Structure of the PUB Domain from Ubiquitin Regulatory X Domain Protein 1 (UBXD1) and Its Interaction with the p97 AAA+ ATPase


Abstract
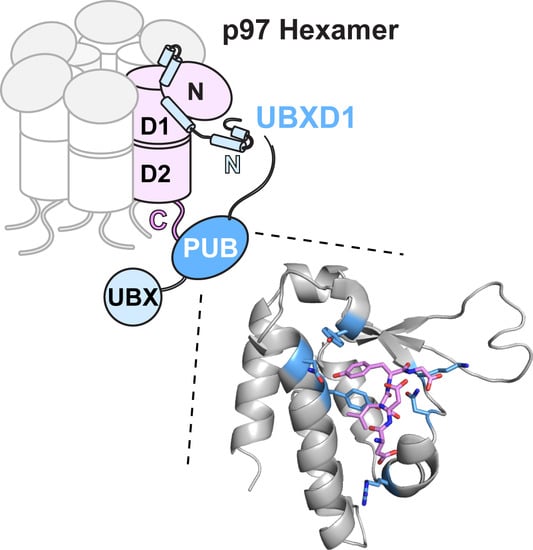
:
1. Introduction
2. Materials and Methods
2.1. Cloning, Protein Expression, and Purification
2.2. Peptides
2.3. Circular Dichroism (CD) Spectroscopy
2.4. MALDI-TOF Mass Spectrometry
2.5. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.5.1. NMR Assignments
2.5.2. NMR Structure Calculation
2.5.3. NMR Titration Experiments
2.5.4. 15N Relaxation Measurements
2.6. Fluorescence Anisotropy
2.7. Isothermal Titration Calorimetry (ITC)
2.8. Alignment of PUB structures and RMSD Calculation
2.9. Model of UBXD1-PUB/p97-PIM Complex
3. Results
3.1. NMR Solution Structure of UBXD1-PUB.
3.2. Interaction of UBXD1-PUB with p97-PIM.
3.3. Structural Model of the UBXD1-PUB/p97-PIM Complex.
3.4. Structure-Based Mutational Analysis Confirms Conserved Binding Pocket.
4. Discussion
4.1. Structural Homology between PUB Domains and Unique Features of UBXD1-PUB
4.2. Determinants of PIM Binding
4.3. PUB Domains as Interaction Hubs in the Ubiquitin–Proteasome System
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A Mighty “Protein Extractor” of the Cell: Structure and Function of the p97/CDC48 ATPase. Front. Mol. Biosci. 2017, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- van den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stach, L.; Freemont, P.S. The AAA+ ATPase p97, a cellular multitool. Biochem. J. 2017, 474, 2953–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shaw, A.; Bates, P.A.; Newman, R.H.; Gowen, B.; Orlova, E.; Gorman, M.A.; Kondo, H.; Dokumo, P.; Lally, J.; et al. Structure of the AAA ATPase p97. Mol. Cell 2000, 6, 1473–1484. [Google Scholar] [CrossRef]
- Davies, J.M.; Brunger, A.T.; Weis, W.I. Improved Structures of Full-Length p97, an AAA ATPase: Implications for Mechanisms of Nucleotide-Dependent Conformational Change. Structure 2008, 16, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blythe, E.E.; Olson, K.C.; Chau, V.; Deshaies, R.J. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem Proteinopathy. Proc. Natl. Acad. Sci. USA 2017, 114, E4380–E4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, N.O.; Rapoport, T.A. Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 2017, 169, 722–735. [Google Scholar] [CrossRef] [Green Version]
- Cooney, I.; Han, H.; Stewart, M.G.; Carson, R.H.; Hansen, D.T.; Iwasa, J.H.; Price, J.C.; Hill, C.P.; Shen, P.S. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science 2019, 365, 502–505. [Google Scholar] [CrossRef]
- Weith, M.; Seiler, J.; van den Boom, J.; Kracht, M.; Hülsmann, J.; Primorac, I.; Del Pino Garcia, J.; Kaschani, F.; Kaiser, M.; Musacchio, A.; et al. Ubiquitin-Independent Disassembly by a p97 AAA-ATPase Complex Drives PP1 Holoenzyme Formation. Mol. Cell 2018, 72, 766–777. [Google Scholar] [CrossRef] [Green Version]
- Buchberger, A.; Schindelin, H.; Hänzelmann, P. Control of p97 function by cofactor binding. FEBS Lett. 2015, 589, 2578–2589. [Google Scholar] [CrossRef] [Green Version]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate Processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019, 365, eaax1033. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sáiz, V.; Buchberger, A. Imbalances in p97 co-factor interactions in human Proteinopathy. EMBO Rep. 2010, 11, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritz, D.; Vuk, M.; Kirchner, P.; Bug, M.; Schütz, S.; Hayer, A.; Bremer, S.; Lusk, C.; Baloh, R.H.; Lee, H.; et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat. Cell Biol. 2011, 13, 1116–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Reim, G.; Hruzova, M.; Goetze, S.; Basler, K. Protection of armadillo/β-Catenin by armless, a novel positive regulator of wingless signaling. PLoS Biol. 2014, 12, e1001988. [Google Scholar] [CrossRef] [Green Version]
- Bento, A.C.; Bippes, C.C.; Kohler, C.; Hemion, C.; Frank, S.; Neutzner, A. UBXD1 is a mitochondrial recruitment factor for p97/VCP and promotes mitophagy. Sci. Rep. 2018, 8, 12415. [Google Scholar] [CrossRef] [Green Version]
- Kern, M.; Fernandez-Sáiz, V.; Schäfer, Z.; Buchberger, A. UBXD1 binds p97 through two independent binding sites. Biochem. Biophys. Res. Commun. 2009, 380, 303–307. [Google Scholar] [CrossRef]
- Stapf, C.; Cartwrightm, E.; Bycroft, M.; Hofmann, K.; Buchberger, A. The general definition of the p97/valosin-containing protein (VCP)-interacting motif (VIM) delineates a new family of p97 cofactors. J. Biol. Chem. 2011, 286, 38670–38678. [Google Scholar] [CrossRef] [Green Version]
- Trusch, F.; Matena, A.; Vuk, M.; Koerver, L.; Knævelsrud, H.; Freemont, P.S.; Meyer, H.; Bayer, P. The N-terminal Region of the Ubiquitin Regulatory X (UBX) Domain-containing Protein 1 (UBXD1) Modulates Interdomain Communication within the Valosin-containing Protein p97. J. Biol. Chem. 2015, 290, 29414–29427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuetz, A.K.; Kay, L.E. A Dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife 2016, 5, e20143. [Google Scholar] [CrossRef] [PubMed]
- Madsen, L.; Andersen, K.M.; Prag, S.; Moos, T.; Semple, C.A.; Seeger, M.; Hartmann-Petersen, R. Ubxd1 is a novel co-factor of the human p97 ATPase. Int. J. Biochem. Cell Biol. 2008, 40, 2927–2942. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Park, H.; Till, E.A.; Lennarz, W.J. The PUB domain: A putative protein-protein interaction domain implicated in the ubiquitin-proteasome pathway. Biochem. Biophys. Res. Commun. 2001, 287, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Zhou, X.; Wang, L.; Li, G.; Schindelin, H.; Lennarz, W.J. Studies on peptide:N-glycanase-p97 interaction suggest that p97 phosphorylation modulates endoplasmic reticulum-associated degradation. Proc. Natl. Acad. Sci. USA 2007, 104, 8785–8790. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhao, G.; Schindelin, H.; Lennarz, W.J. Tyrosine phosphorylation of ATPase p97 regulates its activity during ERAD. Biochem. Biophys. Res. Commun. 2008, 375, 247–251. [Google Scholar] [CrossRef]
- Elliot, P.R.; Nielsen, S.V.; Marco-Casanove, P.; Fiil, B.K.; Keusekotten, K.; Mailand, N.; Freund, S.M.V.; Gyrd-Hansen, M.; Komander, D. Molecular Basis and Regulation of OTULIN-LUBAC Interaction. Mol. Cell 2014, 54, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Leske, D.; Hrdinka, M.; Bagola, K.; Fiil, B.K.; McLaughlin, S.H.; Wagstaff, J.; Volkmar, N.; Christianson, J.C.; Kessler, B.M.; et al. SPATA2 Links CYLD to LUBAC, Activates CYLD, and Controls LUBAC Signaling. Mol. Cell 2016, 63, 990–1005. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.D.; Buchberger, A.; Bycroft, M. The PUB domain functions as a p97 binding module in human peptide N-glycanase. J. Biol. Chem. 2006, 281, 25502–25508. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhao, G.; Zhou, X.; Schindelin, H.; Lennarz, W. The AAA ATPase p97 links peptide N-glycanase to the endoplasmic reticulum-associated E3 ligase autocrine motility factor receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 8348–8353. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, C.; Blom, D.; Ploegh, H.L. A role for N-glycanase in the cytosolic turnover of glycoproteins. EMBO J. 2003, 33, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Walczak, H.; Iwai, K.; Dikic, I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, V.; Akutsu, M.; Olma, M.H.; Gomes, L.C.; Kawasaki, M.; Dikic, I. Binding of OTULIN to the PUB domain of HOIP controls NF-κB signaling. Mol. Cell 2014, 54, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Rivkin, E.; Almeida, S.M.; Ceccarelli, D.F.; Juang, Y.-C.; MacLean, T.A.; Srikumar, T.; Huang, H.; Dunham, W.H.; Fukumura, R.; Xie, G.; et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature 2013, 498, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Schlicher, L.; Brauns-Schubert, P.; Schubert, F.; Maurer, U. SPATA2: More than a missing link. Cell Death Differ. 2017, 24, 1142–1147. [Google Scholar] [CrossRef]
- Custer, S.K.; Neumann, M.; Lu, H.; Wright, A.C.; Taylor, J.P. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet. 2010, 19, 1741–1755. [Google Scholar] [CrossRef]
- Li, J.M.; Wu, H.; Zhang, W.; Blackburn, M.R.; Jin, J. The p97-UFD1L-NPL4 protein complex mediates cytokine-induced IkBa proteolysis. Mol. Cell. Biol. 2014, 34, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Lobley, A.; Whitmore, L.; Wallace, B.A. DICHROWEB: An interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 2002, 18, 211–212. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, L.; Wallace, B.A. DICHROWEB: An online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [Green Version]
- Keller, R. The Computer Aided Resonance Assignment Tutorial, 1st ed.; Cantina Verlag: Goldau, Switzerland, 2004; Available online: http://www.cara.nmr-software.org/downloads/3-85600-112-3.pdf (accessed on 6 August 2018).
- Sattler, M.; Schleucher, J.; Griesinger, C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 93–158. [Google Scholar] [CrossRef]
- Kovacs, H.; Gossert, A. Improved NMR experiments with 13C-isotropic mixing for assignement of aromatic and aliphatic side chains in labeled proteins. J. Biomol. NMR 2014, 58, 101–112. [Google Scholar] [CrossRef]
- Wishart, D.; Bigam, C.; Yao, J.; Abildgaard, F.; Dyson, H.; Oldfield, E.; Markley, J.; Sykes, B. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Cornilescu, G.; Delaglio, F.; Bax, A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 1999, 13, 289–302. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. The 13C Chemical-Shift Index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef]
- Shen, Y.; Vernon, R.; Baker, D.; Bax, A. De novo protein structure generation from incomplete chemical shift assignments. J. Biomol. NMR 2009, 43, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Serrano, P.; Pedrini, B.; Mohanty, B.; Geralt, M.; Herrmann, T.; Wüthrich, K. The J-UNIO protocol for automated protein structure determination by NMR in solution. J. Biomol. NMR 2012, 53, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, T.; Güntert, P.; Wüthrich, K. Protein NMR structure determination with automated NOE identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 2002, 24, 171–189. [Google Scholar] [CrossRef]
- Herrmann, T.; Güntert, P.; Wüthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002, 319, 209–227. [Google Scholar] [CrossRef] [Green Version]
- Güntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Ayed, A.; Mulder, F.A.; Yi, G.S.; Lu, Y.; Kay, L.E.; Arrowsmith, C.H. Latent and active p53 are identical in conformation. Nat. Struct. Biol. 2001, 8, 756–760. [Google Scholar] [CrossRef]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone Dynamics of a Free and a Phosphopeptide-Complexed Src Homology 2 Domain Studied by 15N NMR Relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Kay, L.E.; Nicholson, L.K.; Delaglio, F.; Bax, A.; Torchia, D.A. Pulse sequences for removal of the effects of cross correlation between dipolar and chemical-shift anisotropy relaxation mechanisms on the measurement of heteronuclear T1 and T2 values in proteins. J. Magn. Reson. 1992, 97, 359–375. [Google Scholar] [CrossRef]
- Rossi, P.; Swapna, G.V.; Huang, Y.J.; Aramini, J.M.; Anklin, C.; Conover, K.; Hamilton, K.; Xiao, R.; Acton, T.B.; Ertekin, A.; et al. A microscale protein NMR sample screening pipeline. J. Biomol. NMR 2010, 46, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, Y.; Uekusa, Y.; Sumiyoshi, A.; Sasakawa, H.; Hirao, T.; Suzuki, T.; Kato, K. NMR characterization of the interaction between the PUB domain of peptide:N-glycanase and ubiquitin-like domain of HR23. FEBS Lett. 2012, 586, 1141–1146. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | |
|---|---|---|
| Total structures computed | 100 | |
| Number of structures analyzed | 20 | |
| Constraints | ||
| NOE-based distance constraints | ||
| Total | 1221 | |
| intra-residue [i = j] | 462 | |
| sequential [| I − j | = 1] | 352 | |
| medium range [1 < | i − j | < 5] | 211 | |
| long range [| i − j | ≥5] | 196 | |
| NOE constraints per restrained residue | 10.3 | |
| Dihedral angle constraints | 567 | |
| Total # of restricting constraints | 1788 | |
| Total # of restricting constraints per residue | 15.0 | |
| Restricting long-range constraints per residue | 1.6 | |
| Residual constraint violationsa) | ||
| Distance violations / structure | ||
| 0.1–0.2 Å | 2.3 | |
| 0.2–0.5 Å | 0.2 | |
| >0.5 Å | 0 | |
| RMS of distance violation / constraint | 0.02 Å | |
| Maximum distance violation | 0.21 Å | |
| Dihedral angle violations / structure | ||
| 1°–10° | 2.6 | |
| >10° | 1.05 | |
| RMS of dihedral angle violation / constraint | 1.34° | |
| Maximum dihedral angle violation | 36.2° | |
| RMSD to the mean structure | ||
| all | ordered | |
| All backbone atoms | 1.6 Å | 0.9 Å |
| All heavy atoms | 1.8 Å | 1.3 Å |
| Ramachandran plot statistics b) | ||
| Most favored regions | 85.9% | |
| Additionally allowed regions | 13.3% | |
| Generously allowed regions | 0.2% | |
| Disallowed regions | 0.5% |
| Mutant | KD (µM) |
|---|---|
| wild type (wt) | 13.8 ± 0.3 |
| Y181A | 111 ± 2 |
| Y181F | 28.4 ± 0.6 |
| N184A | 28.7 ± 0.5 |
| N184D | 80 ± 2 |
| K193E | 17.7 ± 0.4 |
| Y194A | 53 ± 2 |
| Y194F | 141 ± 2 |
| K198E | 49.3 ± 0.8 |
| Q200A | 8.3 ± 0.3 |
| N201A | 64.1 ± 0.8 |
| N201D | 280 ± 3 |
| V203A | 25.3 ± 0.5 |
| E206R | 39.4 ± 0.9 |
| R207E | 88 ± 1 |
| wt + p97-fl | 8.4 ± 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blueggel, M.; van den Boom, J.; Meyer, H.; Bayer, P.; Beuck, C. Structure of the PUB Domain from Ubiquitin Regulatory X Domain Protein 1 (UBXD1) and Its Interaction with the p97 AAA+ ATPase. Biomolecules 2019, 9, 876. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9120876
Blueggel M, van den Boom J, Meyer H, Bayer P, Beuck C. Structure of the PUB Domain from Ubiquitin Regulatory X Domain Protein 1 (UBXD1) and Its Interaction with the p97 AAA+ ATPase. Biomolecules. 2019; 9(12):876. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9120876
Chicago/Turabian StyleBlueggel, Mike, Johannes van den Boom, Hemmo Meyer, Peter Bayer, and Christine Beuck. 2019. "Structure of the PUB Domain from Ubiquitin Regulatory X Domain Protein 1 (UBXD1) and Its Interaction with the p97 AAA+ ATPase" Biomolecules 9, no. 12: 876. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9120876