
Craniofacial Phenotypes and Genetics of DiGeorge Syndrome


Department of Signal Gene Regulation, Advanced Therapeutic Sciences, Medical and Dental Sciences, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University (TMDU), Tokyo 113-8510, Japan
J. Dev. Biol. 2022, 10(2), 18; https://0-doi-org.brum.beds.ac.uk/10.3390/jdb10020018
Submission received: 21 April 2022
/
Revised: 11 May 2022
/
Accepted: 11 May 2022
/
Published: 13 May 2022
(This article belongs to the Special Issue Scientific Papers by Developmental Biologists in Japan)
Abstract
:The 22q11.2 deletion is one of the most common genetic microdeletions, affecting approximately 1 in 4000 live births in humans. A 1.5 to 2.5 Mb hemizygous deletion of chromosome 22q11.2 causes DiGeorge syndrome (DGS) and velocardiofacial syndrome (VCFS). DGS/VCFS are associated with prevalent cardiac malformations, thymic and parathyroid hypoplasia, and craniofacial defects. Patients with DGS/VCFS manifest craniofacial anomalies involving the cranium, cranial base, jaws, pharyngeal muscles, ear-nose-throat, palate, teeth, and cervical spine. Most craniofacial phenotypes of DGS/VCFS are caused by proximal 1.5 Mb microdeletions, resulting in a hemizygosity of coding genes, microRNAs, and long noncoding RNAs. TBX1, located on chromosome 22q11.21, encodes a T-box transcription factor and is a candidate gene for DGS/VCFS. TBX1 regulates the fate of progenitor cells in the cranial and pharyngeal apparatus during embryogenesis. Tbx1-null mice exhibit the most clinical features of DGS/VCFS, including craniofacial phenotypes. Despite the frequency of DGS/VCFS, there has been a limited review of the craniofacial phenotypes of DGC/VCFS. This review focuses on these phenotypes and summarizes the current understanding of the genetic factors that impact DGS/VCFS-related phenotypes. We also review DGS/VCFS mouse models that have been designed to better understand the pathogenic processes of DGS/VCFS.
1. Introduction
The 22q11.2 deletion syndrome is one of the most common chromosomal microdeletions, affecting approximately 1 in 4000 live births in humans [1]. A 1.5 to 2.5 Mb hemizygous deletion of chromosome 22q11.2 causes DiGeorge syndrome (DGS; OMIM #188400) and velocardiofacial syndrome (VCFS or Shprintzen VCF syndrome; OMIM #192430) [2]. DGS/VCFS appears to be a genomic disorder distinct from 22q11.2 distal deletion syndrome (OMIM #611867). The clinical phenotype of DGS/VCFS is a complex and variable congenital disability, including cardiovascular defects, thymic hypoplasia, parathyroid hypoplasia, and craniofacial malformations [3]. Craniofacial malformations occur in approximately 60% of patients with DGS/VCFS [4].
TBX1, located on chromosome 22q11.21, encodes a T-box transcription factor and is considered a candidate gene for DGS/VCFS since mutations in TBX1 have been found in patients with DGS/VCFS [5]. Heterozygous Tbx1-mutant (Tbx1+/−) mice exhibit DGS/VCFS-related cardiovascular, parathyroid, and thymic phenotypes, suggesting that TBX1 dosage is critical for cardiovascular, parathyroid and thymic development [6,7,8,9]. Tbx1-null mice exhibit the most clinical features of DGS/VCFS, including craniofacial phenotypes, while Tbx1+/− mice exhibit no significant craniofacial phenotypes [6,7,8,9,10].
There have been some excellent reviews on genetics and cardiovascular anomalies of DGS/VCFS [3,11,12,13]. However, information on the craniofacial anomalies of DGS/VCFS is limited. This review focuses on these phenotypes and summarizes the current understanding of the genetic factors that impact DGS/VCFS-related phenotypes. We also review DGS/VCFS mouse models that have been designed to better understand the pathogenic processes of DGS/VCFS.
2. Craniofacial Phenotypes of Patients with DGS/VCFS
Patients with DGS/VCFS manifest craniofacial anomalies involving the cranium, cranial base, jaws, pharyngeal muscles, ear-nose-throat, palate, teeth, and cervical spine (Figure S1, Table 1 and Table 2). Frequently observed craniofacial phenotypes include velopharyngeal insufficiency (27–92%), enamel hypomineralization (39–41%), hearing loss (33–39%), platybasia (50–91%), and cervical spine anomalies (75%) (Table 1). Delayed development of the hyoid bone has also been reported [14,15].
In addition to morphological anomalies, infants and young children with DGS/VCFS often exhibit a high prevalence of functional difficulties in feeding and speech/language associated with cleft palate, laryngeal anomalies, and velopharyngeal dysfunction [37]. Even after cleft palate closure, children with DGS/VCFS sometimes present communication disorders related to speech-language problems, such as articulation disorders of speech sounds and vocal disorders [37]. They exhibit slower language acquisition than those with other disorders that may be associated with abnormal muscle development.
3. Genetics of DGS/VCFS
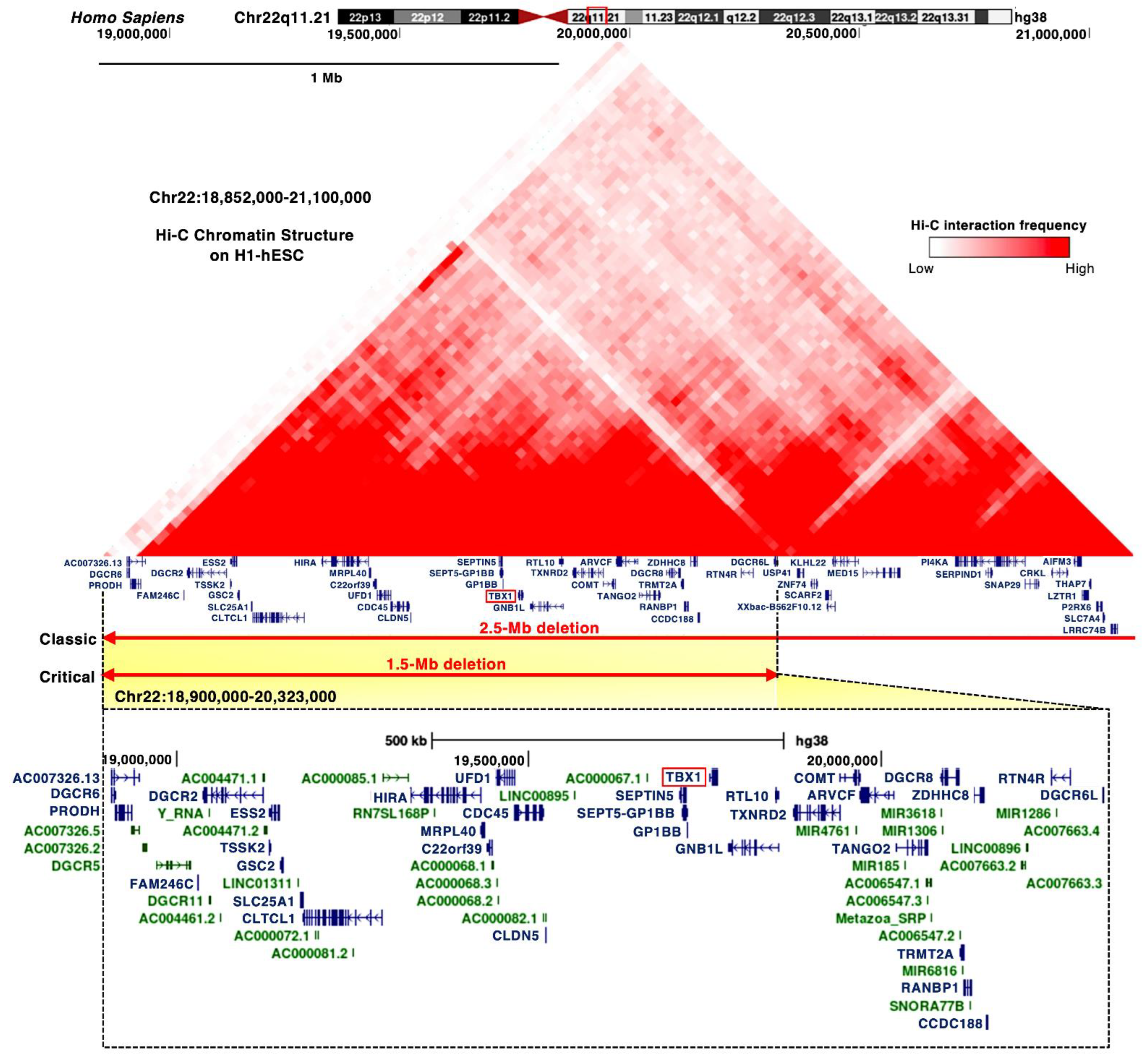
DGS/VCFS is caused by a 1.5 to 2.5 Mb hemizygous deletion of chromosome 22q11.2 (Figure 1). Chromosomal microdeletions at 10p14-p13 (the DGS2 locus) in patients with DGS/VCFS phenotypes are defined as the DGS/VCFS complex 2. In this review, we focus on the 22q11.2 locus, its associated genes, and miRNAs.
Most of the chromosomal deletions of the 22q11.2 locus are de novo, but inherited deletions of the 22q11.2 locus have been reported in 6–28% of patients as autosomal dominant [16,17]. The majority of clinical phenotypes of DGS/VCFS are caused by proximal 1.5 Mb microdeletions [3,22], resulting in a hemizygosity of approximately 30 coding genes, including DGCR6, PRODH, DGCR2, ESS2, TSSK2, GSC2, FAM246C, SLC25A1, CLTCL1, UFD1, HIRA, CDC45, MRPL40, C22orf39, CLDN5, TBX1, SEPTIN5, SEPT5-GP1BB, GP1BB, GNB1L, RTL10, TXNRD2, COMT, ARVCF, TANGO2, TRMT2A, RANBP1, CCDC188, DGCR8, ZDHHC8, RTN4R, DGCR6L, and C007326, as well as microRNAs (miRNAs) and long noncoding RNAs (Figure 1 and Figure S2A). The Hi-C chromatin structure of the 1.5 Mb region indicates interactions between these loci and their neighboring regions (Figure 1).
3.1. TBX1 Gene
The proximal deletion of 1.5 Mb on the 22q11.2 locus includes TBX1 (Figure 1). TBX1 is considered a candidate gene of DGS/VCFS because haploinsufficiency of TBX1 leads to the typical phenotypes of DGS/VCFS, conotruncal anomaly face syndrome (OMIM #217095), and tetralogy of Fallot (OMIM #187500) (Table 3). Identical mutations in TBX1 present among patients resulted in distinct phenotypes, suggesting that genetic and epigenetic changes or environmental factors are involved in the clinical phenotypes [5]. The coding variants in the T-box and C-terminal domains of TBX1 showed high combined annotation-dependent depletion (CADD) scores (Table S1); however, further investigation is required to confirm that the variants cause DGS/VCFS and how they impact the phenotypes.
3.2. DiGeorge Syndrome Critical Region (DGCR)
DGCR8, DGCR6, and DGCR6L map to the commonly deleted 1.5 Mb region in DGS/VCFS (Figure 1). DGCR8 is a nuclear miRNA-binding protein required for miRNA biogenesis. Dgcr8 haploinsufficiency in mice reduces the expression of miRNAs in the brain [45]. DGCR6 and DGCR6L genes encode a protein with a sequence similar to the Drosophila gonadal [46] (Figure S2B). In a chicken model, targeting DGCR6 function resulted in a vascular phenotype [47]. Attenuation of DGCR6 affects the expression of three genes localized within the 1.5 Mb region, upregulating the expression of TBX1 and UFD1 and reducing the expression of HIRA in the heart and pharyngeal arches of the chicken embryos [47]. Thus, the haploinsufficiency of DGCR8 or DGCR6 may be linked to DGS/VCFS phenotypes when targeting DGS/VCFS-related genes and miRNAs.
3.3. MicroRNAs
The deleted 1.5 Mb on the 22q11.2 locus includes several miRNAs, such as miR-185, miR-4716, miR-3618, miR-1286, miR-1306, and miR-6816 (Figure 1). The TargetScan miRNA target prediction program (http://www.targetscan.org accessed on 3 August 2021) identified that the 3′ UTR of TBX1 includes conserved sites for miR-183-5p, miR-96-5p, miR-1271-5p, miR-182-5p, miR-144-3p, miR-139-5p, miR-101-3p, and miR-451. Two miRNAs were confirmed to target the 3′ UTR of TBX1. miR-96-5p represses Tbx1 expression and, in turn, TBX1 suppresses the promoter activity and expression of miR-96 [48]. miR-451a, a tumor suppressor, also directly targets TBX1 [49]. The expression of this gene is upregulated in cutaneous basal cell carcinoma, inversely to miR-451a [49]. miR-17-92 fine-tunes the expression of Tbx1 in craniofacial development, suggesting miR-17-92 as a candidate genetic modifier for Tbx1 [50]. Thus, miRNAs both inside and outside the 22q11.2 locus may influence the severity of the clinical phenotypes of DGS/VCFS.
4. Craniofacial Phenotypes of DGS/VCFS Mouse Models
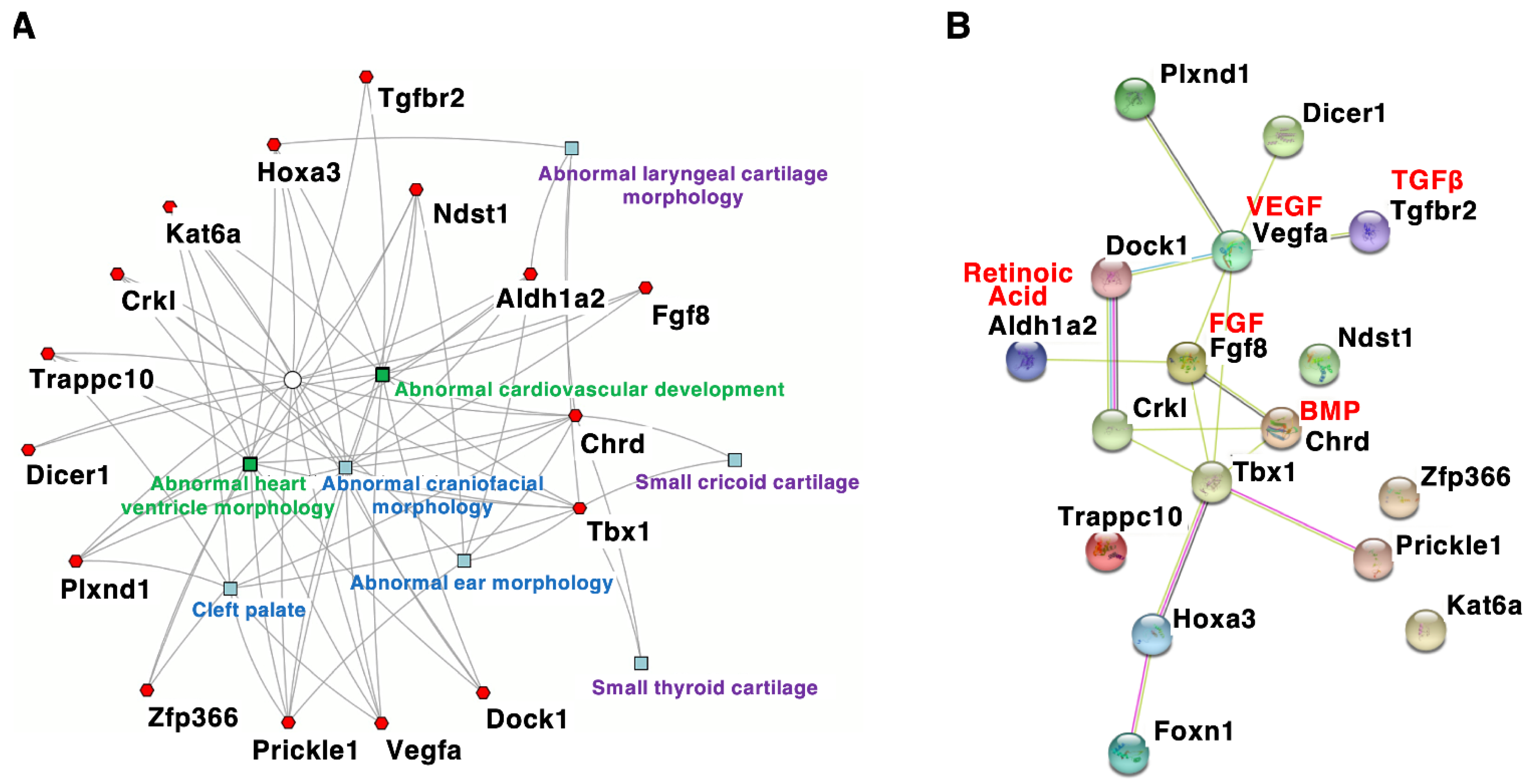
Mouse models with DGS/VCFS help identify additional candidate genes or modifier genes that influence the penetrance and/or severity of DGS/VCFS-related phenotypes. According to the mouse genome informatics (MGI) database (http://www.informatics.jax.org accessed on 3 August 2021), DGS/VCFS-related anomalies concerning Tbx1, Chrd, Tgfbr2, Vegfa, Fgf8, Crkl, Aldh1a2/Raldh2, Hoxa3, Kat6a/Moz/Myst3, Dicer1, Plxnd1, Dock1, Ndst1, Prickle1, Trappc10, Zfp366, and Foxn1 have been reported in genetically altered mice (Table 4 and Table S2). When these genes were analyzed according to biological process, “heart morphogenesis” and “cranial skeletal system development” were enriched (Table S3). Our enrichment analysis using ToppCluster [51] indicated that genes associated with DGS/VCFS phenotypes in mice are specifically enriched in the morphogenesis of craniofacial tissues and heart (Figure 2A). Interestingly, among these genes, only Tbx1 and Chrd were specifically enriched in the morphogenesis of cricoid and thyroid cartilages (Figure 2A). Genes associated with DGS/VCFS phenotypes in mice also indicated that DGS/VCFS-related phenotypes involve the interaction of several signaling pathways, including bone morphogenetic protein (BMP), transforming growth factor (TGF)β, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and retinoic acid signaling pathways (Figure 2B). Genes involved in the genetic pathway of Tbx1 are likely to induce phenotypes similar to Tbx1-null mice (Figure 2B, Table 4 and Table S2). These are described below.
4.1. Tbx1
Craniofacial structures with DGS/VCFS phenotypes are derivatives of the head mesenchyme and the first and second pharyngeal arches [62]. Tbx1 is expressed in the mesoderm, ectoderm, and endoderm of the pharyngeal apparatus and head mesenchyme between embryonic day (E)9.5 and E11.5 in mice [62,63]. At E12.5, Tbx1 is expressed in the oral epithelium, the myogenic core of the tongue, incisor tooth buds, pharyngeal muscles, and otic vesicle epithelium [63,64]. Tbx1-null mice exhibit the most clinical features of DGS/VCFS, while Tbx1+/− mice exhibit no significant craniofacial phenotypes (Table 2, Table 5 and Table S2). Information about ocular phenotypes in Tbx1-mutant mice is limited (Table 2), although these anomalies in patients with DGS/VCFS have been reported [16,17]. The Cre/loxP system has been used with Tbx1 conditional knockout mice to examine the tissue-specific function of TBX1 in craniofacial development (Table 5).
4.1.1. Cleft Palate
During palatogenesis, the palatal shelves develop bilaterally from the internal parts of the maxillary prominences and fuse above the tongue to form an intact oral cavity roof [67,68]. Because the palate consists of a bone-lined hard palate and a bone-free soft palate, cleft palate phenotypes include incomplete and submucosal cleft palates [67,68]. Ablation of Tbx1, which is expressed in the epithelium of the palatal shelves, results in abnormal intraoral epithelial fusions between the palatal shelves and the mandible, resulting in various degrees of the cleft palate phenotype (complete, incomplete, and submucosal cleft palate) [30,34,69]. Expression of Pax9, whose mutations lead to cleft palate and tooth agenesis [70], is downregulated in the palatal shelves and pharyngeal region of Tbx1-null embryos [34,71]. In Tbx1-null palatal shelves, muscle- and bone-related genes are downregulated, whereas neuron- and collagen biosynthesis-related genes are upregulated [72].
4.1.2. Abnormalities in Craniofacial Bones
Tbx1-null mice display craniofacial bone abnormalities, including persistently open fontanelles, micrognathia, a short clavicle, a hypoplastic zygomatic arch, and the absence of the hyoid bone (Table 2 and Table S2). Conditional deletion of Tbx1 in the mesoderm or osteochondral progenitors recapitulates the calvarial and mandibular phenotypes of Tbx1-null mice [35,66], suggesting that Tbx1 is required for morphogenesis and ossification of craniofacial bones. Although Tbx1 expression has not been reported in the neural crest, conditional deletion of Tbx1 here results in a hypoplastic hyoid bone [35] (Table 2 and Table 5). These results indicate that Tbx1 is required for the morphogenesis and ossification of mesoderm- and neural crest-derived membranous bones, although malformations observed in most neural crest-derived bones of Tbx1-null mice are secondary defects induced by non-neural crest cells [35,66]. Interestingly, abnormalities in membranous bones observed in Tbx1-null mice are similar to those of cleidocranial dysplasia (OMIM #119600 and #216330) in humans, exhibiting hypoplastic membranous bones, including abnormal neurocranial morphology, a short clavicle, a hypoplastic zygomatic arch, and hyoid bone [73,74,75]. Cleidocranial dysplasia (OMIM #119600) is caused by heterozygous mutations in RUNX2, which encodes a master transcription factor for osteoblast differentiation [74,75]. Since ablation of Tbx1 affects Runx2 expression in calvarial bones, and TBX1 overexpression induces Runx2 expression in vitro [35], TBX1 may act upstream of Runx2 by maintaining cell populations that express Runx2 at the onset of bone development. In addition, TBX1 could be a candidate gene for recessive inheritance of cleidocranial dysplasia (OMIM #216330).
4.1.3. Abnormalities in the Cranial Base and Cervical Spine
The spheno-occipital synchondrosis (SOS) in the cranial base is a vital growth center for the skull (reviewed in [76]). TBX1 is expressed in the mesoderm-derived cartilage primordium of the SOS and basioccipital bones, and Tbx1 deletion in the mesoderm induces malformed basioccipital bones and precocious ossified SOS. This indicates that Tbx1 is an essential regulator of chondrocyte differentiation and subsequent ossification at the SOS [36]. TBX1 inhibits the transcriptional activity of RUNX2 in vitro as well as the expression of RUNX2 target genes in SOS [36]. Tbx1-null mice also exhibit endochondral bone abnormalities in the atlas, axis, and xiphoid process [6,35]. There is potential to examine the phenotypes of cranial synchondroses in DGS/VCFS patients, as abnormalities in the SOS and basioccipital bones may induce cranial phenotypes of DGS/VCFS, such as dolichocephaly, basilar impression, and platybasia.
4.1.4. Dental Anomalies
Dental abnormalities (single central incisors, enamel hypoplasia, and small teeth) have been reported in many patients [18,28]. Accordingly, in approximately 30% of Tbx1-null mice, the upper incisors are absent [6]. Tbx1 is expressed in the cervical loops, which contain the dental stem cell niche in mice. The cervical loop region of the incisor is either severely reduced or completely absent in Tbx1-null mice, and cultured incisors of Tbx1-null mice are hypoplastic and lack enamel [77]. Ablation of Tbx1 in the epithelium results in smaller teeth than in the wild type, suggesting that TBX1 regulates the proliferation of dental progenitor cells [48].
4.1.5. Muscle Hypotonia
Branchiomeric muscles are derived from the mesoderm of the pharyngeal arch. In Tbx1-null and Tbx1flox/-;Mesp1-Cre embryos, the masseter, pterygoid, and temporalis muscles are intermittently absent [78,79]. Accordingly, muscle-related genes are also downregulated in Tbx1-null palatal shelves [72]. Tbx1 acts upstream of critical transcription factors to form branchiomeric muscles. These include LIM homeobox protein 2 (Lhx2), transcription factor 21 (Tcf21/capsulin), musculin (Msc), myogenic factor 5 (Myf5), myogenic differentiation 1 (Myod1), myocyte enhancer factor 2C (Mef2c), and GATA binding protein 4 (Gata4) [79,80,81,82]. Tbx1 is in the downstream genetic pathways of Tcf21, paired-like homeodomain transcription factor 2 (Pitx2), and ISL LIM homeobox 1 (Isl1) [80,83,84]. Thus, TBX1 regulates the pattern and development of branchiomeric muscles through the transcriptional regulation of myogenic genes.
4.2. Chordin (Chrd) and Transforming Growth Factor, Beta Receptor II (Tgfbr2)
Mice lacking the Chrd gene encoding chordin, an antagonist of bone morphogenetic proteins (BMPs), exhibit recapitulating phenotypes in Tbx1-null mice [32,52] (Table S2). Chrd-null neonates exhibit most craniofacial phenotypes in the cranium, cranial base, maxilla, mandible, ears, and hyoid bone (Table S2). Both Tbx1 and Fgf8 were reduced in the endoderm of Chrd-null mice, indicating that Chrd acts upstream of Tbx1 and Fgf8 [52]. Tbx1 acts upstream of SMAD family member 7 (Smad7), an inhibitory Smad within the BMP/TGFβ pathway, to regulate vascular smooth muscle and extracellular matrix investment of the fourth arch artery [85]. Conditional deletion of Tgfbr2, which encodes TGFβ receptor 2, in the neural crest resulted in DGS/VCFS-related cardiovascular defects [53]. These findings suggest a potential role of BMP/TGFβ signaling in the pathogenesis of DGS/VCFS.
4.3. Vascular Endothelial Growth Factor A (Vegfa)
VEGFA is an essential cytokine in angiogenesis and vascular development during embryogenesis [86]. Vegfa-null neonates exhibit a few aspects of DGS/VCFS-related craniofacial anomalies, including unfused cranial sutures, absent incisors, and short mandibles, as well as cardiovascular abnormalities [54] (Table S2). The deletion of Vegfa in mice reduces Tbx1 expression, and the knockdown of vegfaa/vegfa levels in zebrafish enhances the pharyngeal arch malformations induced by tbx1 knockdown [54]. In humans, low expression of the VEGFA haplotype increases the risk of a cardiac phenotype of DGS/VCFS, indicating that expression levels of VEGFA affect the severity of DGS/VCFS phenotypes [87]. These results suggest that VEGFA modifies DGS/VCFS-related phenotypes by regulating TBX1 expression.
4.4. Fibroblast Growth Factor 8 (Fgf8) and FGF Receptor 2 (Fgfr2)
Ablation of Fgf8 induces craniofacial, cardiovascular, thymic, and parathyroid phenotypes [55,88]. Fgf8-null neonates exhibit a few aspects of DGS/VCFS-related craniofacial anomalies, including cleft palate and abnormal outer ear morphology [55,88] (Table S2). Fgf8+/−;Tbx1+/− double heterozygous embryos show an increased penetrance of cardiovascular defects compared with Tbx1-heterozygous embryos [89]. Tissue-specific deletion of Fgf8 in Tbx1-expressing domains results in cardiovascular anomalies [90]. TBX1 activates the Fgf8 enhancer during cardiac development [9]. Deletion of the Fgfr2 gene that encodes FGF receptor 2 decreases Tbx1 expression in the dental epithelium, indicating a genetic link between FGF signaling and Tbx1 in tooth development [91]. In addition, a Tbx1-Six1/Eya1-Fgf8 genetic pathway is crucial for craniofacial morphogenesis [92,93]. These findings demonstrate that the FGF pathway and Tbx1 interact genetically during pharyngeal arch development.
4.5. CRK like Proto-Oncogene, Adaptor Protein (Crkl)
CRKL maps to the 2.5 Mb region commonly deleted in DGS/VCFS (Figure 1). Variants in a predicted enhancer of CRKL are significantly associated with the risk of congenital heart defects in DGS/VCFS [94]. Approximately 12% of Crkl-null mice show mild cranial bone defects, such as small cranium and poor membranous ossification of the nasal bones [56]. Compound heterozygosity of Crkl and Tbx1 in mice has revealed that Crkl deletion enhances DGS/VCFS-related abnormalities compared with Tbx1-heterozygous embryos [56], suggesting that Tbx1 and Crkl genes act in the same genetic pathway. CRKL encodes an adaptor protein that promotes the intracellular response of FGF signaling. Crkl+/−;Fgf8+/− double heterozygous mice showed DGS/VCFS-related defects [95]. Thus, CRKL mutations cause or modify DGS/VCFS-related phenotypes and/or penetrance as a contiguous gene syndrome.
4.6. Aldehyde Dehydrogenase Family 1, Subfamily A2 (Aldh1a2/Raldh2)
Retinoic acid (RA), an active vitamin A derivative, is essential for various developmental processes in vertebrates. High levels of RA act as morphogens that cause phenocopies of DGS/VCFS by downregulating Tbx1 expression in the pharyngeal apparatus [96,97]. RA levels are balanced by the RA-synthesizing enzyme aldehyde dehydrogenase (ALDH) and the Cyp26 RA-catabolizing enzyme [98,99]. Mouse embryos hypomorphic for Aldh1a2/Raldh2 display DGS/VCFS-related cardiovascular, thymic, and parathyroid malformations [57]. Haploinsufficiency of Aldh1a2/Raldh2 results in reduced embryonic synthesis of RA, increased levels of Tbx1, and accelerated recovery from arterial growth delay in Tbx1-heterozygous mice [100]. An inhibitor of the Cyp26 enzyme induces a phenocopy of DGS/VCFS in chick embryos [101]. In Tbx1-null mice, upregulated expression of Aldh1a2/Raldh2 and downregulated expression of Cyp26a1 have been observed [71].
Further interactions occur between RA signaling, Crkl, and Tbx1. The penetrance of thymic hypoplasia is reduced in Crkl+/−;Tbx1+/−;Aldh1a2+/− triple heterozygous embryos compared to Crkl+/−;Tbx1+/− mutants, suggesting that reducing the amount of RA may rescue the DGS/VCFS-related phenotype [102]. Thus, the levels of RA in embryogenesis could contribute to the phenotypic variability of DGS/VCFS.
4.7. Homeobox A3 (Hoxa3)
RA exposure increases the expression of Hoxa3, a gene which encodes a homeobox transcription factor, in the neural tube and pharyngeal apparatus [103]. Interestingly, Hoxa3-null neonates show some aspects of the abnormalities of DGS/VCFS [58,104] (Table S2). Thus, HOXA3 may be a genetic modifier of DGS/VCFS-related abnormalities.
4.8. Kat6a/Moz/Myst3 (Lysine Acetyltransferase 6A) and Epigenetic Modifiers
Homozygous mutation of Kat6a/Moz/Myst3, which encodes a histone acetyltransferase, leads to cardiovascular defects seen in DGS/VCFS and reduces Tbx1 expression [59]. Treatment of pregnant mice with a histone demethylase inhibitor reportedly increased the methylation levels of histone H3 lysine K4 (H3K4) and partially rescued the cardiovascular phenotypes of Tbx1-heterozygous mice [105]. TBX1 regulates genes transcribed at a low level by recruiting lysine methyltransferase (KMT2C) and controlling monomethylation of H3K4 (H3K4me1) enrichment on chromatin [105]. In addition, TBX1 transcriptionally targets Wnt5a by interacting with SMARCD1/BAF60a, a component of the SWI/SNF-like BAF chromatin remodeling complex, along with the H3K4 monomethyltransferase SETD7 [106]. Microduplication in KANSL1, which encodes a member of the histone acetyltransferase complex, is associated with heart anomalies in individuals with DGS/VCFS [107]. In T cells of patients with DGS/VCFS, the status of transcriptional activation (H3K4me3 and H3K27ac) is globally increased [108]. Thus, epigenetic changes are involved in DGS/VCFS-related phenotypes.
4.9. Sonic Hedgehog (Shh)
Shh encodes an SHH signaling molecule. In humans, SHH mutations lead to holoprosencephaly 3 (OMIM #142945), microphthalmia with coloboma (OMIM #611638), and single median maxillary central incisor (OMIM #147250). Shh-null embryos exhibit conotruncal and pharyngeal arch artery defects similar to those observed in DGS/VCFS and Tbx1-null embryos [109]. Tbx1 expression is reduced in Shh-null embryos, and ectopic expression of Shh can result in the upregulation of Tbx1, suggesting that Shh is a possible modifier for DGS/VCFS [62,110]. Shh is also required for the expression of the Fox family of transcription factor genes, forkhead box A2 (Foxa2) and forkhead box C2 (Foxc2), in the head mesenchyme and the pharyngeal endoderm [62]. FOXA2 and FOXC2 bind to regulatory regions in the mouse and human TBX1 loci [111].
4.10. Paired-like Homeodomain Transcription Factor 2 (Pitx2)
Pitx2 gene encodes a bicoid-like homeodomain transcription factor. Pitx2-null mice show craniofacial defects, such as the arrest of tooth development, abnormal morphology of maxilla and mandible, and cleft palate. In humans, PITX2 mutations lead to Axenfeld–Rieger syndrome, type 1 (OMIM #180500). Patients with Axenfeld–Rieger syndrome manifest dental and craniofacial anomalies involving the maxilla, mandible, and cranial base [112]. Both Tbx1 and Pitx2 are expressed in the early dental epithelium, oral epithelium, and secondary heart field [64,113,114]. Tbx1+/−;Pitx2+/− double heterozygous embryos exhibit increased penetrance of an extra premolar-like tooth [115] and DGS/VCFS-related cardiovascular anomalies [114]. TBX1 directly activates the Pitx2c enhancer through the synergistic action of the homeobox-containing transcription factor NK2 homeobox 5 (NKX2-5) [114]. TBX1 also interacts with PITX2 and represses PITX2 transcriptional activity [48,115]. Thus, PITX2 may be a genetic modifier of DGS/VCFS-related abnormalities.
5. Discussion
The penetrance and severity of congenital anomalies are related to genetic and environmental factors. Recent studies have revealed the function of TBX1 and modifiers that impact the severity and penetrance of DGS/VCFS. Studies of DGS/VCFS mouse models have provided insights into signaling pathways and genes that interact with TBX1 and/or affect the DGS/VCFS phenotypes. In addition, mouse models with DGS/VCFS may help us to identify additional DGS/VCFS-related phenotypes. For example, there is potential to examine the phenotypes of cranial synchondroses, cranium, zygomatic arches, and pharyngeal muscles in DGS/VCFS patients. We also noted that information about ocular phenotypes in Tbx1-mutant mice is limited, although these anomalies in patients with DGS/VCFS have been reported [16,17]. Crosstalk with key embryonic signals, especially BMP, TGFβ, VEGFA, FGF, RA, and SHH, critically regulates DGS/VCFS-related pharyngeal development. Genes involved in these signaling pathways may modify the phenotypic spectrum of DGS/VCFS. Given the broad spectrum of DGS/VCFS disease phenotypes, other genes essential to craniofacial development could modify the phenotypic spectrum. Genetically engineered mice are useful for studying disease phenotypes; however, ablation of essential genes involved in cardiovascular development may cause early embryonic lethality, which would prevent observation of craniofacial phenotypes. For example, ablation of Ufd1, whose human ortholog has been mapped to the 1.5 Mb region, causes early embryonic lethality before organogenesis in mice [116]. It is also essential to identify novel proteins that interact with TBX1 and examine whether interacting partners may influence the phenotypes of mouse models.
6. Conclusions
Studies of Tbx1-mutant mice have provided insights into the underlying pathogenesis of DGS/VCFS and the knowledge to diagnose patients with DGS/VCFS. Genes, miRNAs, and epigenetics could change Tbx1 expression. Polymorphisms, variations, and mutations in TBX1 may induce the penetrance and severity of DGS/VCFS-like craniofacial phenotypes. The molecular basis of the variant sequence of TBX1 will further define how TBX1 contributes to the craniofacial and other phenotypes of DGS/VCFS. Since interactions with TBX1 and other molecules in transcriptional complexes or chromatin remodeling are crucial for TBX1 function, identifying and understanding these genetic and epigenetic modifiers individually for each patient may direct therapeutics to minimize the severity.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/jdb10020018/s1, Figure S1: Craniofacial and skeletal phenotypes of DGS/VCFS. Figure S2: Human genes in the proximal deletion of 1.5 Mb on the 22q11.2 locus. Table S1: Craniofacial and skeletal phenotypes of DGS/VCFS and Tbx1-null mice. Table S2: Craniofacial and skeletal phenotypes in mouse models of DGS/VCFS. Table S3: Classification of mouse genes associated with DGS/VCFS.
Author Contributions
N.F. contributed to the conceptual idea, performed the database searches, analyzed the data, and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI [20K09901].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We would like to thank Hiroshi Kurosaka, Cedric Boeckx, and Mizuki Funato for a critical reading of the manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- Tézenas Du Montcel, S.; Mendizabai, H.; Aymé, S.; Lévy, A.; Philip, N. Prevalence of 22q11 microdeletion. J. Med. Genet. 1996, 33, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Rivera, E.; Liu, Y.P.; Verbitsky, M.; Anderson, B.R.; Capone, V.P.; Otto, E.A.; Yan, Z.; Mitrotti, A.; Martino, J.; Steers, N.J.; et al. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N. Engl. J. Med. 2017, 376, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; de la Morena, M.T.; van Oers, N.S.C. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front. Genet. 2019, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J.; Cohen, M.M., Jr.; Hennekam, R.C.M. Syndromes of the Head and the Neck; Oxford University Press: New York, NY, USA, 2001; pp. 850–853. [Google Scholar]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286–291. [Google Scholar] [CrossRef]
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101. [Google Scholar] [CrossRef]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Yamagishi, H.; Maeda, J.; McAnally, J.; Yamagishi, C.; Srivastava, D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development 2004, 131, 5491–5502. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Kochilas, L.; Nowotschin, S.; Arnold, J.S.; Aggarwal, V.S.; Epstein, J.A.; Brown, M.C.; Adams, J.; Morrow, B.E. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum. Mol. Genet. 2004, 13, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Baldini, A. Dissecting contiguous gene defects: TBX1. Curr. Opin. Genet. Dev. 2005, 15, 279–284. [Google Scholar] [CrossRef]
- Aggarwal, V.S.; Morrow, B.E. Genetic modifiers of the physical malformations in velo-cardio-facial syndrome/DiGeorge syndrome. Dev. Disabil. Res. Rev. 2008, 14, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papangeli, I.; Scambler, P. The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Heliovaara, A.; Ranta, R.; Rautio, J. Pharyngeal morphology in children with submucous cleft palate with and without surgery. Eur. Arch. Oto-Rhino-Laryngol. Head Neck 2005, 262, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Heliövaara, A.; Hurmerinta, K. Craniofacial cephalometric morphology in children with CATCH 22 syndrome. Orthod. Craniofac. Res. 2006, 9, 186–192. [Google Scholar] [CrossRef]
- Ryan, A.K.; Goodship, J.A.; Wilson, D.I.; Philip, N.; Levy, A.; Seidel, H.; Schuffenhauer, S.; Oechsler, H.; Belohradsky, B.; Prieur, M.; et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: A European collaborative study. J. Med. Genet. 1997, 34, 798–804. [Google Scholar] [CrossRef] [Green Version]
- McDonald-McGinn, D.M.; Kirschner, R.; Goldmuntz, E.; Sullivan, K.; Eicher, P.; Gerdes, M.; Moss, E.; Solot, C.; Wang, P.; Jacobs, I.; et al. The Philadelphia story: The 22q11.2 deletion: Report on 250 patients. Genet. Couns. 1999, 10, 11–24. [Google Scholar]
- Klingberg, G.; Oskarsdóttir, S.; Johannesson, E.L.; Norén, J.G. Oral manifestations in 22q11 deletion syndrome. Int. J. Paediatr. Dent. 2002, 12, 14–23. [Google Scholar]
- Ricchetti, E.T.; States, L.; Hosalkar, H.S.; Tamai, J.; Maisenbacher, M.; McDonald-McGinn, D.M.; Zackai, E.H.; Drummond, D.S. Radiographic study of the upper cervical spine in the 22q11.2 deletion syndrome. J. Bone Joint Surg. Am. 2004, 86, 1751–1760. [Google Scholar] [CrossRef]
- Herman, S.B.; Guo, T.; McGinn, D.M.M.; Bolsa, A.; Shanske, A.L.; Bassett, A.S.; Chow, E.W.C.; Bowser, M.; Sheridan, M.; Beemer, F.; et al. Overt cleft palate phenotype and TBX1 genotype correlations in velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Am. J. Med. Genet. A 2012, 158A, 2781–2787. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, M.; Nabi, S.; Husein, M.; Mohamed, M.E.; Tay, K.Y.; McKillop, S. Cervical spine abnormalities in 22q11.2 deletion syndrome. Cleft Palate-Craniofacial J. 2014, 51, 230–233. [Google Scholar] [CrossRef]
- Jackson, O.; Crowley, T.B.; Sharkus, R.; Smith, R.; Jeong, S.; Solot, C.; McDonald-Mcginn, D. Palatal evaluation and treatment in 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 2019, 179, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.S.; Chow, E.W.C.; Husted, J.; Weksberg, R.; Caluseriu, O.; Webb, G.D.; Gatzoulis, M.A. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am. J. Med. Genet. A 2005, 138, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loos, E.; Verhaert, N.; Willaert, A.; Devriendt, K.; Swillen, A.; Hermans, R.; Op de Beeck, K.; Hens, G. Malformations of the middle and inner ear on CT imaging in 22q11 deletion syndrome. Am. J. Med. Genet. Part A 2016, 170, 2975–2983. [Google Scholar] [CrossRef] [PubMed]
- Verheij, E.; Kist, A.L.; Mink van der Molen, A.B.; Stegeman, I.; van Zanten, G.A.; Grolman, W.; Thomeer, H.G.X.M. Otologic and audiologic findings in 22q11.2 deletion syndrome. Eur. Arch. Oto-Rhino-Laryngol. Head Neck 2017, 274, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Ford, L.C.; Sulprizio, S.L.; Rasgon, B.M. Otolaryngological manifestations of velocardiofacial syndrome: A retrospective review of 35 patients. Laryngoscope 2000, 110, 362–367. [Google Scholar] [CrossRef]
- Kobrynski, L.J.; Sullivan, K.E. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes. Lancet 2007, 370, 1443–1452. [Google Scholar] [CrossRef]
- Oberoi, S.; Vargervik, K. Velocardiofacial syndrome with single central incisor. Am. J. Med. Genet. A 2005, 132A, 194–197. [Google Scholar] [CrossRef]
- Zhang, Z.; Huynh, T.; Baldini, A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development 2006, 133, 3587–3595. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.S.; Werling, U.; Braunstein, E.M.; Liao, J.; Nowotschin, S.; Edelmann, W.; Hebert, J.M.; Morrow, B.E. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development 2006, 133, 977–987. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.S.; Braunstein, E.M.; Ohyama, T.; Groves, A.K.; Adams, J.C.; Brown, M.C.; Morrow, B.E. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum. Mol. Genet. 2006, 15, 1629–1639. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Klingensmith, J. Chordin is a modifier of tbx1 for the craniofacial malformations of 22q11 deletion syndrome phenotypes in mouse. PLoS Genet. 2009, 5, e1000395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, F.; Novoa, A.; Jerome-Majewska, L.A.; Papaioannou, V.E.; Mallo, M. Tbx1 is required for proper neural crest migration and to stabilize spatial patterns during middle and inner ear development. Mech. Dev. 2005, 122, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum. Mol. Genet. 2012, 21, 2524–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Hum. Mol. Genet. 2015, 24, 424–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, N.; Srivastava, D.; Shibata, S.; Yanagisawa, H. TBX1 Regulates Chondrocyte Maturation in the Spheno-occipital Synchondrosis. J. Dent. Res. 2020, 99, 1182–1191. [Google Scholar] [CrossRef]
- Solot, C.B.; Sell, D.; Mayne, A.; Baylis, A.L.; Persson, C.; Jackson, O.; McDonald-McGinn, D.M. Speech-Language Disorders in 22q11.2 Deletion Syndrome: Best Practices for Diagnosis and Management. Am. J. Speech-Lang. Pathol. 2019, 28, 984–999. [Google Scholar] [CrossRef] [Green Version]
- Jaouadi, A.; Tabebi, M.; Abdelhedi, F.; Abid, D.; Kamoun, F.; Chabchoub, I.; Maatoug, S.; Doukali, H.; Belghuith, N.; Ksentini, M.A.; et al. A novel TBX1 missense mutation in patients with syndromic congenital heart defects. Biochem. Biophys. Res. Commun. 2018, 499, 563–569. [Google Scholar] [CrossRef]
- Zweier, C.; Sticht, H.; Aydin-Yaylagül, I.; Campbell, C.E.; Rauch, A. Human TBX1 Missense Mutations Cause Gain of Function Resulting in the Same Phenotype as 22q11.2 Deletions. Am. J. Hum. Genet. 2007, 80, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Tanaka, H.; Higuchi, Y.; Hayashi, Y.; Kobayashi, K.; Tsukahara, H. Novel heterozygous mutation in TBX1 in an infant with hypocalcemic seizures. Clin. Pediatr. Endocrinol. 2018, 27, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Alghamdi, M.; Al Khalifah, R.; Al Homyani, D.K.; Alkhamis, W.H.; Arold, S.T.; Ekhzaimy, A.; El-Wetidy, M.; Kashour, T.; Halwani, R. A novel TBX1 variant causing hypoparathyroidism and deafness. J. Endocr. Soc. 2020, 4, bvz028. [Google Scholar] [CrossRef]
- Ogata, T.; Niihori, T.; Tanaka, N.; Kawai, M.; Nagashima, T.; Funayama, R.; Nakayama, K.; Nakashima, S.; Kato, F.; Fukami, M.; et al. TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia. PLoS ONE 2014, 9, e91598. [Google Scholar] [CrossRef] [PubMed]
- Paylor, R.; Glaser, B.; Mupo, A.; Ataliotis, P.; Spencer, C.; Sobotka, A.; Sparks, C.; Choi, C.H.; Oghalai, J.; Curran, S.; et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 7729–7734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, R.; Hofbeck, M.; Zweier, C.; Koch, A.; Zink, S.; Trautmann, U.; Hoyer, J.; Kaulitz, R.; Singer, H.; Rauch, A. Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. J. Med. Genet. 2010, 47, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.-S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Edelmann, L.; Stankiewicz, P.; Spiteri, E.; Pandita, R.K.; Shaffer, L.; Lupski, J.R.; Morrow, B.E.; Lupski, J. Two functional copies of the DGCR6 gene are present on human chromosome 22q11 due to a duplication of an ancestral locus. Genome Res. 2001, 11, 208–217. [Google Scholar] [CrossRef]
- Hierck, B.P.; Molin, D.G.M.; Boot, M.J.; Poelmann, R.E.; Gittenberger-De Groot, A.C. A chicken model for DGCR6 as a modifier gene in the DiGeorge critical region. Pediatr. Res. 2004, 56, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Moreno, M.; Eliason, S.; Cao, H.; Li, X.; Yu, W.; Bidlack, F.B.; Margolis, H.C.; Baldini, A.; Amendt, B.A. TBX1 protein interactions and microRNA-96-5p regulation controls cell proliferation during craniofacial and dental development: Implications for 22q11.2 deletion syndrome. Hum. Mol. Genet. 2015, 24, 2330–2348. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Jiang, P. MicroRNA-451a acts as tumor suppressor in cutaneous basal cell carcinoma. Mol. Genet. Genom. Med. 2018, 6, 1001–1009. [Google Scholar] [CrossRef]
- Wang, J.; Bai, Y.; Li, H.; Greene, S.B.; Klysik, E.; Yu, W.; Schwartz, R.J.; Williams, T.J.; Martin, J.F. MicroRNA-17-92, a direct Ap-2alpha transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet. 2013, 9, e1003785. [Google Scholar] [CrossRef]
- Kaimal, V.; Bardes, E.E.; Tabar, S.C.; Jegga, A.G.; Aronow, B.J. ToppCluster: A multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010, 38, W96-102. [Google Scholar] [CrossRef] [Green Version]
- Bachiller, D.; Klingensmith, J.; Shneyder, N.; Tran, U.; Anderson, R.; Rossant, J.; De Robertis, E.M. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development 2003, 130, 3567–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurdak, H.; Ittner, L.M.; Lang, K.S.; Leveen, P.; Suter, U.; Fischer, J.A.; Karlsson, S.; Born, W.; Sommer, L. Inactivation of TGFβ signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005, 19, 530–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stalmans, I.; Lambrechts, D.; De Smet, F.; Jansen, S.; Wang, J.; Maity, S.; Kneer, P.; von der Ohe, M.; Swillen, A.; Maes, C.; et al. VEGF: A modifier of the del22q11 (DiGeorge) syndrome? Nat. Med. 2003, 9, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Abu-Issa, R.; Smyth, G.; Smoak, I.; Yamamura, K.; Meyers, E.N. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 2002, 129, 4613–4625. [Google Scholar] [CrossRef]
- Guris, D.L.; Fantes, J.; Tara, D.; Druker, B.J.; Imamoto, A. Mice lacking the homologue of the human 22q11.2 gene CRLK phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 2001, 27, 293–298. [Google Scholar] [CrossRef]
- Vermot, J.; Niederreither, K.; Garnier, J.M.; Chambon, P.; Dollé, P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1763–1768. [Google Scholar] [CrossRef] [Green Version]
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature 1991, 350, 473–479. [Google Scholar] [CrossRef]
- Voss, A.K.; Vanyai, H.K.; Collin, C.; Dixon, M.P.; McLennan, T.J.; Sheikh, B.N.; Scambler, P.; Thomas, T. MOZ Regulates the Tbx1 Locus, and Moz Mutation Partially Phenocopies DiGeorge Syndrome. Dev. Cell 2012, 23, 652–663. [Google Scholar] [CrossRef] [Green Version]
- Sheehy, N.T.; Cordes, K.R.; White, M.P.; Ivey, K.N.; Srivastava, D. The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development 2010, 137, 4307–4316. [Google Scholar] [CrossRef] [Green Version]
- Gershwin, M.E. DiGeorge syndrome: Congenital thymic hypoplasia. Animal model: Congenitally athymic (nude) mouse. Am. J. Pathol. 1977, 89, 809–812. [Google Scholar]
- Yamagishi, H.; Maeda, J.; Hu, T.; McAnally, J.; Conway, S.J.; Kume, T.; Meyers, E.N.; Yamagishi, C.; Srivastava, D. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 2003, 17, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, D.L.; Garvey, N.; Hancock, S.; Alexiou, M.; Agulnik, S.I.; Gibson-Brown, J.J.; Cebra-Thomas, J.; Bollag, R.J.; Silver, L.M.; Papaioannou, V.E. Expression of the T-box family genes, Tbx1-Tbx5, during early mouse development. Dev. Dyn. 1996, 206, 379–390. [Google Scholar] [CrossRef]
- Zoupa, M.; Seppala, M.; Mitsiadis, T.; Cobourne, M.T. Tbx1 is expressed at multiple sites of epithelial-mesenchymal interaction during early development of the facial complex. Int. J. Dev. Biol. 2006, 50, 504–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Cerrato, F.; Xu, H.; Vitelli, F.; Morishima, M.; Vincentz, J.; Furuta, Y.; Ma, L.; Martin, J.F.; Baldini, A.; et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development 2005, 132, 5307–5315. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, V.S.; Carpenter, C.; Freyer, L.; Liao, J.; Petti, M.; Morrow, B.E. Mesodermal Tbx1 is required for patterning the proximal mandible in mice. Dev. Biol. 2010, 344, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Bush, J.O.; Jiang, R. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development 2012, 139, 828. [Google Scholar] [CrossRef] [Green Version]
- Funato, N. Molecular basis of cleft palates in mice. World J. Biol. Chem. 2015, 6, 121. [Google Scholar] [CrossRef]
- Goudy, S.; Law, A.; Sanchez, G.; Baldwin, H.S.; Brown, C. Tbx1 is necessary for palatal elongation and elevation. Mech. Dev. 2010, 127, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Peters, H.; Neubuser, A.; Kratochwil, K.; Balling, R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998, 12, 2735–2747. [Google Scholar] [CrossRef] [Green Version]
- Ivins, S.; Van Beuren, K.L.; Roberts, C.; James, C.; Lindsay, E.; Baldini, A.; Ataliotis, P.; Scambler, P.J. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Dev. Biol. 2005, 285, 554–569. [Google Scholar] [CrossRef] [Green Version]
- Funato, N.; Yanagisawa, H. Deletion of the T-box transcription factor gene, Tbx1, in mice induces differential expression of genes associated with cleft palate in humans. Arch. Oral Biol. 2018, 95, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.; Beddington, R.S.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Mundlos, S.; Otto, F.; Mundlos, C.; Mulliken, J.B.; Aylsworth, A.S.; Albright, S.; Lindhout, D.; Cole, W.G.; Henn, W.; Knoll, J.H.; et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997, 89, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Funato, N. New Insights Into Cranial Synchondrosis Development: A Mini Review. Front. Cell Dev. Biol. 2020, 8, 706. [Google Scholar] [CrossRef]
- Catón, J.; Luder, H.U.; Zoupa, M.; Bradman, M.; Bluteau, G.; Tucker, A.S.; Klein, O.; Mitsiadis, T.A. Enamel-free teeth: Tbx1 deletion affects amelogenesis in rodent incisors. Dev. Biol. 2009, 328, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.G.; Jerome-Majewska, L.A.; Papaioannou, V.E. The del22q11.2 candidate gene Tbx1 regulates branchiomeric myogenesis. Hum. Mol. Genet. 2004, 13, 2829–2840. [Google Scholar] [CrossRef]
- Kong, P.; Racedo, S.E.; Macchiarulo, S.; Hu, Z.; Carpenter, C.; Guo, T.; Wang, T.; Zheng, D.; Morrow, B.E. Tbx1 is required autonomously for cell survival and fate in the pharyngeal core mesoderm to form the muscles of mastication. Hum. Mol. Genet. 2014, 23, 4215–4231. [Google Scholar] [CrossRef] [Green Version]
- Harel, I.; Maezawa, Y.; Avraham, R.; Rinon, A.; Ma, H.Y.; Cross, J.W.; Leviatan, N.; Hegesh, J.; Roy, A.; Jacob-Hirsch, J.; et al. Pharyngeal mesoderm regulatory network controls cardiac and head muscle morphogenesis. Proc. Natl. Acad. Sci. USA. 2012, 109, 18839–18844. [Google Scholar] [CrossRef] [Green Version]
- Grifone, R.; Jarry, T.; Dandonneau, M.; Grenier, J.; Duprez, D.; Kelly, R.G. Properties of branchiomeric and somite-derived muscle development in Tbx1 mutant embryos. Dev. Dyn. 2008, 237, 3071–3078. [Google Scholar] [CrossRef]
- Pane, L.S.; Zhang, Z.; Ferrentino, R.; Huynh, T.; Cutillo, L.; Baldini, A. Tbx1 is a negative modulator of Mef2c. Hum. Mol. Genet. 2012, 21, 2485–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, F.; Sun, X.; Liu, W.; Ai, D.; Klysik, E.; Lu, M.F.; Hadley, J.; Antoni, L.; Chen, L.; Baldini, A.; et al. Pitx2 promotes development of splanchnic mesoderm-derived branchiomeric muscle. Development 2006, 133, 4891–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.P.; Gross, M.K.; Kioussi, C. Cranial muscle defects of Pitx2 mutants result from specification defects in the first branchial arch. Proc. Natl. Acad. Sci. USA 2007, 104, 5907–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papangeli, I.; Scambler, P.J. Tbx1 genetically interacts with the transforming growth factor-β/bone morphogenetic protein inhibitor Smad7 during great vessel remodeling. Circ. Res. 2013, 112, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Vieira, J.M.; Ruhrberg, C.; Schwarz, Q. VEGF receptor signaling in vertebrate development. Organogenesis 2010, 6, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Lambrechts, D.; Devriendt, K.; Driscoll, D.A.; Goldmuntz, E.; Gewillig, M.; Vlietinck, R.; Collen, D.; Carmeliet, P. Low expression VEGF haplotype increases the risk for tetralogy of Fallot: A family based association study. J. Med. Genet. 2005, 42, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.U.; Fotheringham, L.K.; Brewer, J.A.; Muglia, L.J.; Tristani-Firouzi, M.; Capecchi, M.R.; Moon, A.M. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development 2002, 129, 4591–4603. [Google Scholar] [CrossRef]
- Vitelli, F.; Taddei, I.; Morishima, M.; Meyers, E.N.; Lindsay, E.A.; Baldini, A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development 2002, 129, 4605–4611. [Google Scholar] [CrossRef]
- Brown, C.B.; Wenning, J.M.; Lu, M.M.; Epstein, D.J.; Meyers, E.N.; Epstein, J.A. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev. Biol. 2004, 267, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Mitsiadis, T.A.; Tucker, A.S.; De Bari, C.; Cobourne, M.T.; Rice, D.P.C. A regulatory relationship between Tbx1 and FGF signaling during tooth morphogenesis and ameloblast lineage determination. Dev. Biol. 2008, 320, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-Y.; Chen, W.-T.; Lee, H.-C.; Yang, P.-H.; Yang, H.-J.; Tsai, H.-J. The transcription factor Six1a plays an essential role in the craniofacial myogenesis of zebrafish. Dev. Biol. 2009, 331, 152–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Sun, Y.; Zhou, B.; Adam, R.M.; Li, X.; Pu, W.T.; Morrow, B.E.; Moon, A.; Li, X. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J. Clin. Investig. 2011, 121, 1585–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Diacou, A.; Johnston, H.R.; Musfee, F.I.; McDonald-McGinn, D.M.; McGinn, D.; Crowley, T.B.; Repetto, G.M.; Swillen, A.; Breckpot, J.; et al. Complete Sequence of the 22q11.2 Allele in 1,053 Subjects with 22q11.2 Deletion Syndrome Reveals Modifiers of Conotruncal Heart Defects. Am. J. Hum. Genet. 2020, 106, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Guris, D.L.; Seo, J.H.; Li, L.; Hammond, J.; Talbot, A.; Imamoto, A. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev. Cell 2006, 10, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, F.W.; Wilk, A.L.; Kelsey, F.O. Teratogen update: Vitamin A congeners. Teratology 1986, 33, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.; Ivins, S.M.; James, C.T.; Scambler, P.J. Retinoic acid down-regulatesTbx1 expression in vivo and in vitro. Dev. Dyn. 2005, 232, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Wendling, O.; Dennefeld, C.; Chambon, P.; Mark, M. Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development 2000, 127, 1553–1562. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Le Roux, I.; Schuhbaur, B.; Chambon, P.; Dollé, P. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development 2003, 130, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Ryckebüsch, L.; Bertrand, N.; Mesbah, K.; Bajolle, F.; Niederreither, K.; Kelly, R.G.; Zaffran, S. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circ. Res. 2010, 106, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.; Ivins, S.; Cook, A.C.; Baldini, A.; Scambler, P.J. Cyp26 genes a1, b1 and c1 are down-regulated in Tbx1 null mice and inhibition of Cyp26 enzyme function produces a phenocopy of DiGeorge Syndrome in the chick. Hum. Mol. Genet. 2006, 15, 3394–3410. [Google Scholar] [CrossRef] [Green Version]
- Guris, D.L.; Duester, G.; Papaioannou, V.E.; Imamoto, A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev. Cell 2006, 10, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, G.B.; Manley, N.; Maggio-Price, L. Retinoic acid-induced thymic abnormalities in the mouse are associated with altered pharyngeal morphology, thymocyte maturation defects, and altered expression of Hoxa3 and Pax1. Teratology 1998, 58, 263–275. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, P.; Wells, L.; Amemiya, C.T.; Condie, B.G.; Manley, N.R. Mouse and zebrafish Hoxa3 orthologues have nonequivalent in vivo protein function. Proc. Natl. Acad. Sci. USA 2010, 107, 10555–10560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulcoli, F.G.; Franzese, M.; Liu, X.; Zhang, Z.; Angelini, C.; Baldini, A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat. Commun. 2016, 7, 11688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Fulcoli, F.G.; Ferrentino, R.; Martucciello, S.; Illingworth, E.A.; Baldini, A. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 2012, 8, e1002571. [Google Scholar] [CrossRef] [Green Version]
- León, L.E.; Benavides, F.; Espinoza, K.; Vial, C.; Alvarez, P.; Palomares, M.; Lay-Son, G.; Miranda, M.; Repetto, G.M. Partial microduplication in the histone acetyltransferase complex member KANSL1 is associated with congenital heart defects in 22q11.2 microdeletion syndrome patients. Sci. Rep. 2017, 7, 1795. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, L.; Song, L.; Maurer, K.; Zhao, X.; Zackai, E.H.; McGinn, D.E.; Crowley, T.B.; McGinn, D.M.M.; Sullivan, K.E. Chromatin Modifications in 22q11.2 Deletion Syndrome. J. Clin. Immunol. 2021, 41, 1853–1864. [Google Scholar] [CrossRef]
- Washington Smoak, I.; Byrd, N.A.; Abu-Issa, R.; Goddeeris, M.M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E.N. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Garg, V.; Yamagishi, C.; Hu, T.; Kathiriya, I.S.; Yamagishi, H.; Srivastava, D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev. Biol. 2001, 235, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Kou, I.; Otomo, N.; Takeda, K.; Momozawa, Y.; Lu, H.-F.; Kubo, M.; Kamatani, Y.; Ogura, Y.; Takahashi, Y.; Nakajima, M.; et al. Genome-wide association study identifies 14 previously unreported susceptibility loci for adolescent idiopathic scoliosis in Japanese. Nat. Commun. 2019, 10, 3685. [Google Scholar] [CrossRef]
- Brooks, J.K.; Coccaro, P.J.; Zarbin, M.A. The Rieger anomaly concomitant with multiple dental, craniofacial, and somatic midline anomalies and short stature. Oral Surg. Oral Med. Oral Pathol. 1989, 68, 717–724. [Google Scholar] [CrossRef]
- Mucchielli, M.L.; Mitsiadis, T.A.; Raffo, S.; Brunet, J.F.; Proust, J.P.; Goridis, C. Mouse Otlx2/RIEG expression in the odontogenic epithelium precedes tooth initiation and requires mesenchyme-derived signals for its maintenance. Dev. Biol. 1997, 189, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowotschin, S.; Liao, J.; Gage, P.J.; Epstein, J.A.; Campione, M.; Morrow, B.E. Tbx1 affects asymmetric cardiac morphogenesis by regulating Pitx2 in the secondary heart field. Development 2006, 133, 1565–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Florez, S.; Amen, M.; Huynh, T.; Skobe, Z.; Baldini, A.; Amendt, B.A. Tbx1 regulates progenitor cell proliferation in the dental epithelium by modulating Pitx2 activation of p21. Dev. Biol. 2010, 347, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, E.A.; Botta, A.; Jurecic, V.; Carattini-Rivera, S.; Cheah, Y.C.; Rosenblatt, H.M.; Bradley, A.; Baldini, A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 1999, 401, 379–383. [Google Scholar] [CrossRef]
Figure 1.
Proximal deletions of chromosome 22q11.2 are responsible for the clinical features of DGS/VCFS. Snapshot of the UCSC Genome Browser (http://genome.ucsc.edu accessed on 3 August 2021) in the hg38 assembly showing the genomic context in the proximal deletions of chromosome 22q11.2. Top, the 25 kb resolution Hi-C data in H1 human embryonic stem cell line (H1-hESC). Bottom, the coding (blue) and noncoding RNAs (green), including miRNAs and long noncoding RNAs, are shown.
Figure 1.
Proximal deletions of chromosome 22q11.2 are responsible for the clinical features of DGS/VCFS. Snapshot of the UCSC Genome Browser (http://genome.ucsc.edu accessed on 3 August 2021) in the hg38 assembly showing the genomic context in the proximal deletions of chromosome 22q11.2. Top, the 25 kb resolution Hi-C data in H1 human embryonic stem cell line (H1-hESC). Bottom, the coding (blue) and noncoding RNAs (green), including miRNAs and long noncoding RNAs, are shown.

Figure 2.
Interaction network of genes associated with DGS/VCFS phenotypes in mice. (A) A gene-based network where each gene connects to a feature. The network was constructed using ToppCluster (https://toppcluster.cchmc.org/ accessed on 6 May 2022). Mouse phenotypes are shown in the network. (B) The protein–protein interaction network was constructed using the STRING tool (https://string-db.org/ accessed on 6 May 2022). Genes associated with DGS/VCFS phenotypes in mice (Table 4) were the input. Different colors represent different types of evidence of a connection between proteins.
Figure 2.
Interaction network of genes associated with DGS/VCFS phenotypes in mice. (A) A gene-based network where each gene connects to a feature. The network was constructed using ToppCluster (https://toppcluster.cchmc.org/ accessed on 6 May 2022). Mouse phenotypes are shown in the network. (B) The protein–protein interaction network was constructed using the STRING tool (https://string-db.org/ accessed on 6 May 2022). Genes associated with DGS/VCFS phenotypes in mice (Table 4) were the input. Different colors represent different types of evidence of a connection between proteins.


{kind=link}
{kind=link}
Table 1.
Craniofacial anomalies in patients with DGS/VCFS.
| Phenotypes | Features | Frequency |
|---|---|---|
| Palatal anomalies | Overt cleft palate | 7–11% |
| Submucous cleft palate | 5–23% | |
| Bifid uvula | 5–10% | |
| Velopharyngeal insufficiency | 27–92% | |
| Dental anomalies | Tooth agenesis | 15% |
| Hypoplasia of primary teeth | 32% | |
| Hypoplasia of permanent teeth | 10% | |
| Enamel hypomineralization of primary teeth | 39% | |
| Enamel hypomineralization of permanent teeth | 41% | |
| Ear-nose-throat abnormalities | Hearing loss | 33–39% |
| Otitis media with effusion | 2% | |
| Tracheomalacia/laryngomalacia | 2% | |
| Laryngeal web | 1% | |
| Ocular abnormalities | Hooding of the upper lid | 41% |
| Ptosis | 9% | |
| Hooding of the lower lid | 6% | |
| Epicanthal folds | 3% | |
| Distichiasis | 3% | |
| Cranial base anomalies | Platybasia | 50–91% |
| Basilar impression | 3% | |
| Cervical spine anomalies | Atlas (C1) anomalies | 75% |
| Axis (C2) anomalies | 59% | |
| Fusion of C2–C3 | 34% |
Table 2.
Craniofacial and skeletal phenotypes of DGS/VCFS and Tbx1-null mice.
| DGS/VCFS | Tbx1-Null Mice | |
|---|---|---|
| Cranium | Dolichocephaly | Small cranium |
| Abnormal skull morphology | Hypoplastic parietal bone | |
| Malar flattening | Hypoplastic interparietal bone | |
| Long face | Unfused cranial sutures between frontal and parietal bones | |
| Temporal bone hypoplasia | ||
| Absent zygomatic arch | ||
| Abnormal zygomatic arch morphology | ||
| Cranial Base | Platybasia | Abnormal fusion of the basioccipital and basisphenoid bones |
| Basilar impression | Abnormal presphenoid bone morphology | |
| Abnormal basioccipital bone morphology | ||
| Palate | Cleft palate | Cleft palate |
| Submucous cleft palate | Submucous cleft palate | |
| Bifid uvula | Bifid uvula | |
| Highly arched palate | ||
| Velopharyngeal insufficiency | ||
| Mandible | Retrognathia | Absent mandibular coronoid process |
| Short mandible | Short mandible | |
| Micrognathia | Micrognathia | |
| Teeth | Enamel hypoplasia | Abnormal upper incisor morphology |
| Single central incisor | Absent upper incisors | |
| Small teeth | ||
| Abnormality of the dentition | ||
| Carious teeth | ||
| Muscles | Pharyngeal hypotonia | Absent masseter muscle |
| Absent pterygoid muscle | ||
| Absent temporalis muscle | ||
| Eyes | Hypertelorism/telecanthus | Hypertelorism |
| Downslanted palpebral fissures | ||
| Proptosis | ||
| Strabismus | ||
| Abnormal eyelid morphology | ||
| Epicanthus | ||
| Microphthalmia | ||
| External Ears | Small earlobe | Ear lobe hypoplasia |
| Low-set ears | Lowered ear position | |
| Abnormally folded pinna | Abnormal ear shape | |
| Preauricular pit | Absent outer ear | |
| Anotia | ||
| Middle and Inner Ears | Chronic otitis media | Abnormal middle ear ossicle morphology |
| Conductive hearing loss | Absent middle ear ossicles | |
| Sensorineural hearing loss | Abnormal stapes morphology | |
| Auditory canal stenosis | Abnormal incus morphology | |
| Pulsatile tympanic membrane | Abnormal malleus morphology | |
| Thickened tympanic membrane | Absent stapes | |
| Tympanic membrane retraction | Abnormal external auditory canal morphology | |
| Decreased tympanic ring size | ||
| Nose | Prominent nasal bridge | Short snout |
| Abnormal nasal morphology | ||
| Underdeveloped nasal alae | ||
| Choanal atresia | ||
| Throat | Abnormal thorax morphology | Small thyroid cartilage |
| Abnormality of the pharynx | Small cricoid cartilage | |
| Abnormal thyroid cartilage morphology | ||
| Pharynx hypoplasia | ||
| Hyoid bones | Delayed development of the hyoid bone | Hyoid bone hypoplasia |
| Invisible hyoid ossification center | Abnormal hyoid bone morphology | |
| Cervical spine | Dysmorphic C1 | Abnormal cervical atlas (C1) morphology |
| Anterior arch cleft of C1 | Absent arcus anterior of C1 | |
| Open posterior arch C1 | ||
| Fusion of C1–C2 | ||
| Fusion of C2–C3 | ||
| Upswept C2 lamina | ||
| Platyspondyly | ||
| Others | Short clavicle | |
| References | [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28] | [6,7,8,9,10,29,30,31,32,33,34,35,36] |
Table 3.
DGS/VCFS-associated variants of TBX1.
| Mutation | Domain | Condition | Craniofacial Anomalies | References |
|---|---|---|---|---|
| c.89_284del | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 971780 |
| c.199_224del | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 949172 |
| c.292A>T | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 526036 |
| c.385G>A | T-box | Tetralogy of Fallot | No | ClinVar Variant: 488618 |
| c.443T>A (F148Y) | T-box | Conotruncal anomaly face syndrome | Yes | [5] |
| c.503T>C | T-box | DiGeorge syndrome Velocardiofacial syndrome (Shprintzen syndrome) Tetralogy of Fallot | Yes | ClinVar Variant: 973222 |
| c.569C > A (P190Q) | T-box | Congenital heart defects | No | [38] |
| c.582C>G (H194Q) | T-box | Velocardiofacial syndrome | Yes | [39] |
| c.928G>A (G310S) | C-terminal | DiGeorge syndrome | Yes | [5] |
| c.967_977dup AACCCCGTGGC | C-terminal | Thymic hypoplasia Postaxial polydactyly of the right fifth toe | No | [40] |
| c.1158_1159delinsT | C-terminal | Hypoparathyroidism and hypocalcemia Facial asymmetry Deafness | Yes | [41] |
| c.1223delC | C-terminal | Conotruncal anomaly face syndrome Velocardiofacial syndrome | Yes | [5] |
| c.1253delA | C-terminal | DiGeorge syndrome | Yes | [42] |
| c.1320-1342del23bp | C-terminal | Velocardiofacial syndrome | Yes/No | [43] |
| c.1399-1428dup30 | C-terminal | Tetralogy of Fallot Scoliosis Facial asymmetry Upslanting palpebral fissures Absent pulmonary valve Isolated left pulmonary artery | Yes | [44] |
ClinVar (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar accessed on 3 August 2021).
Table 4.
Craniofacial phenotypes of DGS/VCFS mouse model.
| Gene Symbol | Induced Mutation Type | Cranium | Palate | Teeth | Muscles | Ear-Nose-Throat | Hyoid Bones | Cardio-Vascular |
|---|---|---|---|---|---|---|---|---|
| Tbx1 | Null | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Chrd | Null | Yes | Yes | nr | nr | Yes | Yes | Yes |
| Tgfbr2 | Deletion (Wnt1-Cre) | Yes | Yes | nr | nr | nr | nr | Yes |
| Vegfa | Null | Yes | Yes | Yes | nr | nr | nr | Yes |
| Fgf8 | Hypomorphic allele | Yes | Yes | Yes | nr | Yes | Yes | Yes |
| Crkl | Null | Yes | nr | nr | nr | Yes | nr | Yes |
| Aldh1a2 | Hypomorphic allele | nr | nr | nr | nr | Yes | Yes | Yes |
| Hoxa3 | Null | nr | Yes | nr | Yes | Yes | Yes | Yes |
| Kat6a | Null | nr | Yes | nr | nr | Yes | nr | Yes |
| Dicer1 | Deletion (Wnt1-Cre) | Yes | nr | nr | nr | nr | nr | Yes |
| Plxnd1 | Single point mutation | nr | Yes | nr | nr | Yes | nr | Yes |
| Dock1 | Undefined | nr | nr | nr | nr | Yes | nr | Yes |
| Ndst1 | Single point mutation | nr | nr | nr | nr | Yes | nr | Yes |
| Prickle1 | Single point mutation | Yes | Yes | nr | nr | Yes | nr | Yes |
| Trappc10 | Undefined | Yes | Yes | nr | nr | nr | nr | Yes |
| Zfp366 | Single point mutation | nr | nr | nr | nr | Yes | nr | Yes |
| Foxn1 | Intragenic deletion | nr | nr | nr | nr | Yes | nr | Yes |
Mouse models of DiGeorge syndrome with phenotypic similarity to human diseases can be found in the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org accessed on 3 August 2021). Data were summarized from the following references [6,7,8,9,10,29,30,31,32,33,34,35,36,52,53,54,55,56,57,58,59,60,61]. nr, not reported. A detailed description is provided in Table S2.
Table 5.
Selected craniofacial phenotypes of Tbx1-mutant neonates.
| Tbx1-Mutant Mice | Craniofacial Phenotypes | ||||||
|---|---|---|---|---|---|---|---|
| Mutation Type | Tissue/Cell | Cranium | Cranial Base | Palate | Mandible | Hyoid Bone | Cervical Spine |
| Tbx1+/− | Entire body | Normal | Normal | Normal | Normal | Normal | Normal |
| Tbx1-null | Entire body | Abnormal | Abnormal | CP | Hypoplastic | Hypoplastic | Abnormal |
| Deletion (Foxg1-Cre) | Pharyngeal tissues * | Abnormal | Abnormal | CP | Hypoplastic | Hypoplastic | NA |
| Deletion (KRT14-Cre) | Epithelium | Normal | Normal | Anterior CP | Normal | Normal | Normal |
| Deletion (Mesp1-Cre) | Mesoderm | Abnormal | Abnormal | NA | Hypoplastic | Hypoplastic | Abnormal |
| Deletion (Twist2-Cre) | Osteochondral progenitors | Abnormal | Abnormal | Normal | Normal | Hypoplastic | Abnormal |
| Deletion (Wnt1-Cre) | Neural crest | Normal | Normal | Normal | Normal | Hypoplastic | Normal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Funato, N. Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. J. Dev. Biol. 2022, 10, 18. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb10020018
AMA Style
Funato N. Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. Journal of Developmental Biology. 2022; 10(2):18. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb10020018
Chicago/Turabian StyleFunato, Noriko. 2022. "Craniofacial Phenotypes and Genetics of DiGeorge Syndrome" Journal of Developmental Biology 10, no. 2: 18. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb10020018
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.