Heteroplasmic Variants of Mitochondrial DNA in Atherosclerotic Lesions of Human Aortic Intima

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Method
2.1. Human Aortic Intimal Tissue Autopsy Samples
2.2. DNA Extraction and mtDNA Enrichment
2.3. Whole mtDNA Sequencing
2.4. Measurement of mtDNA Copy Number
2.5. Bioinformatical and Statistical Analysis of Sequencing Data
- both of two variant nucleotides at the SNP site should be presented in the reads mapped to rCRS;
- the coverage of heteroplasmic site should be within the limit of 0.5–2.0-fold of the mean coverage depth;
- SNPs on homopolymeric sites were excluded from further analysis;
- heteroplasmic site should be covered with forward and reverse reads.
3. Results
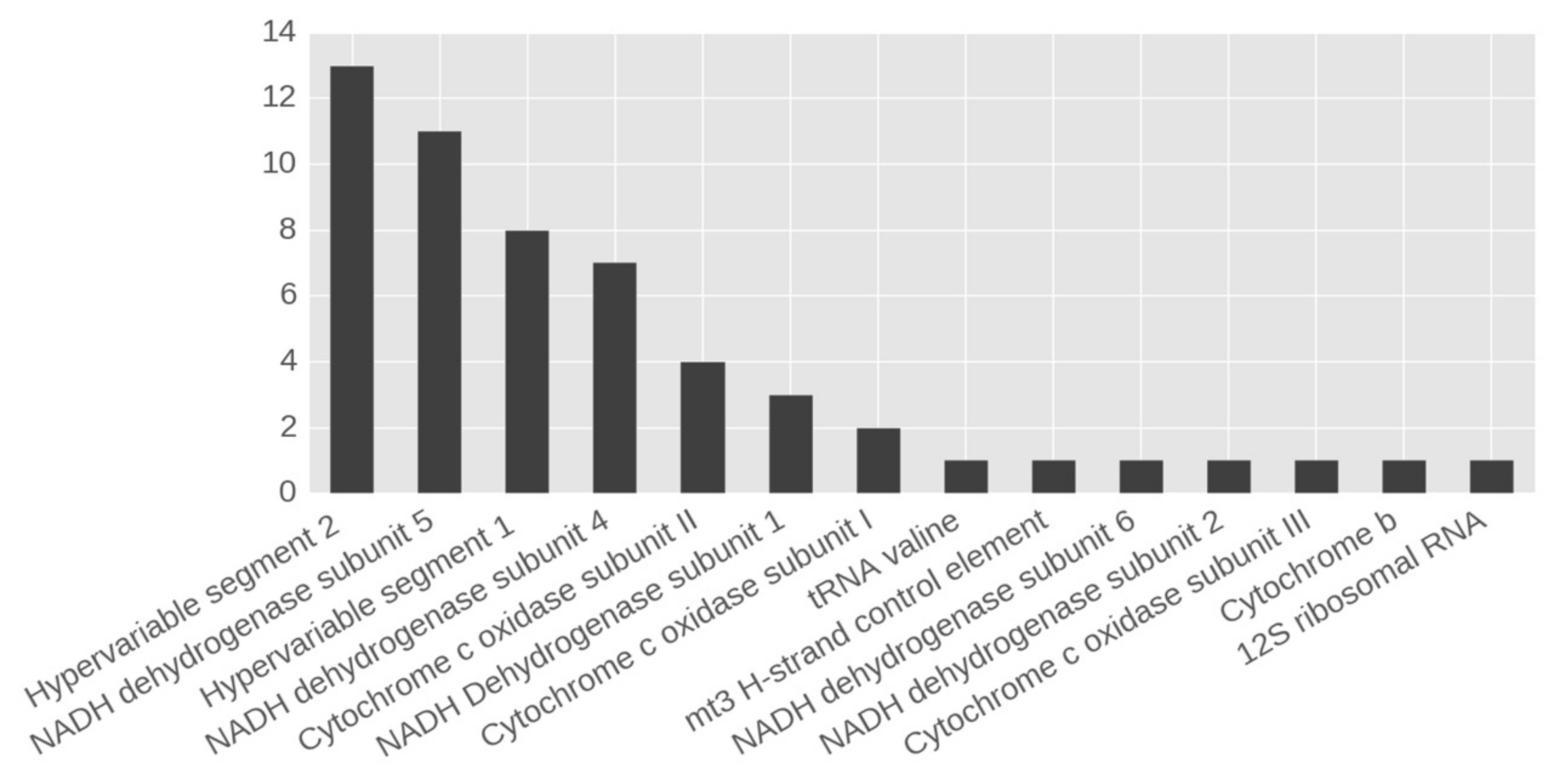
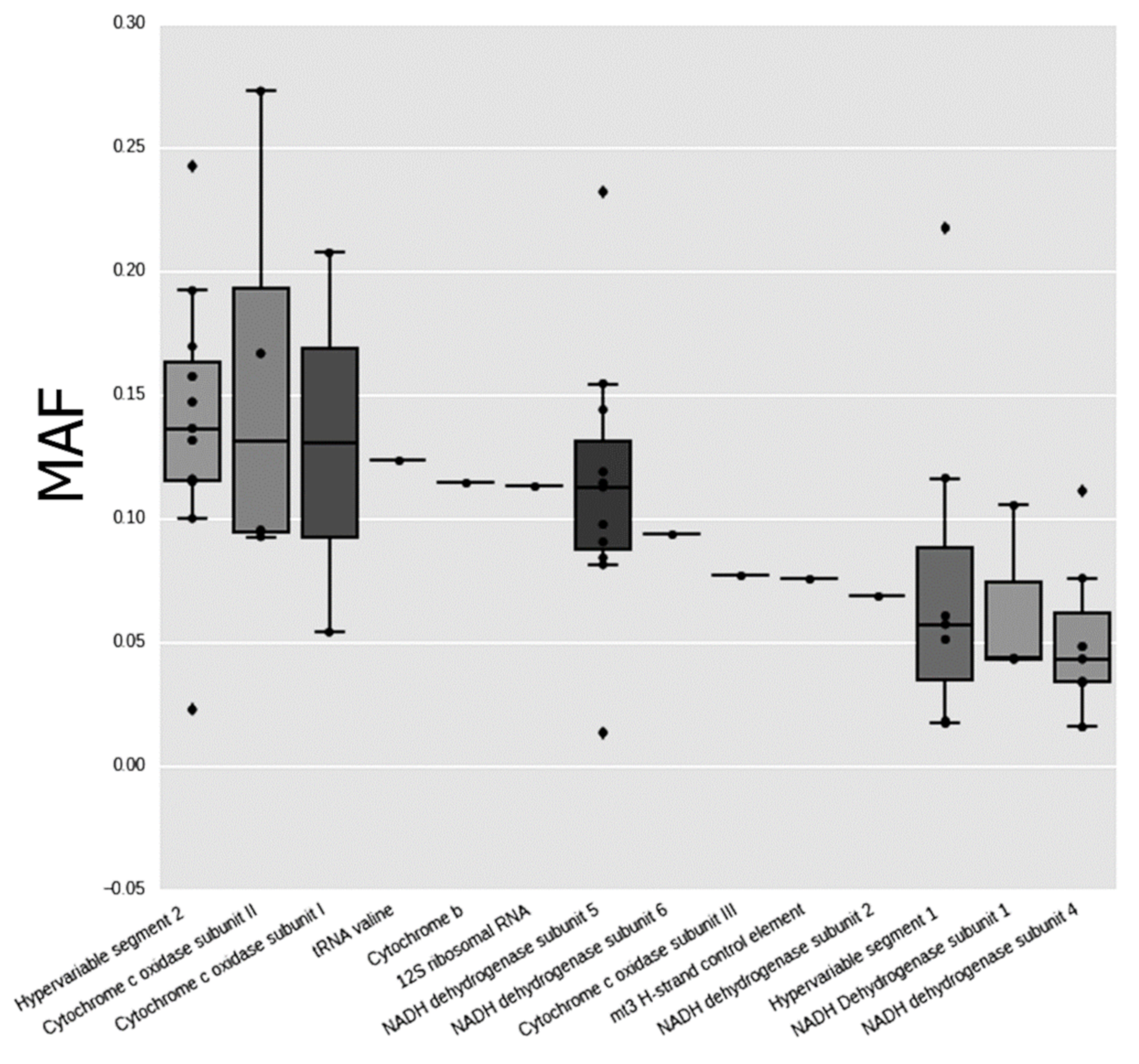
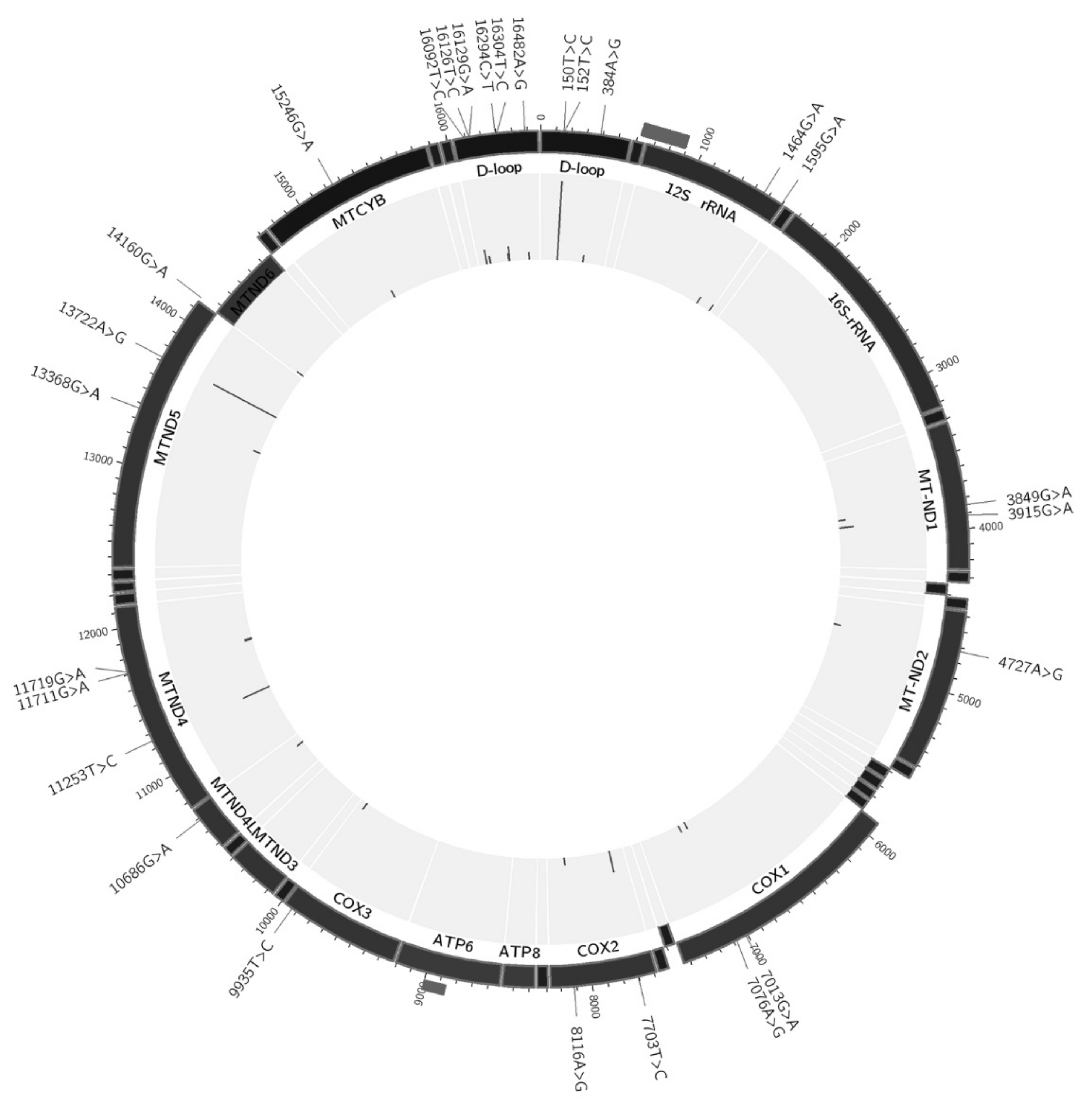
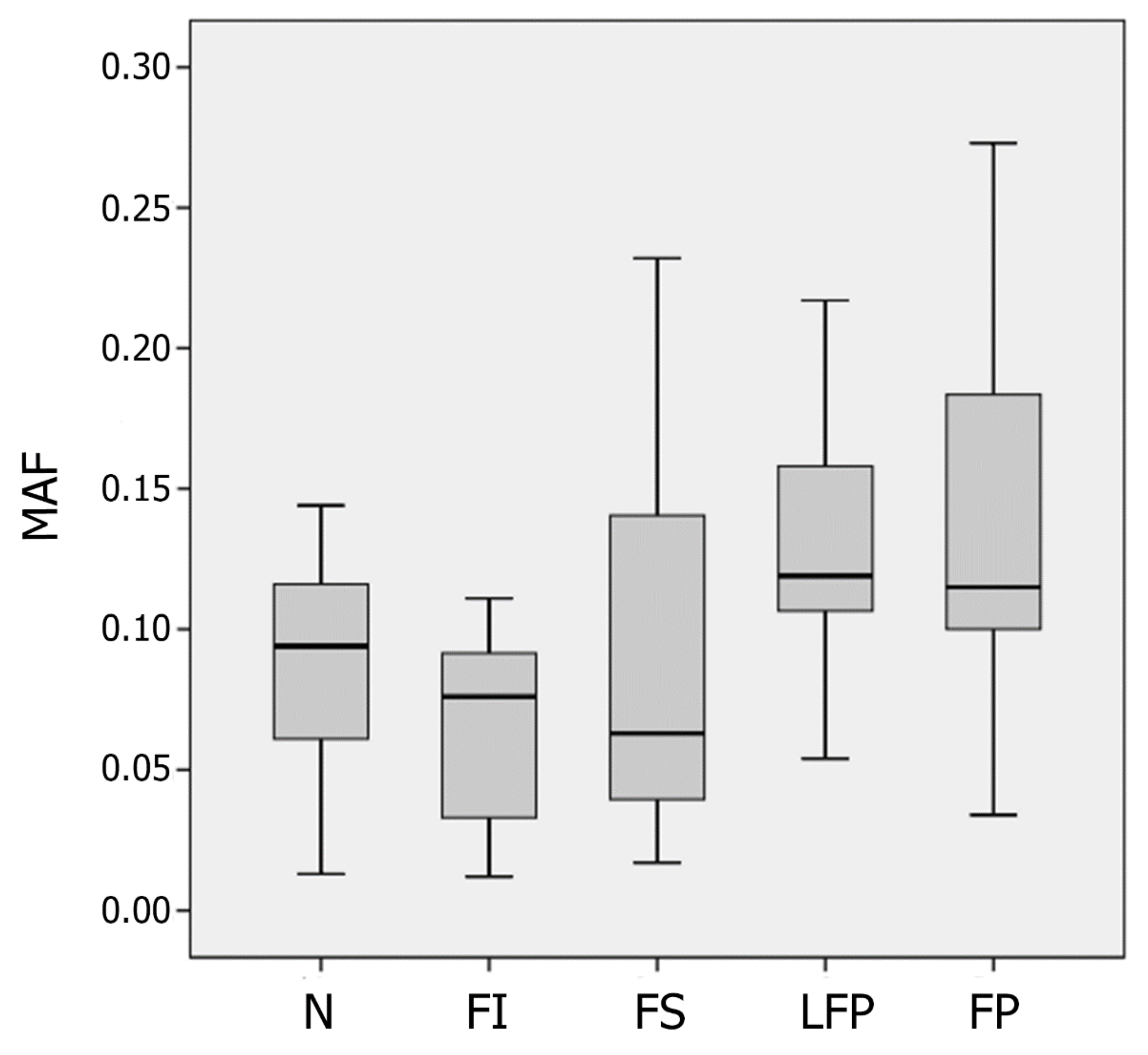
3.1. Heteroplasmic Variants of mtDNA
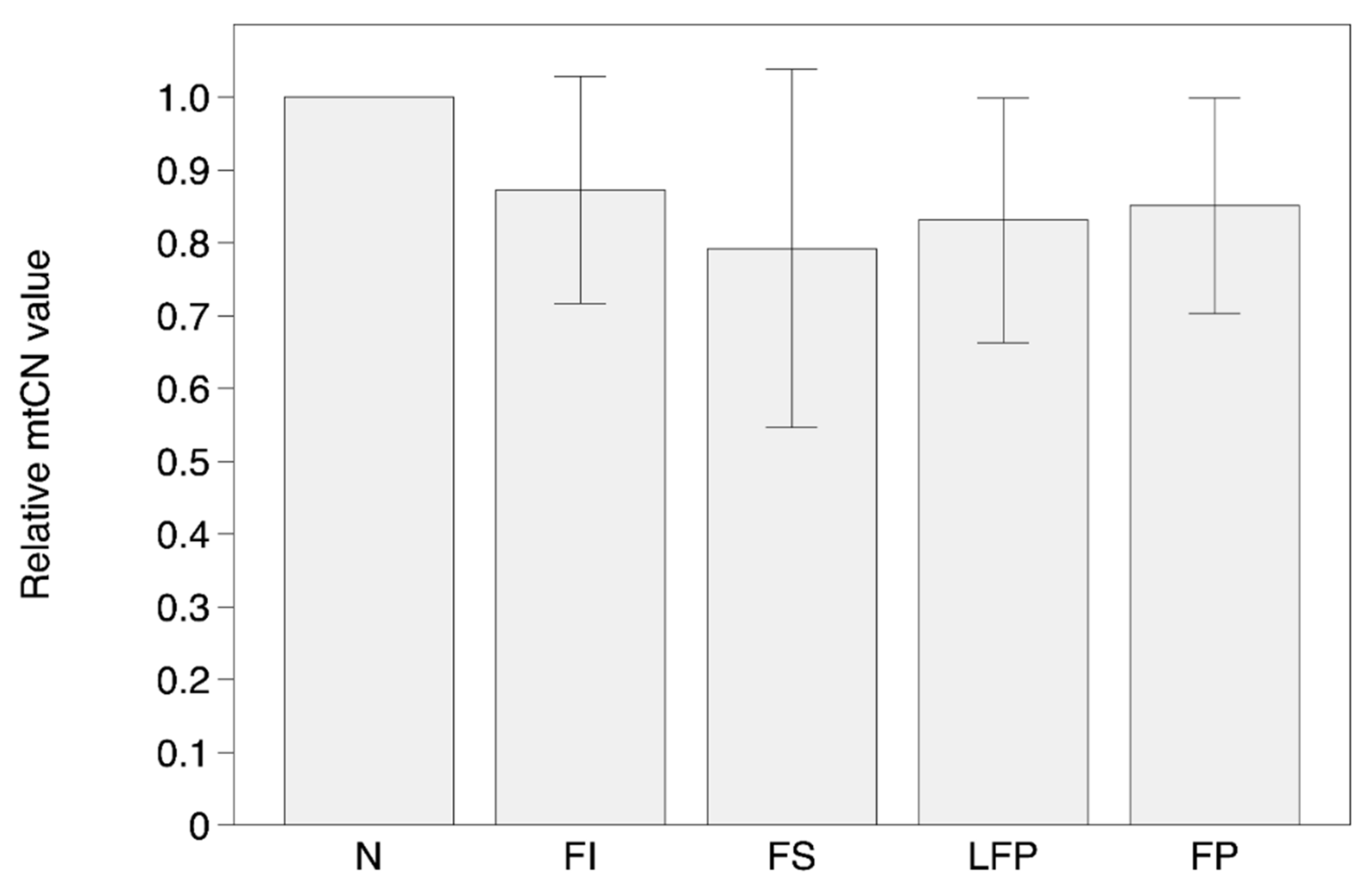
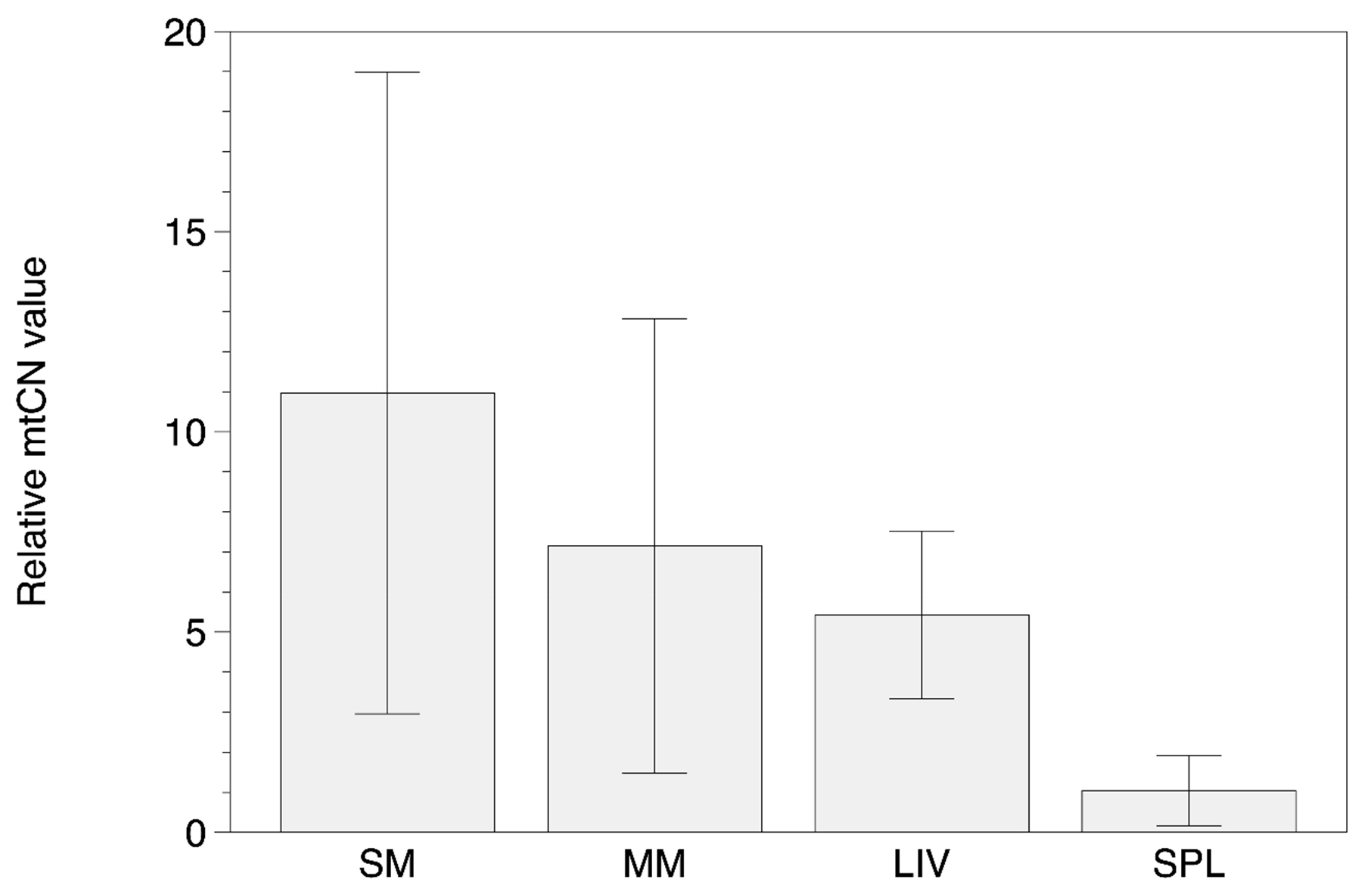
3.2. MtDNA Copy Number
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheng, C.; Tempel, D.; van Haperen, R.; van der Baan, A.; Grosveld, F.; Daemen, M.J.; Krams, R.; de Crom, R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 2006, 113, 2744–2753. [Google Scholar] [CrossRef]
- Lehoux, S.; Castier, Y.; Tedgui, A. Molecular mechanisms of the vascular responses to haemodynamic forces. J. Intern. Med. 2006, 259, 381–392. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Dendritic cells: A double-edge sword in atherosclerotic inflammation. Curr. Pharm. Des. 2015, 21, 1118–1123. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef]
- Hulsmans, M.; van Dooren, E.; Holvoet, P. Mitochondrial reactive oxygen species and risk of atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 264–276. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Just, R.S.; Irwin, J.A.; Parson, W. Mitochondrial DNA heteroplasmy in the emerging field of massively parallel sequencing. Forensic. Sci. Int. Genet. 2015, 18, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Palculict, M.E.; Zhang, V.W.; Wong, L.J.; Wang, J. Comprehensive mitochondrial genome analysis by massively parallel sequencing. Methods Mol. Biol. 2016, 1351, 3–17. [Google Scholar] [CrossRef]
- Li, M.; Schröder, R.; Ni, S.; Madea, B.; Stoneking, M. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 2491–2496. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Xie, X.; Wang, Y.; Gao, Y.; Xie, X.; Yang, J.; Ye, J. Association between leukocyte mitochondrial DNA content and risk of coronary heart disease: A case-control study. Atherosclerosis 2014, 237, 220–226. [Google Scholar] [CrossRef]
- Ashar, F.N.; Zhang, Y.; Longchamps, R.J.; Lane, J.; Moes, A.; Grove, M.L.; Mychaleckyj, J.C.; Taylor, K.D.; Coresh, J.; Rotter, J.I.; et al. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol. 2017, 2, 1247–1255. [Google Scholar] [CrossRef]
- Zhang, Y.; Guallar, E.; Ashar, F.N.; Longchamps, R.J.; Castellani, C.A.; Lane, J.; Grove, M.L.; Coresh, J.; Sotoodehnia, N.; Ilkhanoff, L.; et al. Association between mitochondrial DNA copy number and sudden cardiac death: Findings from the Atherosclerosis Risk in Communities study (ARIC). Eur. Heart J. 2017, 38, 3443–3448. [Google Scholar] [CrossRef]
- Liu, L.P.; Cheng, K.; Ning, M.A.; Li, H.H.; Wang, H.C.; Li, F.; Chen, S.Y.; Qu, F.L.; Guo, W.Y. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis 2017, 261, 105–110. [Google Scholar] [CrossRef]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 6, 274–286. [Google Scholar] [CrossRef]
- Lombès, A.; Auré, K.; Bellanné-Chantelot, C.; Gilleron, M.; Jardel, C. Unsolved issues related to human mitochondrial diseases. Biochimie 2014, 100, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Vizarra, E.; Enríquez, J.A.; Pérez-Martos, A.; Montoya, J.; Fernández-Silva, P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 2011, 11, 207–213. [Google Scholar] [CrossRef]
- Li, M.; Rothwell, R.; Vermaat, M.; Wachsmuth, M.; Schröder, R.; Laros, J.F.; van Oven, M.; de Bakker, P.I.; Bovenberg, J.A.; van Duijn, C.M.; et al. Transmission of human mtDNA heteroplasmy in the Genome of the Netherlands families: Support for a variable-size bottleneck. Genome Res. 2016, 26, 417–426. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Chistiakov, D.A.; Bobryshev, Y.V.; Postnov, A.Y.; Orekhov, A.N. Mitochondrial mutations in atherosclerosis: New solutions in research and possible clinical applications. Curr. Pharm. Des. 2013, 19, 5942–5953. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Mitrofanov, K.Y.; Zhelankin, A.V.; Sazonova, M.A.; Postnov, A.Y.; Revin, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Quantitative assessment of heteroplasmy of mitochondrial genome: Perspectives in diagnostics and methodological pitfalls. Biomed. Res. Int. 2014, 2014, 292017. [Google Scholar] [CrossRef]
- Jenuth, J.P.; Peterson, A.C.; Shoubridge, E.A. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat. Genet. 1997, 16, 93–95. [Google Scholar] [CrossRef]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Larsson, N.G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 2008, 9, 657–662. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Zhelankin, A.V.; Postnov, A.Y.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. Biomed. Res. Int. 2015, 2015, 825468. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288. [Google Scholar] [CrossRef]
- Li, M.; Schönberg, A.; Schaefer, M.; Schroeder, R.; Nasidze, I.; Stoneking, M. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 2010, 87, 237–249. [Google Scholar] [CrossRef]
- Payne, B.A.; Wilson, I.J.; Yu-Wai-Man, P.; Coxhead, J.; Deehan, D.; Horvath, R.; Taylor, R.W.; Samuels, D.C.; Santibanez-Koref, M.; Chinnery, P.F. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 2013, 22, 384–390. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W., Jr.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef]
- Venegas, V.; Halberg, M.C. Measurement of mitochondrial DNA copy number. Methods Mol. Biol. 2012, 837, 327–335. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Mikkelsen, M.; Frank-Hansen, R.; Hansen, A.J.; Morling, N. Massively parallel pyrosequencing of the mitochondrial genome with the 454 methodology in forensic genetics. Forensic. Sci. Int. Genet. 2014, 12, 30–37. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Homo Sapiens Mitochondrion, Complete Genome. NCBI Reference Sequence: NC_012920.1. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/nuccore/NC_012920.1?report=fasta&to=16569. (accessed on 20 August 2019).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2 [q-bio.GN]. [Google Scholar]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: http://www.scipy.org (accessed on 30 June 2019).
- McKinney, W. Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; van der Walt, S., Millman, J., Eds.; SciPy: Austin, TX, USA; pp. 51–56. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.; Botvinnik, O.; O’ Kane, D.; Hobson, P.; Ostblom, J.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Cole, J.B.; et al. Mwaskom/Seaborn: v0.9.0 (July 2018); Zenodo: Geneva, Switzerland, 2018. [Google Scholar] [CrossRef]
- den Dunnen, J.T. Describing sequence variants using HGVS nomenclature. Methods Mol. Biol. 2017, 1492, 243–251. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Mitochondrial mutations are associated with atherosclerotic lesions in the human aorta. Clin. Dev. Immunol. 2012, 2012, 832464. [Google Scholar] [CrossRef]
- Wachsmuth, M.; Huebner, A.; Li, M.; Madea, B.; Stoneking, M. Age-related and heteroplasmy-related variation in human mtDNA copy number. PLoS Genet. 2016, 12, e1005939. [Google Scholar] [CrossRef]
- van Osch, F.H.; Voets, A.M.; Schouten, L.J.; Gottschalk, R.W.; Simons, C.C.; van Engeland, M.; Lentjes, M.H.; van den Brandt, P.A.; Smeets, H.J.; Weijenberg, M.P. Mitochondrial DNA copy number in colorectal cancer: Between tissue comparisons, clinicopathological characteristics and survival. Carcinogenesis 2015, 36, 1502–1510. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lee, W.C.; Liao, S.C.; Lee, L.C.; Su, Y.J.; Lee, C.T.; Chen, J.B. Mitochondrial DNA copy number correlates with oxidative stress and predicts mortality in nondiabetic hemodialysis patients. J. Nephrol. 2011, 24, 351–358. [Google Scholar] [CrossRef]
- Chien, M.C.; Huang, W.T.; Wang, P.W.; Liou, C.W.; Lin, T.K.; Hsieh, C.J.; Weng, S.W. Role of mitochondrial DNA variants and copy number in diabetic atherogenesis. Genet. Mol. Res. 2012, 11, 3339–3348. [Google Scholar] [CrossRef]
- Miller, F.J.; Rosenfeldt, F.L.; Zhang, C.; Linnane, A.W.; Nagley, P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: Lack of change of copy number with age. Nucleic Acids Res. 2003, 31, e61. [Google Scholar] [CrossRef]
- Samuels, D.; Li, C.; Li, B.; Song, Z.; Torstenson, E.; Boyd Clay, H.; Rokas, A.; Thornton-Wells, T.A.; Moore, J.H.; Hughes, T.M.; et al. Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 2013, 9, e1003929. [Google Scholar] [CrossRef]
- Hofmann, S.; Jaksch, M.; Bezold, R.; Mertens, S.; Aholt, S.; Paprotta, A.; Gerbitz, K.D. Population genetics and disease susceptibility: Characterization of central European haplogroups by mtDNA gene mutations, correlation with D loop variants and association with disease. Hum. Mol. Genet. 1997, 6, 1835–1846. [Google Scholar] [CrossRef]
- Jeronimo, C.; Nomoto, S.; Caballero, O.L.; Usadel, H.; Henrique, R.; Varzim, G.; Oliveira, J.; Lopes, C.; Fliss, M.S.; Sidransky, D. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 2001, 20, 5195–5198. [Google Scholar] [CrossRef] [Green Version]
- Zaki, E.A.; Freilinger, T.; Klopstock, T.; Baldwin, E.E.; Heisner, K.R.; Adams, K.; Dichgans, M.; Wagler, S.; Boles, R.G. Two common mitochondrial DNA polymorphisms are highly associated with migraine headache and cyclic vomiting syndrome. Cephalalgia 2009, 29, 719–728. [Google Scholar] [CrossRef]
- Alvarez-Iglesias, V.; Mosquera-Miguel, A.; Cerezo, M.; Quintans, B.; Zarrabeitia, M.T.; Cusco, I.; Lareu, M.V.; García, O.; Pérez-Jurado, L.; Carracedo, A.; et al. New population and phylogenetic features of the internal variation within mitochondrial DNA macro-haplogroup R0. PLoS ONE 2009, 4, e5112. [Google Scholar] [CrossRef]
- Wong, L.J.; Liang, M.H.; Kwon, H.; Park, J.; Bai, R.K.; Tan, D.J. Comprehensive scanning of the entire mitochondrial genome for mutations. Clin. Chem. 2002, 48, 1901–1912. [Google Scholar]
- Ericson, N.G.; Kulawiec, M.; Vermulst, M.; Sheahan, K.; O’Sullivan, J.; Salk, J.J.; Bielas, J.H. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 2012, 8, e1002689. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Andreeva, E.R.; Andrianova, I.V.; Bobryshev, Y.V. Peculiarities of cell composition and cell proliferation in different type atherosclerotic lesions in carotid and coronary arteries. Atherosclerosis 2010, 212, 436–443. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Andreeva, E.R.; Mikhailova, I.A.; Andrianova, I.V.; Moisenovich, M.M.; Khapchaev, S.; Agapov, I.I.; Sobenin, I.A.; Lusta, K.A.; Orekhov, A.N. Correlation between lipid deposition, immune-inflammatory cell content and MHC class II expression in diffuse intimal thickening of the human aorta. Atherosclerosis 2011, 219, 171–183. [Google Scholar] [CrossRef]
- Summerhill, V.; Orekhov, A. Pericytes in atherosclerosis. Adv. Exp. Med. Biol. 2019, 1147, 279–297. [Google Scholar] [CrossRef]
- Andreeva, E.R.; Pugach, I.M.; Orekhov, A.N. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis 1997, 135, 19–27. [Google Scholar] [CrossRef]
- Yu, E.P.; Bennett, M.R. Mitochondrial DNA damage and atherosclerosis. Trends Endocrinol. Metab. 2014, 25, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Sinyov, V.V.; Sazonova, M.A.; Ryzhkova, A.I.; Galitsyna, E.V.; Melnichenko, A.A.; Postnov, A.Y.; Orekhov, A.N.; Grechko, A.V.; Sobenin, I.A. Potential use of buccal epithelium for genetic diagnosis of atherosclerosis using mtDNA mutations. Vessel Plus 2017, 1, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef] [Green Version]
- Itsara, L.S.; Kennedy, S.R.; Fox, E.J.; Yu, S.; Hewitt, J.J.; Sanchez-Contreras, M.; Cardozo-Pelaez, F.; Pallanck, L.J. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014, 10, e1003974. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Melnichenko, A.A.; Postnov, A.Y.; Orekhov, A.N.; Sobenin, I.A. Cybrid models of pathological cell processes in different diseases. Oxid. Med. Cell Longev. 2018, 2018, 4647214. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Khasanova, Z.B.; Nikitina, N.A.; Karagodin, V.P.; Orekhov, A.N.; Sobenin, I.A. Creation of cultures containing mutations linked with cardiovascular diseases using transfection and genome editing. Curr. Pharm. Des. 2019, 25, 693–699. [Google Scholar] [CrossRef]
- Zhang, E.; Zhang, C.; Su, Y.; Cheng, T.; Shi, C. Newly developed strategies for multifunctional mitochondria-targeted agents in cancer therapy. Drug Discov. Today 2011, 16, 140–146. [Google Scholar] [CrossRef]
- Fogleman, S.; Santana, C.; Bishop, C.; Miller, A.; Capco, D.G. CRISPR/Cas9 and mitochondrial gene replacement therapy: Promising techniques and ethical considerations. Am. J. Stem. Cells 2016, 5, 39–52. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case No. (ID) | Age | Gender | Pathologic Diagnosis | The Number of Tissue Samples | ||||
|---|---|---|---|---|---|---|---|---|
| N | FI | FS | LFP | FP | ||||
| as_01 | 85 | f | macrofocal atherosclerosis | 2 | 1 | 2 | 3 | 3 |
| as_02 | 83 | m | pulmonary heart disease, thromboembolia of small pulmonary arteries, macrofocal cardiosclerosis | 2 | 2 | 2 | 3 | 3 |
| as_03 | 86 | f | pulmonary heart disease, bilateral confluent bronchopneumonia, diffuse microfocal cardiosclerosis | 2 | 2 | 2 | 3 | 3 |
| as_04 | 87 | f | pulmonary artery thromboembolia, macrofocal cardiosclerosis | 2 | 3 | 3 | 2 | 2 |
| as_05 | 60 | m | gastrorrhagia | 3 | 2 | 2 | 2 | 3 |
| as_06 | 83 | m | diffuse microfocal cardiosclerosis, cardiohepatic insufficiency, right-focal abscessed confluent pneumonia | 2 | 2 | 2 | 3 | 3 |
| as_07 | 83 | m | right kidney cancer, cancerous cachexia, macrofocal atherosclerosis | 2 | 3 | 2 | 2 | 3 |
| Total number of samples | 15 | 15 | 15 | 18 | 20 | |||
| Position | Gene/Region | Nucleotide Change | Mutation Type (for Protein-Coding Genes) and Aminoacid Change | Total Number of Hetero-Plasmy Cases | Mean MAF | SD |
|---|---|---|---|---|---|---|
| 150 | Hypervariable segment 2 | C > T | --- | 2 | 0.0175 | 0.0078 |
| 152 | Hypervariable segment 2 | T > C | --- | 11 | 0.1528 | 0.0407 |
| 384 | mt3 H-strand control element | A > G | --- | 1 | 0.0760 | |
| 1464 | 12S ribosomal RNA | G > A | --- | 1 | 0.1130 | |
| 1595 | tRNA valine | G > A | --- | 1 | 0.1240 | |
| 3849 | NADH Dehydrogenase subunit 1 | G > A | syn:L-L | 1 | 0.1050 | |
| 3915 | NADH Dehydrogenase subunit 1 | G > A | syn:G-G | 2 | 0.0430 | 0.0000 |
| 4727 | NADH dehydrogenase subunit 2 | A > G | syn:M-M | 1 | 0.0690 | |
| 7013 | Cytochrome c oxidase subunit I | G > A | syn:T-T | 1 | 0.0540 | |
| 7076 | Cytochrome c oxidase subunit I | A > G | syn:G-G | 1 | 0.2070 | |
| 7703 | Cytochrome c oxidase subunit II | T > C | non-syn:Y-H | 3 | 0.1183 | 0.0422 |
| 8116 | Cytochrome c oxidase subunit II | A > G | syn:G-G | 1 | 0.2730 | |
| 9935 | Cytochrome c oxidase subunit III | T > C | syn:H-H | 1 | 0.0770 | |
| 10686 | NADH dehydrogenase subunit 4 | G > A | non-syn:V-M | 1 | 0.0760 | |
| 11253 | NADH dehydrogenase subunit 4 | T > C | non-syn:I-T | 4 | 0.0398 | 0.0069 |
| 11711 | NADH dehydrogenase subunit 4 | G > A | non-syn:A-T | 1 | 0.1110 | |
| 11719 | NADH dehydrogenase subunit 4 | G > A | syn:G-G | 1 | 0.0160 | |
| 13368 | NADH dehydrogenase subunit 5 | G > A | syn:G-G | 1 | 0.0130 | |
| 13722 | NADH dehydrogenase subunit 5 | A > G | syn:L-L | 10 | 0.1229 | 0.0453 |
| 14160 | NADH dehydrogenase subunit 6 | G > A | non-syn:R-W | 1 | 0.0940 | |
| 15246 | Cytochrome b | G > A | non-syn:G-D | 1 | 0.1140 | |
| 16092 | Hypervariable segment 1 | T > C | --- | 2 | 0.0410 | 0.0141 |
| 16126 | Hypervariable segment 1 | T > C | --- | 1 | 0.0170 | |
| 16129 | Hypervariable segment 1 | G > A | --- | 1 | 0.2170 | |
| 16294 | Hypervariable segment 1 | C > T | --- | 1 | 0.1160 | |
| 16304 | Hypervariable segment 1 | T > C | --- | 2 | 0.0395 | 0.0304 |
| 16482 | Hypervariable segment 1 | A > G | --- | 1 | 0.0570 |
| Parameter | The Type of Atherosclerotic Lesion | ||||
|---|---|---|---|---|---|
| N | FI | FS | LFP | FP | |
| The number of minor variants | 68 | 70 | 106 | 113 | 134 |
| The number of major variants | 786 | 1049 | 965 | 732 | 801 |
| Case No. (ID) | Mean MAF | |
|---|---|---|
| N+FI | FS+FP+LFP | |
| as_01 | 0 | 0.115 |
| as_02 | 0.097 | 0.107 |
| as_03 | 0.074 | 0.133 |
| as_04 | 0.060 | 0.111 |
| as_05 | 0.098 | 0.136 |
| as_06 | 0.039 | 0.055 |
| as_07 | 0.019 | 0.211 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobenin, I.A.; Zhelankin, A.V.; Khasanova, Z.B.; Sinyov, V.V.; Medvedeva, L.V.; Sagaidak, M.O.; Makeev, V.J.; Kolmychkova, K.I.; Smirnova, A.S.; Sukhorukov, V.N.; et al. Heteroplasmic Variants of Mitochondrial DNA in Atherosclerotic Lesions of Human Aortic Intima. Biomolecules 2019, 9, 455. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9090455
Sobenin IA, Zhelankin AV, Khasanova ZB, Sinyov VV, Medvedeva LV, Sagaidak MO, Makeev VJ, Kolmychkova KI, Smirnova AS, Sukhorukov VN, et al. Heteroplasmic Variants of Mitochondrial DNA in Atherosclerotic Lesions of Human Aortic Intima. Biomolecules. 2019; 9(9):455. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9090455
Chicago/Turabian StyleSobenin, Igor A., Andrey V. Zhelankin, Zukhra B. Khasanova, Vasily V. Sinyov, Lyudmila V. Medvedeva, Maria O. Sagaidak, Vsevolod J. Makeev, Kira I. Kolmychkova, Anna S. Smirnova, Vasily N. Sukhorukov, and et al. 2019. "Heteroplasmic Variants of Mitochondrial DNA in Atherosclerotic Lesions of Human Aortic Intima" Biomolecules 9, no. 9: 455. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9090455