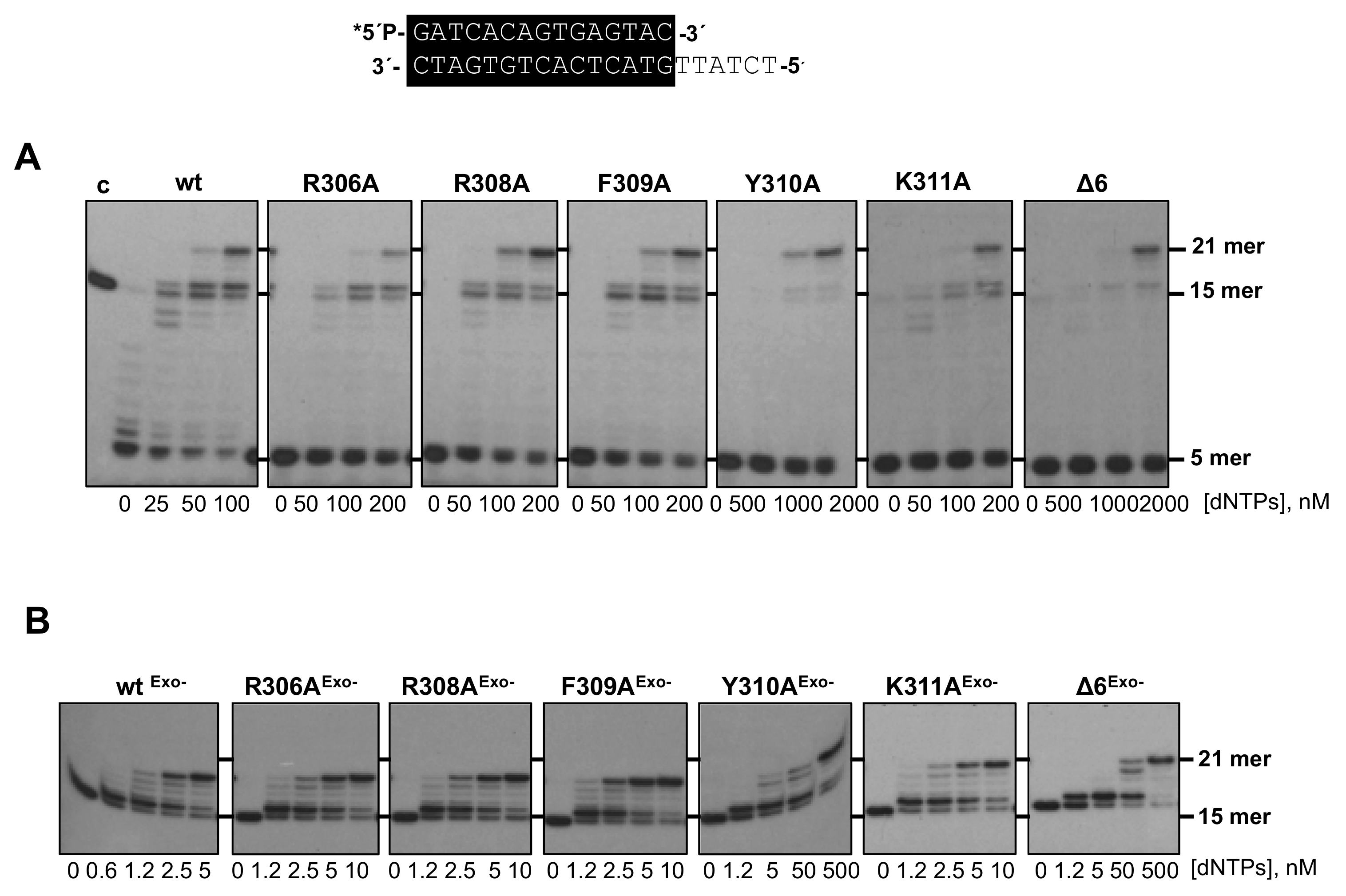
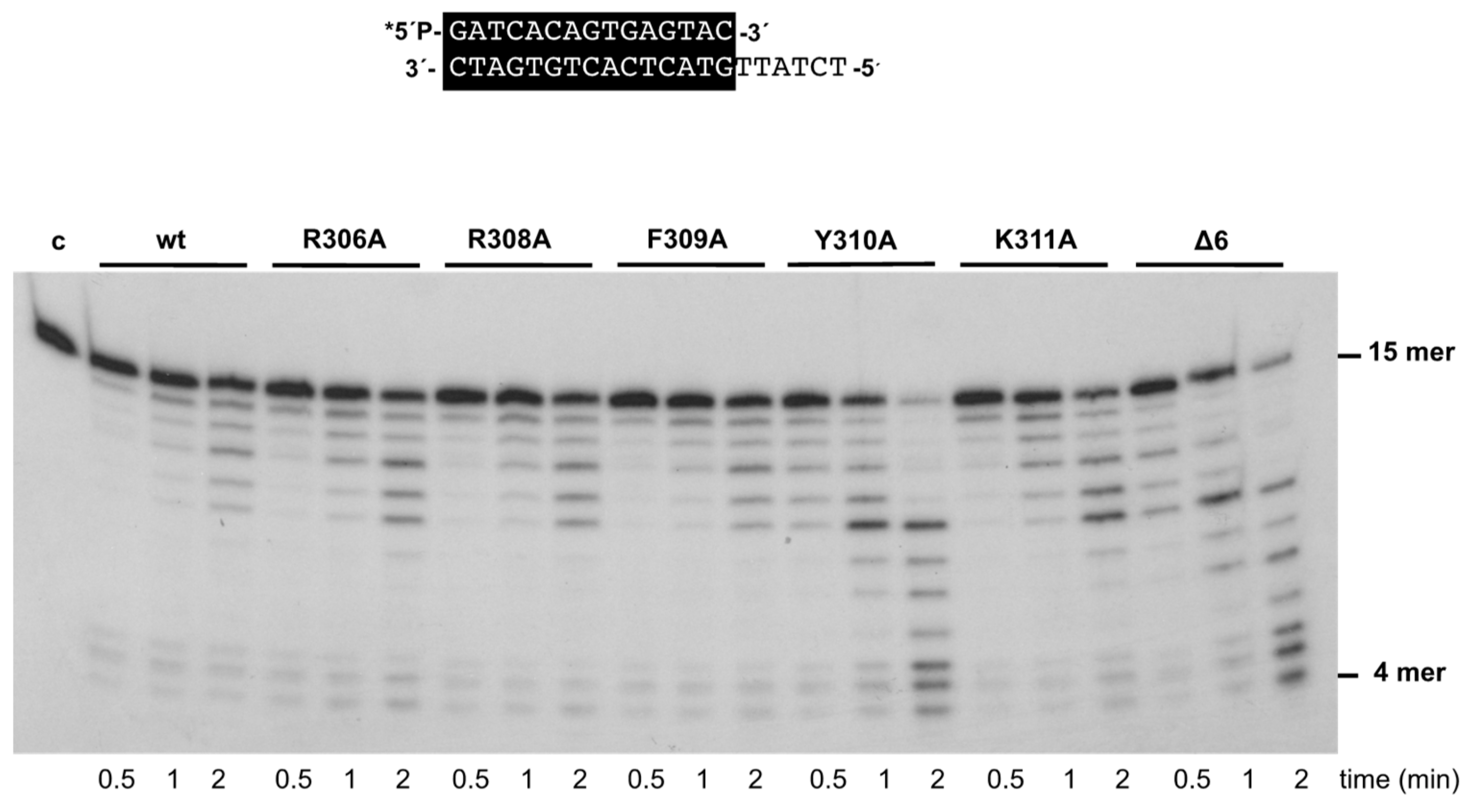
3.2. Changes at Phi29 DNA Polymerase TPR1 Loop Residues Impair the Stabilization of the DNA Primer Terminus at the Polymerization Active Site
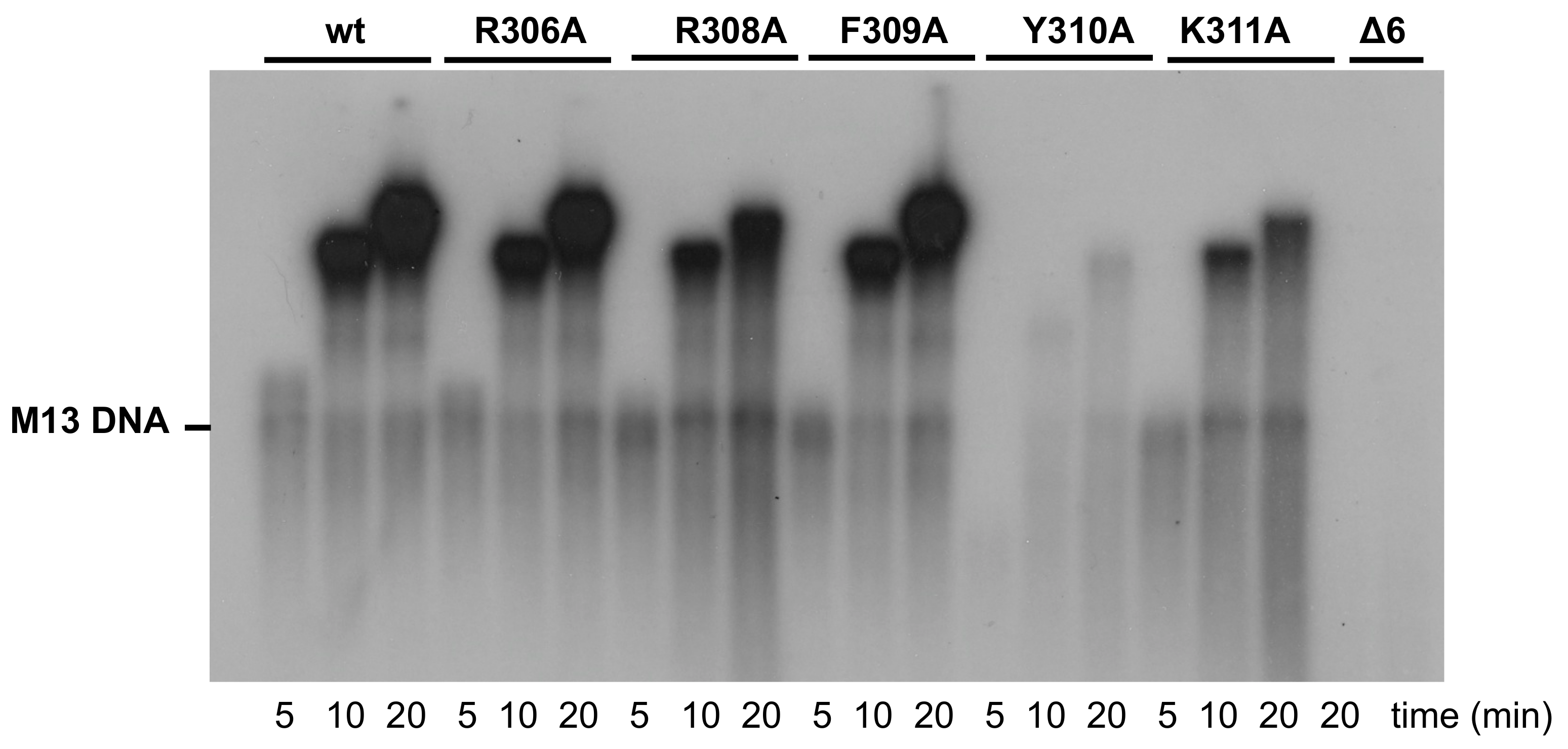
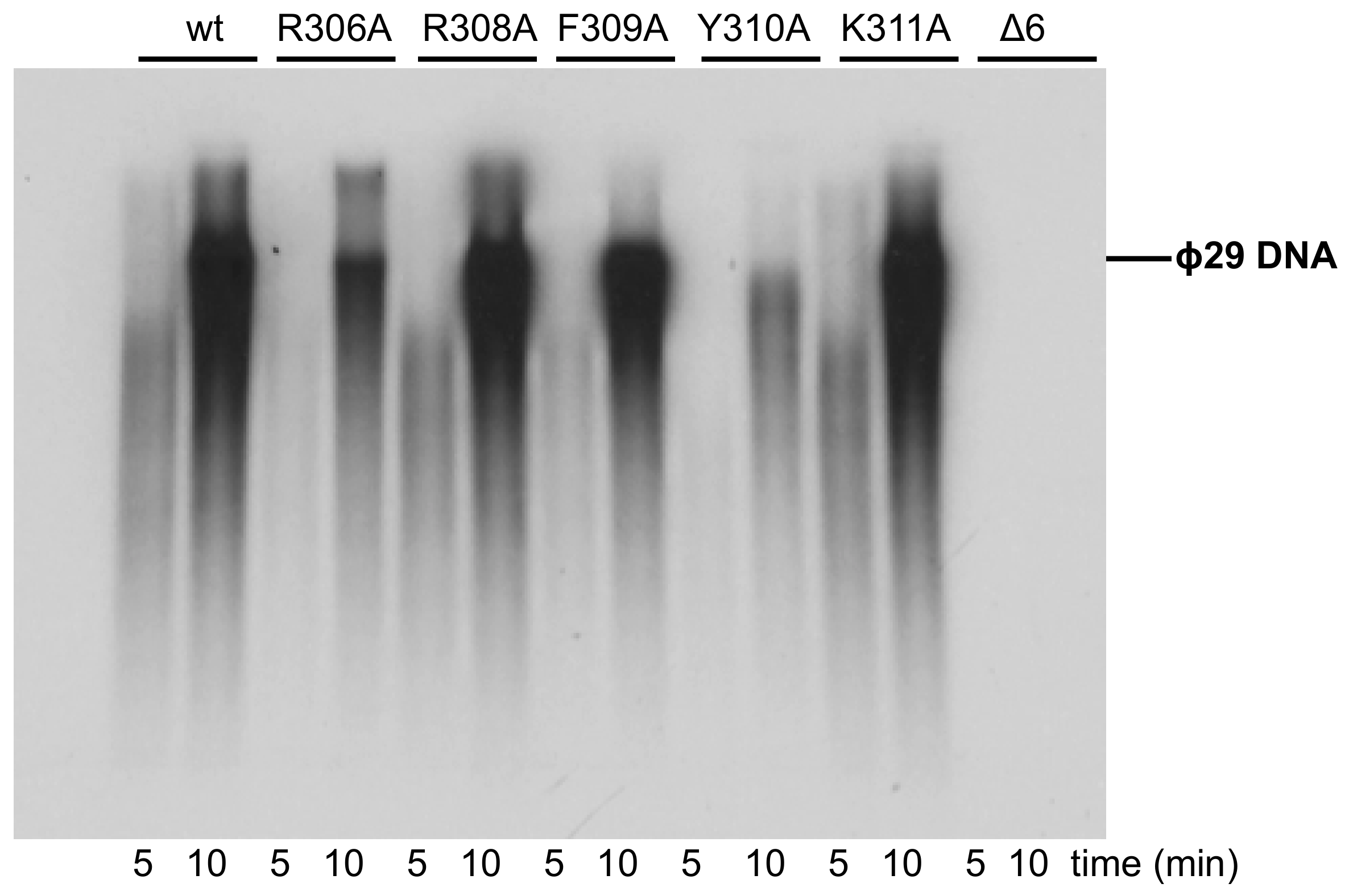
3.3. Processive DNA Synthesis Coupled to Strand Displacement by Phi29 DNA Polymerase Variants
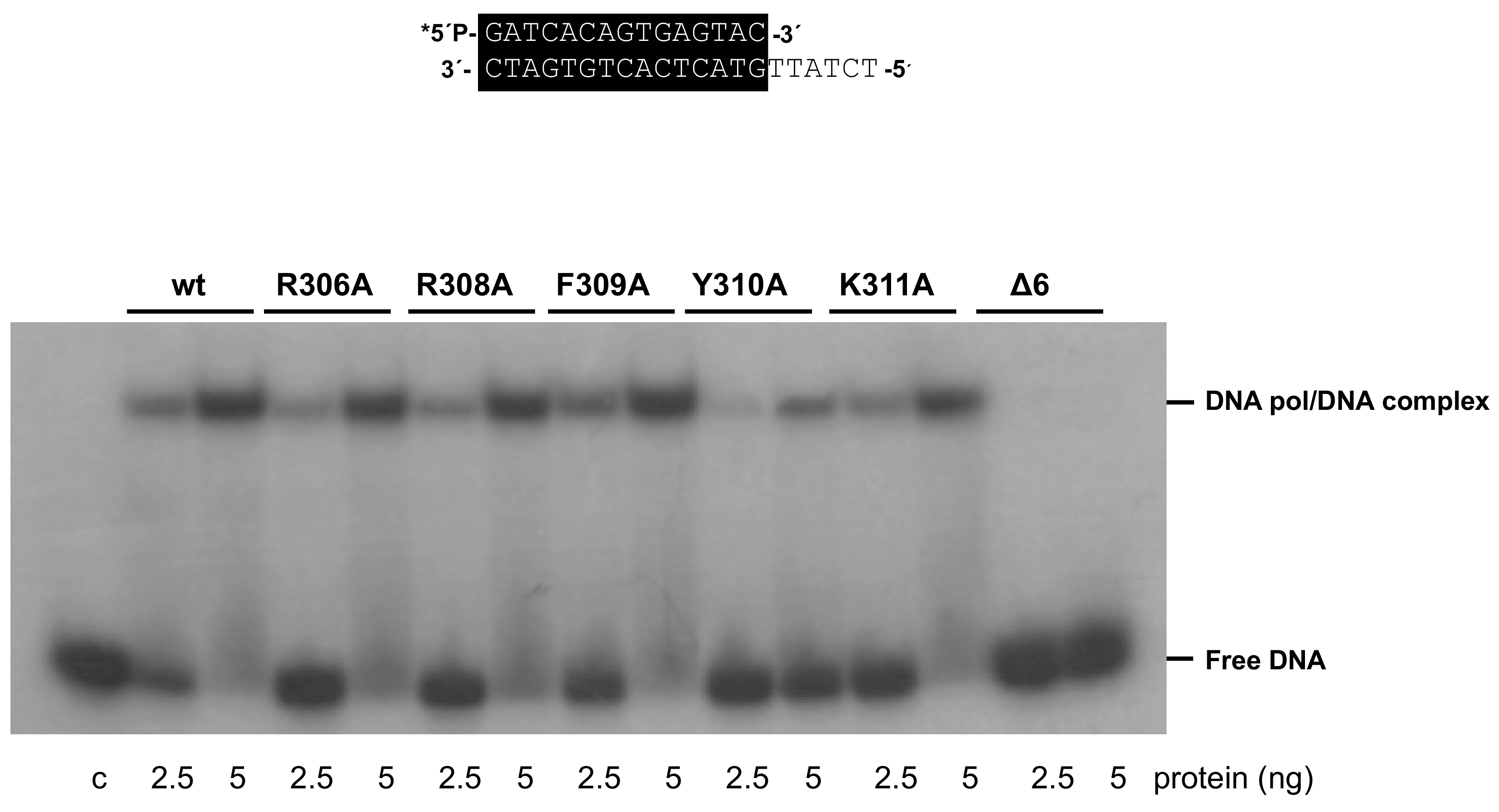
3.4. Mutations at Phi29 DNA Polymerase Terminal Protein Region 1 Loop Affect the Interaction with the Terminal Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kornberg, A.; Baker, T.A. DNA Replication, 2nd ed.; W.H. Freeman and Company: New York, NY, USA, 1992. [Google Scholar]
- Watson, J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef]
- Salas, M. Protein-priming of DNA replication. Annu. Rev. Biochem. 1991, 60, 39–71. [Google Scholar] [CrossRef]
- Salas, M. Mechanisms of initiation of linear DNA replication in prokaryotes. Genet. Eng. 1999, 21, 159–171. [Google Scholar]
- Salas, M.; Miller, J.; Leis, J.; DePamphilis, M. Mechanisms for Priming DNA Synthesis; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1996; pp. 131–176. [Google Scholar]
- Gutiérrez, J.; Vinós, J.; Prieto, I.; Méndez, E.; Hermoso, J.M.; Salas, M. Signals in the ϕ29 DNA-terminal protein template for the initiation of phage φ 29 DNA replication. Virology 1986, 155, 474–483. [Google Scholar]
- Méndez, J.; Blanco, L.; Esteban, J.A.; Bernad, A.; Salas, M. Initiation of ϕ29 DNA replication occurs at the second 3′ nucleotide of the linear template: a sliding-back mechanism for protein-primed DNA replication. Proc. Natl. Acad. Sci. USA 1992, 89, 9579–9583. [Google Scholar] [CrossRef] [PubMed]
- Méndez, J.; Blanco, L.; Salas, M. Protein-primed DNA replication: a transition between two modes of priming by a unique DNA polymerase. EMBO J. 1997, 16, 2519–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, L.; Bernad, A.; Lázaro, J.M.; Martín, G.; Garmendia, C.; Salas, M. Highly efficient DNA synthesis by the phage ϕ29 DNA polymerase. Symmetrical mode of DNA replication. J. Biol. Chem. 1989, 264, 8935–8940. [Google Scholar]
- Kamtekar, S.; Berman, A.J.; Wang, J.; Lázaro, J.M.; de Vega, M.; Blanco, L.; Salas, M.; Steitz, T.A. Insights into strand displacement and processivity from the crystal structure of the protein-primed DNA polymerase of bacteriophage φ29. Mol. Cell 2004, 16, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Dufour, E.; Méndez, J.; Lázaro, J.M.; de Vega, M.; Blanco, L.; Salas, M. An aspartic acid residue in TPR-1, a specific region of protein-priming DNA polymerases, is required for the functional interaction with primer terminal protein. J. Mol. Biol. 2000, 304, 289–300. [Google Scholar] [CrossRef]
- Pérez-Arnaiz, P.; Longás, E.; Villar, L.; Lázaro, J.M.; Salas, M.; de Vega, M. Involvement of phage φ29 DNA polymerase and terminal protein subdomains in conferring specificity during initiation of protein-primed DNA replication. Nucleic Acids Res. 2007, 35, 7061–7073. [Google Scholar] [CrossRef] [PubMed]
- Dufour, E.; Rodríguez, I.; Lázaro, J.M.; de Vega, M.; Salas, M. A conserved insertion in protein-primed DNA polymerases is involved in primer terminus stabilisation. J. Mol. Biol. 2003, 331, 781–794. [Google Scholar] [CrossRef]
- Rodríguez, I.; Lázaro, J.M.; Blanco, L.; Kamtekar, S.; Berman, A.J.; Wang, J.; Steitz, T.A.; Salas, M.; de Vega, M. A specific subdomain in ϕ29 DNA polymerase confers both processivity and strand-displacement capacity. Proc. Natl. Acad. Sci. USA 2005, 102, 6407–6412. [Google Scholar] [CrossRef] [PubMed]
- Redrejo-Rodríguez, M.; Muñoz-Espín, D.; Holguera, I.; Mencía, M.; Salas, M. Nuclear and nucleoid localization are independently conserved functions in bacteriophage terminal proteins. Mol. Microbiol. 2013, 90, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Kamtekar, S.; Berman, A.J.; Wang, J.; Lázaro, J.M.; de Vega, M.; Blanco, L.; Salas, M.; Steitz, T.A. The phi29 DNA polymerase:protein-primer structure suggests a model for the initiation to elongation transition. EMBO J. 2006, 25, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Del Prado, A.; Lázaro, J.M.; Longas, E.; Villar, L.; de Vega, M.; Salas, M. Insights into the Determination of the Templating Nucleotide at the Initiation of ϕ29 DNA Replication. J. Biol. Chem. 2015, 290, 27138–27145. [Google Scholar] [CrossRef] [PubMed]
- Bernad, A.; Blanco, L.; Lázaro, J.M.; Martín, G.; Salas, M. A conserved 3′-5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell 1989, 59, 219–228. [Google Scholar] [CrossRef]
- Lázaro, J.M.; Blanco, L.; Salas, M. Purification of bacteriophage ϕ29 DNA polymerase. Methods Enzymol. 1995, 262, 42–49. [Google Scholar]
- Mencía, M.; Gella, P.; Camacho, A.; de Vega, M.; Salas, M. Terminal protein-primed amplification of heterologous DNA with a minimal replication system based on phage ϕ29. Proc. Natl. Acad. Sci. USA 2011, 108, 18655–18660. [Google Scholar] [CrossRef]
- Kumar, J.K.; Chiu, E.T.; Tabor, S.; Richardson, C.C. A unique region in bacteriophage T7 DNA polymerase important for exonucleolytic hydrolysis of DNA. J. Biol. Chem. 2004, 279, 42018–42025. [Google Scholar] [CrossRef]
- Carthew, R.W.; Chodosh, L.A.; Sharp, P.A. An RNA polymerase II transcription factor binds to an upstream element in the adenovirus major late promoter. Cell 1985, 43, 439–448. [Google Scholar] [CrossRef]
- McDonell, M.W.; Simon, M.N.; Studier, F.W. Analysis of restriction fragments of T7 DNA and determination of molecular weights by electrophoresis in neutral and alkaline gels. J. Mol. Biol. 1977, 110, 119–146. [Google Scholar] [CrossRef]
- de Vega, M.; Blanco, L.; Salas, M. ϕ29 DNA polymerase residue Ser122, a single-stranded DNA ligand for 3′-5′ exonucleolysis, is required to interact with the terminal protein. J. Biol. Chem. 1998, 273, 28966–28977. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A.; Lázaro, J.M.; Blanco, L.; Salas, M. ϕ29 DNA polymerase active site. Residue Asp249 of conserved amino acid motif "Dx2SLYP" is critical for synthetic activities. J. Biol. Chem. 1993, 268, 24106–24113. [Google Scholar] [PubMed]
- Blanco, L.; Salas, M. Mutational analysis of bacteriophage ϕ29 DNA polymerase. Methods Enzymol. 1995, 262, 283–294. [Google Scholar] [PubMed]
- Garmendia, C.; Bernad, A.; Esteban, J.A.; Blanco, L.; Salas, M. The bacteriophage ϕ29 DNA polymerase, a proofreading enzyme. J. Biol. Chem. 1992, 267, 2594–2599. [Google Scholar] [PubMed]
- de Vega, M.; Lázaro, J.M.; Salas, M.; Blanco, L. Primer-terminus stabilization at the 3′-5′ exonuclease active site of ϕ29 DNA polymerase. Involvement of two amino acid residues highly conserved in proofreading DNA polymerases. EMBO J. 1996, 15, 1182–1192. [Google Scholar] [CrossRef]
- Truniger, V.; Lázaro, J.M.; Salas, M.; Blanco, L. A DNA binding motif coordinating synthesis and degradation in proofreading DNA polymerases. EMBO J. 1996, 15, 3430–3441. [Google Scholar] [CrossRef]
- Méndez, J.; Blanco, L.; Lázaro, J.M.; Salas, M. Primer-terminus stabilization at the ϕ29 DNA polymerase active site. Mutational analysis of conserved motif Tx2GR. J. Biol. Chem. 1994, 269, 30030–30038. [Google Scholar]
- Blanco, L.; Salas, M. Replication of phage ϕ29 DNA with purified terminal protein and DNA polymerase: synthesis of full-length ϕ29 DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 6404–6408. [Google Scholar] [CrossRef]
- del Prado, A.; Villar, L.; de Vega, M.; Salas, M. Involvement of residues of the ϕ29 terminal protein intermediate and priming domains in the formation of a stable and functional heterodimer with the replicative DNA polymerase. Nucleic Acids Res. 2012, 40, 3886–3897. [Google Scholar] [CrossRef]
- Blasco, M.A.; Méndez, J.; Lázaro, J.M.; Blanco, L.; Salas, M. Primer terminus stabilization at the φ29 DNA polymerase active site. Mutational analysis of conserved motif KxY. J. Biol. Chem. 1995, 270, 2735–2740. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phi29 DNA Polymerase Mutants | ||||||||
|---|---|---|---|---|---|---|---|---|
| Assay | Substrate | Wild-Type | R306A | R308A | F309A | Y310A | K311A | Δ6 |
| Exonuclease | dsDNA | 100 | 106 ± 11 | 99 ± 10 | 90 ± 6 | 164 ± 11 | 108 ± 7 | 131 ± 9 |
| Retarding | dsDNA | 100 | 82 ± 23 | 113 ± 49 | 89 ± 34 | 29 ± 6 | 92 ± 41 | 7 ± 4 |
| Replication | M13-DNA | 100 | 92 ± 4 | 39 ± 3 | 87 ± 6 | 3 ± 1 | 18 ± 1 | n.d. |
| Initiation | TP-DNA | 100 | 8 ± 1 | 49 ± 9 | 25 ± 2 | 30 ± 2 | 100 ± 19 | 3 ± 1 |
| Replication | TP-DNA | 100 | 20 ± 6 | 57 ± 16 | 34 ± 12 | 11 ± 3 | 73 ± 23 | n.d. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Prado, A.; Santos, E.; Lázaro, J.M.; Salas, M.; de Vega, M. The Loop of the TPR1 Subdomain of Phi29 DNA Polymerase Plays a Pivotal Role in Primer-Terminus Stabilization at the Polymerization Active Site. Biomolecules 2019, 9, 648. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9110648
del Prado A, Santos E, Lázaro JM, Salas M, de Vega M. The Loop of the TPR1 Subdomain of Phi29 DNA Polymerase Plays a Pivotal Role in Primer-Terminus Stabilization at the Polymerization Active Site. Biomolecules. 2019; 9(11):648. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9110648
Chicago/Turabian Styledel Prado, Alicia, Eugenia Santos, José M. Lázaro, Margarita Salas, and Miguel de Vega. 2019. "The Loop of the TPR1 Subdomain of Phi29 DNA Polymerase Plays a Pivotal Role in Primer-Terminus Stabilization at the Polymerization Active Site" Biomolecules 9, no. 11: 648. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9110648