
Detection of Chromosomal Segments Introgressed from Wild Species of Carrot into Cultivars: Quantitative Trait Loci Mapping for Morphological Features in Backcross Inbred Lines

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
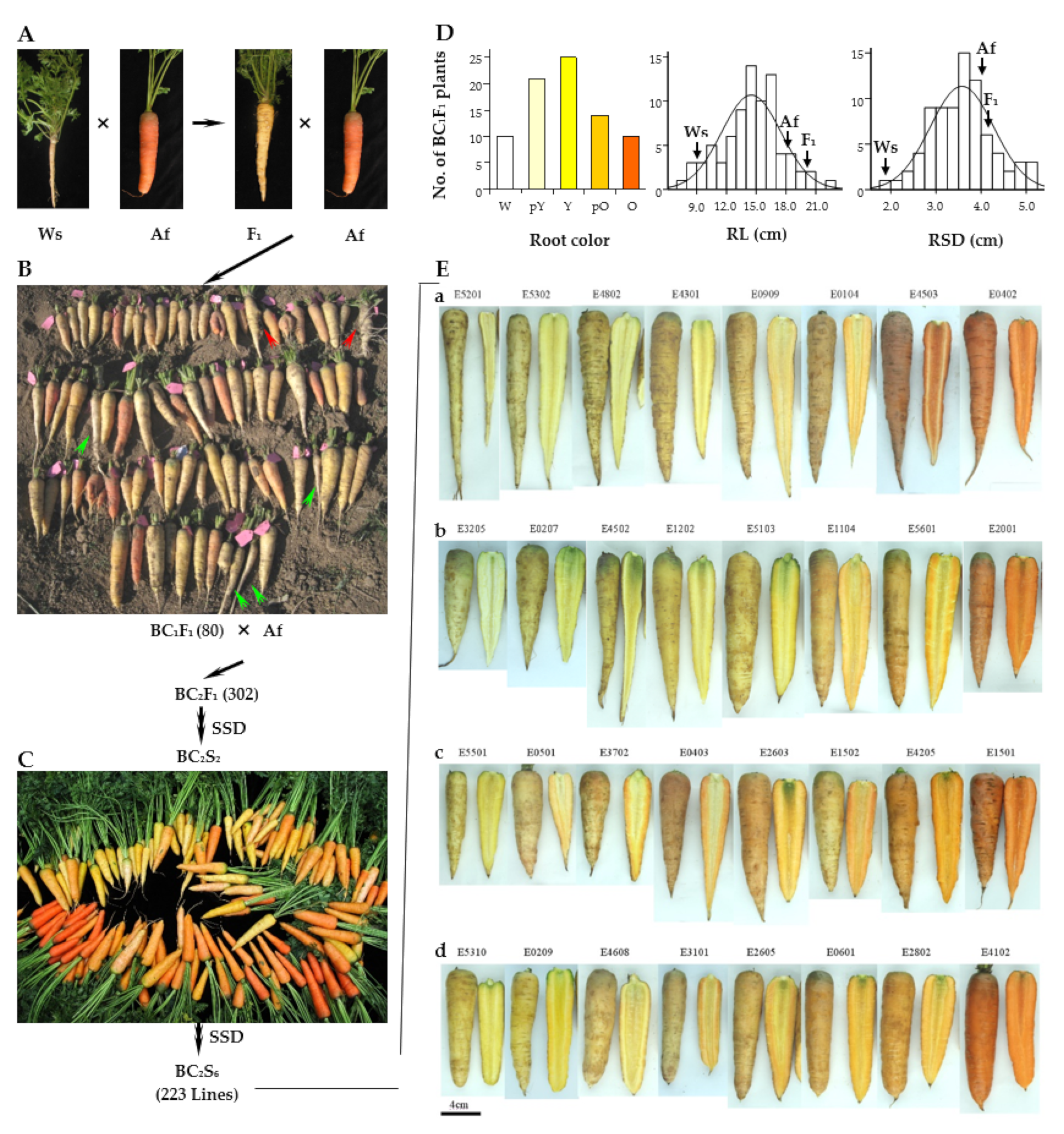
2.1. Plant Materials and Morphological Evaluation
2.2. Genome Resequencing of the BIL Population
2.3. SNP Identification and Mapping in the BIL Population
2.4. QTL Analysis
3. Results
3.1. Morphology of the F1 Hybrid and the Backcross Progeny Plants
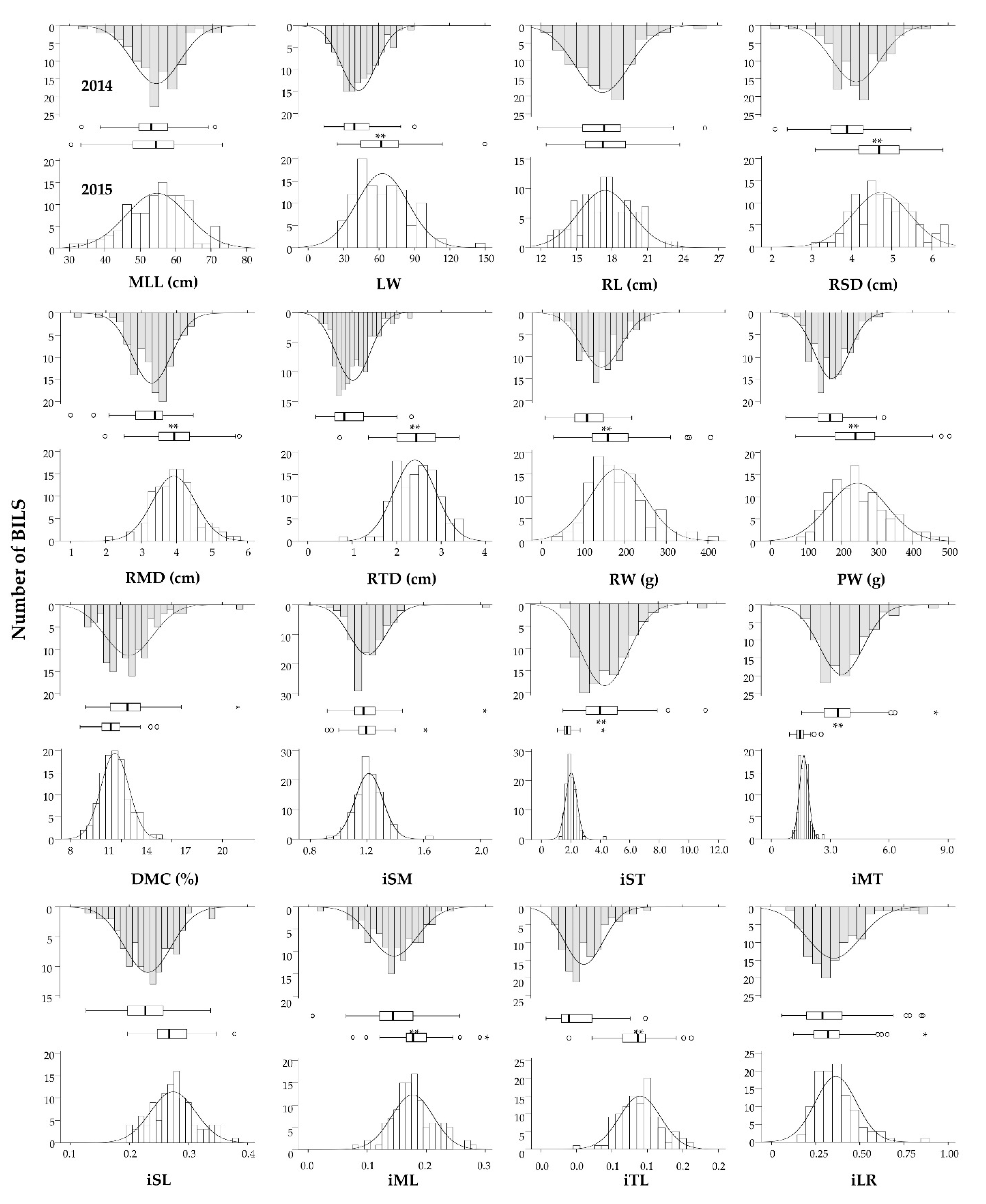
3.2. Variation in Morphological Traits in the BIL Population
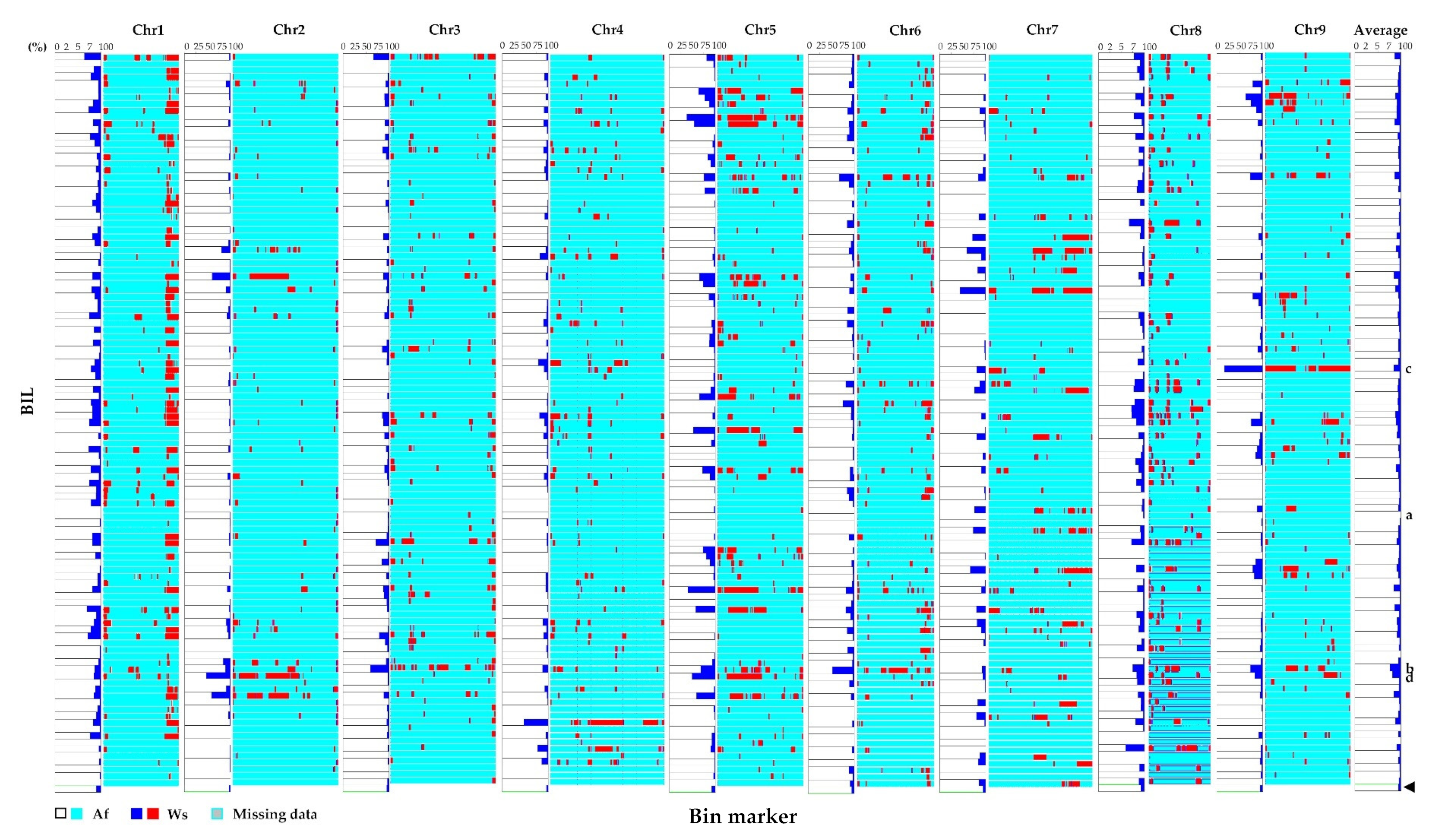
3.3. Genotyping and Construction of the Bin Map in the BIL Population
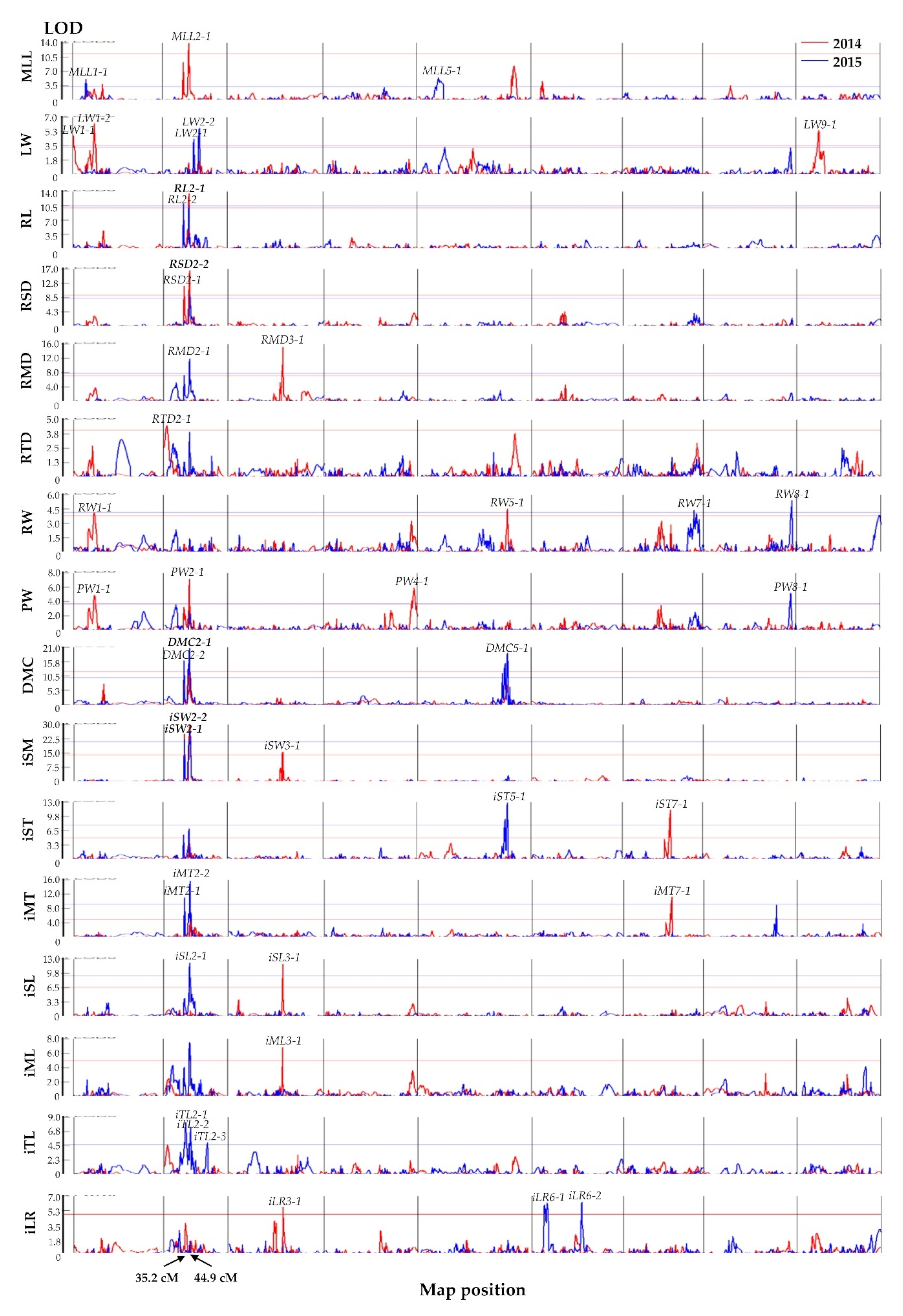
3.4. QTL Analysis of BIL Morphological Traits
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rubatzky, V.E.; Simon, P.W.; Quiros, C.F. Carrots and Related Vegetable Umbelliferae; University Press: Cambridge, UK, 1999. [Google Scholar]
- She, M.; Pu, F.; Pan, Z.; Mark, F.W.; John, F.M.C.; Ingrid, H.S.; Eugene, V.K.; Loy, R.P.; Michael, G.P. APIACEAE (UMBELLIFERAE). Flora China 2005, 14, 1–205. [Google Scholar]
- IPGRI. Descriptors for Wild and Cultivated Carrots (Daucus carota L.); International Plant Genetic Resources Institute: Rome, Italy, 1998. [Google Scholar]
- Iorizzo, M.; Senalik, D.; Ellison, S.; Grzebelus, D.; Cavagnaro, P.F.; Allender, C.; Bunet, J.; Spooner, D.M.; Deynze, A.V.; Simon, P.W. Genetic structure and domestication of carrot (Daucus carota subsp. sativus) (Apiaceae). Am. J. Bot. 2013, 100, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Iorizzo, M.; Ellison, S.; Senalik, D.; Zeng, P.; Satapoomin, P.; Huang, J.; Bowman, M.; Iovene, M.; Sanseverino, W.; Cavagnaro, P.; et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 2016, 48, 657–666. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.G.; Kong, X.P.; Liu, L.J.; Ou, C.G.; Sun, T.T.; Zhao, Z.W.; Miao, Z.J.; Rong, J.; Zhuang, F.Y. The unique origin of orange carrot cultivars in China. Euphytica 2016, 212, 37–49. [Google Scholar] [CrossRef]
- Simon, P.W. Domestication, historical development, and modern breeding of carrot. Plant Breed. Rev. 2000, 19, 157–190. [Google Scholar]
- Small, E. Numerical taxonomic analysis of Daucus carota complex. Can. J. Bot. 1978, 56, 248–276. [Google Scholar] [CrossRef]
- Heywood, V.H. Relationship and evolution in the Daucus carota complex. Isr. J. Bot. 1983, 32, 51–65. [Google Scholar]
- Rong, J.; Lanmers, Y.; Strasburg, J.L.; Schidlo, N.S.; Ariyurek, Y.; de Jong, T.J.; Klinkhamer, P.G.; Smulders, M.J.; Vrieling, K. New insights into domestication of carrot from root transcriptome analyses. BMC Genom. 2014, 15, 895. [Google Scholar] [CrossRef] [Green Version]
- Grzebelus, D.; Baranski, R.; Spalik, K.; Allender, C.; Simon, P.W. Daucus. In Wild Crop Relatives: Genomic and Breeding Resources; Springer: Berlin/Heidelberg, Germany, 2011; pp. 91–113. [Google Scholar]
- Li, Y.; Yang, L.; Pathak, M.; Li, D.; He, X.; Weng, Y. Fine genetic mapping of cp: A recessive gene for compact (dwarf) plant architecture in cucumber, Cucumis sativus L. Theor. Appl. Genet. 2011, 123, 973–983. [Google Scholar] [CrossRef]
- Lv, H.; Fang, Z.; Yang, L.; Zhang, Y.; Wang, Q.; Liu, Y.; Zhuang, M.; Yang, Y.; Xie, B.; Liu, B.; et al. Mapping and analysis of a novel candidate Fusarium wilt resistance gene FOC1 in Brassica oleracea. BMC Genom. 2014, 15, 1094. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Feng, Q.; Yua, H.; Huang, X.; Zhao, Q.; Xinga, Y.; Yua, S.; Han, B.; Zhang, Q. Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 10578–10583. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Li, M.W.; Xie, M.; Liu, X.; Ni, M.; Shao, G.; Song, C.; Kay-Yuen, Y.A.; Tao, Y.; Wong, F.L.; et al. Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat. Commun. 2014, 5, 4340. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhang, C.; Zhou, Y.; Hao, Z.; Wang, Z.; Zeng, X.; Di, H.; Li, M.; Zhang, D.; Yong, H.; et al. Genetic dissection of maize plant architecture with an ultra-high density bin map based on recombinant inbred lines. BMC Genom. 2016, 17, 178. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef]
- Han, K.; Jeong, H.J.; Yang, H.B.; Kang, S.M.; Kwon, J.K.; Kim, S.; Choi, D.; Kang, B.C. An ultra-high-density bin map facilitates high-throughput QTL mapping of horticultural traits in pepper(Capsicum annuum). DNA Res. 2016, 23, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Cavagnaro, P.F.; Iorizzo, M.; Yildiz, M.; Senalik, D.; Parsons, J.; Ellison, S.; Simon, P.W. A gene-derived SNP-based high resolution linkage map of carrot including the location of QTL conditioning root and leaf anthocyanin pigmentation. BMC Genom. 2014, 15, 1118. [Google Scholar] [CrossRef] [Green Version]
- Ellison, S.; Senalik, D.; Bostan, H.; Iorizzo, M.; Simon, P.W. Fine mapping, transcriptome analysis, and marker development for Y2, the gene that conditions β-carotene accumulation in carrot (Daucus carota L.). G3 2017, 7, 2665–2675. [Google Scholar] [CrossRef] [Green Version]
- Macko-Podgórni, A.; Machaj, G.; Stelmach, K.; Senalik, D.; Grzebelus, E.; Iorizzo, M.; Simon, P.W.; Grzebelus, D. Characterization of a genomic region under selection in cultivated carrot (Daucus carota subsp. sativus) reveals a candidate domestication gene. Front. Plant Sci. 2017, 8, 12. [Google Scholar]
- Turner, S.D.; Ellison, S.L.; Senalik, D.A.; Simon, P.W.; Spalding, E.P.; Miller, N.D. An automated image analysis pipeline enables genetic studies of shoot and root morphology in carrot (Daucus carota L.). Front. Plant Sci. 2018, 9, 1703. [Google Scholar] [CrossRef] [Green Version]
- Bannoud, F.; Carvajal, S.; Ellison, S.; Senalik, D.; Gomez Talquenca, S.; Iorizzo, M.; Simon, P.W.; Cavagnaro, P.F. Genetic and transcription profile analysis of tissue-specific anthocyanin pigmentation in carrot root phloem. Genes 2021, 12, 1464. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.G.; Zhao, Z.W.; Hu, H.; Pei, H.X.; Zhuang, F.Y. Genetic and Heterosis analysis for root width and length in carrot (Daucus carota L.). Acta Hortic. Sin. 2009, 36, 115–120. [Google Scholar]
- Rosenfeld, H.J.; Dalen, K.S.; Haffner, K. The growth and development of carrot roots. Gartenbauwissenschaft 2002, 67, 11–16. [Google Scholar]
- Thompson, R. Some factors affecting carrot root shape and size. Euphytica 1969, 18, 277–285. [Google Scholar] [CrossRef]
- Thomas, T.H. Effect of root restriction and growth regulator treatments on the growth of carrot (Daucus carota L.) seedlings. Plant Growth Regul. 1993, 13, 95–101. [Google Scholar] [CrossRef]
- Wu, X.J.; Wang, G.L.; Song, X.; Xu, Z.S.; Wang, F.; Xiong, A.S. Regulation of auxin accumulation and perception at different developmental stages in carrot. Plant Growth Regul. 2016, 80, 243–251. [Google Scholar] [CrossRef]
- Wang, G.L.; Que, F.; Xu, Z.S.; Wang, F.; Xiong, A.S. Exogenous gibberellin altered morphology, anatomic and transcriptional regulatory networks of hormones in carrot root and shoot. BMC Plant Biol. 2015, 15, 290. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Que, F.; Xu, Z.S.; Wang, F.; Xiong, A.S. Exogenous gibberellin enhances secondary xylem development and lignification in carrot taproot. Protoplasma 2017, 254, 839–848. [Google Scholar] [CrossRef]
- Macko-Podgórni, A.; Stelmach, K.; Kwolek, K.; Machaj, G.; Ellison, S.; Senalik, D.A.; Simon, P.W.; Grzebelus, D. Mining for candidate genes controlling secondary growth of the carrot storage root. Int. J. Mol. Sci. 2020, 21, 4263. [Google Scholar] [CrossRef]
- Jeuken, M.J.W.; Pelgrom, K.; Stam, P.; Lindhout, P. Efficient QTL detection for nonhost resistance in wild lettuce: Backcross inbred lines versus F2 population. Theor. Appl. Genet. 2008, 116, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Ou, C.G.; Mao, J.H.; Liu, L.J.; Li, C.J.; Ren, H.F.; Zhao, Z.W.; Zhuang, F.Y. Characterizing genes associated with flowering time in carrot (Daucus carota L.) using transcriptome analysis. Plant Biol. 2017, 19, 286–297. [Google Scholar] [CrossRef]
- Liu, L.J.; Ou, C.G.; Chen, S.M.; Shen, Q.; Liu, B.; Li, M.; Zhao, Z.W.; Kong, X.P.; Yan, X.P.; Zhuang, F.Y. The response of COL and FT homologues to photoperiodic regulation in carrot (Daucus carota L.). Sci. Rep. 2020, 10, 9984. [Google Scholar] [CrossRef]
- Briard, M.; Clerc, V.L.; Grzebelus, D.; Senalik, D.; Simon, P.W. Modified protocols for rapid carrot genomic DNA extraction and AFLPTM analysis using silver stain or radioisotopes. Plant Mol. Biol. Rep. 2000, 18, 235–241. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Angel, G.D.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. JoinMap 4.0: Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma B.V.: Wageningen, The Netherlands, 2006. [Google Scholar]
- Wang, S. Windows QTL Cartographer 2.5; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 2011; Available online: http://statgen.ncsu.edu/qtlcart/WQTLCart.htm (accessed on 1 January 2022).
- Que, F.; Hou, X.L.; Wang, G.L.; Xu, Z.S.; Tan, G.F.; Li, T.; Wang, Y.H.; Khadr, A.; Xiong, A.S. Advances in research on the carrot, an important root vegetable in the Apiaceae family. Hortic. Res. 2019, 6, 69. [Google Scholar] [CrossRef] [Green Version]
- Ellison, S. Carrot domestication. In The Carrot Genome; Springer: Berlin/Heidelberg, Germany, 2019; pp. 77–91. [Google Scholar]
- Simon, P.W.; Freeman, R.E.; Vieira, J.V.; Boiteux, L.S.; Briard, M.; Nothnagel, T.; Michalik, B.; Kwon, Y.S. “Carrot”. In Vegetables II: Fabaceae, Liliaceae, Solanaceae, and Umbelliferae; Prohens, J., Nuez, F., Eds.; Springer: New York, NY, USA, 2008; pp. 327–357. [Google Scholar]
- Zou, G.; Zhai, G.; Feng, Q.; Yan, S.; Wang, A.; Zhao, Q.; Shao, J.; Zhang, Z.; Zou, J.; Han, B.; et al. Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J. Exp. Bot. 2012, 63, 5451–5462. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Zhang, H.; Cao, C.; Han, J.; Li, H.; Ren, Z. QTL Mapping for cucumber fruit size and shape with populations from long and round fruited inbred lines. Hortic. Plant J. 2020, 6, 132–144. [Google Scholar] [CrossRef]
- Santos, C.A.F.; Simon, P.W. QTL analyses reveal clustered loci for accumulation of major provitamin a carotenes and lycopene in carrot roots. Mol. Gen. Genom. 2002, 268, 122–129. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, J.; Liu, X.; Wang, W.; Liu, D.; Teng, Z.; Fang, X.; Tan, Z.; Tang, S.; Yang, J.; et al. Fine mapping and RNA-Seq unravels candidate genes for a major QTL controlling multiple fiber quality traits at the T1 region in upland cotton. BMC Genom. 2016, 17, 295. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | MLL | LW | RL | RSD | RMD | RTD | RW | PW | DMC |
|---|---|---|---|---|---|---|---|---|---|
| MLL | 0.46 *** | 0.18 | 0.06 | −0.01 | 0.08 | 0.10 | 0.20 * | −0.21 * | |
| LW | 0.62 *** | 0.57 *** | 0.60 *** | 0.49 *** | 0.31 ** | 0.63 *** | 0.78 *** | −0.07 | |
| RL | 0.13 | 0.26 ** | 0.51 *** | 0.42 *** | 0.29 ** | 0.70 *** | 0.72 *** | 0.09 | |
| RSD | 0.29 ** | 0.30 ** | 0.22 * | 0.87 *** | 0.61 *** | 0.85 *** | 0.88 *** | −0.17 | |
| RMD | 0.25 ** | 0.17 | 0.08 | 0.86 *** | 0.75 *** | 0.85 *** | 0.83 *** | −0.30 ** | |
| RTD | 0.21 * | −0.04 | −0.25 ** | 0.30 ** | 0.57 *** | 0.41 *** | 0.63 *** | −0.37 *** | |
| RW | 0.29 ** | 0.28 ** | 0.46 *** | 0.88 *** | 0.86 *** | 0.68 *** | 0.98 *** | −0.17 | |
| PW | 0.44 *** | 0.55 *** | 0.48 *** | 0.83 *** | 0.80 *** | 0.35 *** | 0.96 *** | −0.25 ** | |
| DMC | −0.12 | −0.01 | 0.12 | −0.36 *** | −0.44 *** | −0.40 *** | −0.29 ** | −0.16 |
| Chr a | Number of SNPs | Number of Bins | Genetic Distance (cm) | Average Distance between the Neighbor Bins (cm) | Max Gap (cm) |
|---|---|---|---|---|---|
| 1 | 19,430 | 184 | 160.48 | 0.87 | 13.54 |
| 2 | 27,142 | 249 | 114.70 | 0.46 | 16.88 |
| 3 | 28,062 | 267 | 170.46 | 0.64 | 12.02 |
| 4 | 22,429 | 281 | 167.26 | 0.60 | 5.02 |
| 5 | 7986 | 224 | 202.93 | 0.91 | 7.96 |
| 6 | 12,070 | 194 | 162.37 | 0.84 | 10.60 |
| 7 | 17,491 | 250 | 143.02 | 0.58 | 3.93 |
| 8 | 5564 | 141 | 166.27 | 1.18 | 14.33 |
| 9 | 14,602 | 237 | 148.97 | 0.63 | 8.59 |
| Average | 17,197 | 225 | 159.61 | - | 10.32 |
| Total | 154,776 | 2027 | 1436.43 | 0.71 | - |
| Trait | Year | QTL | Chr | LOD | Position (cm) | QTL Region (cm) | Additive Effect | Phenotypic Variance (%) |
|---|---|---|---|---|---|---|---|---|
| MLL | 2014 | MLL2-1 | 2 | 13.9 | 44.9 | 44.4–45.0 | 23.1 | 25.7 |
| 2015 | MLL1-1 | 1 | 5.2 | 22.8 | 21.8–23.6 | 11.8 | 13.2 | |
| 2015 | MLL5-1 | 5 | 5.5 | 37.3 | 35.0–38.3 | 4.4 | 13.9 | |
| LW | 2014 | LW1-1 | 1 | 7.0 | 0.5 | 0.0–1.6 | −7.2 | 12.7 |
| 2014 | LW1-2 | 1 | 6.3 | 37.9 | 36.6–40.7 | 11.7 | 17.3 | |
| 2014 | LW9-1 | 9 | 5.6 | 39.4 | 37.5–39.9 | −11.3 | 15.2 | |
| 2015 | LW2-1 | 2 | 4.4 | 54.1 | 53.5–54.6 | −21.9 | 13.1 | |
| 2015 | LW2-2 | 2 | 5.7 | 64.2 | 63.8–64.8 | −30.0 | 16.6 | |
| RL | 2014/2015 | RL2-1 | 2 | 13.5/10.7 | 44.9 | 44.4–45.0 | 8.6/8.2 | 33.7/30.5 |
| 2015 | RL2-2 | 2 | 11.1 | 35.2 | 34.2–35.6 | 10.7 | 40.4 | |
| RSD | 2014 | RSD2-1 | 2 | 12.0 | 35.2 | 34.2–35.4 | 2.3 | 39.1 |
| 2014/2015 | RSD2-2 | 2 | 16.6/9.7 | 44.9 | 44.4–45.0 | 2.3/2.2 | 39.1/24.6 | |
| RMD | 2014 | RMD3-1 | 3 | 15.0 | 96.3 | 95.8–96.7 | 1.5 | 38.9 |
| 2015 | RMD2-1 | 2 | 11.8 | 44.9 | 44.4–45.0 | 2.0 | 29.5 | |
| RTD | 2014 | RTD2-1 | 2 | 4.5 | 4.9 | 1.6–6.8 | 0.2 | 12.6 |
| RW | 2014 | RW1-1 | 1 | 4.1 | 35.5 | 34.4–38.1 | 27.3 | 10.5 |
| 2014 | RW5-1 | 5 | 4.5 | 158.8 | 157.7–159.5 | 49.3 | 13.2 | |
| 2015 | RW7-1 | 7 | 4.4 | 124.9 | 124.0–125.5 | −44.5 | 11.4 | |
| 2015 | RW8-1 | 8 | 5.4 | 155.9 | 155.2–156.6 | −104.7 | 15.9 | |
| PW | 2014 | PW1-1 | 1 | 4.9 | 36.5 | 35.1–38.0 | 33.6 | 13.2 |
| 2014 | PW2-1 | 2 | 7.1 | 44.9 | 44.4–45.0 | 123.2 | 18.9 | |
| 2014 | PW4-1 | 4 | 5.9 | 160.1 | 158.3–161.5 | −34.2 | 15.3 | |
| 2015 | PW8-1 | 8 | 5.2 | 154.9 | 154.4–156.1 | −89.2 | 14.1 | |
| DMC | 2014/2015 | DMC2-1 | 2 | 12.3/20.9 | 44.9 | 44.4–45.0 | 0.1/0.1 | 23.4/37.2 |
| 2015 | DMC2-2 | 2 | 16.1 | 35.2 | 34.2–35.5 | 0.1 | 37.2 | |
| 2015 | DMC5-1 | 5 | 18.9 | 159.7 | 158.8–159.9 | −0.1 | 32.4 | |
| iSM | 2014/2015 | iSM2-1 | 2 | 25.1/22.3 | 35.2 | 34.2–35.4 | 1.1/0.6 | 66.9/55.7 |
| 2014/2015 | iSM2-2 | 2 | 29.7/27.0 | 44.9 | 44.4–45.0 | 1.1/0.6 | 66.9/55.7 | |
| 2014 | iSM3-1 | 3 | 15.5 | 94.9 | 94.4–94.9 | −0.5 | 24.9 | |
| iST | 2014 | iST7-1 | 7 | 11.2 | 84.5 | 84.0–84.9 | −3.9 | 30.9 |
| 2015 | iST5-1 | 5 | 12.8 | 159.7 | 158.8–159.9 | −1.2 | 27.4 | |
| iMT | 2014 | iMT7-1 | 7 | 11.2 | 84.5 | 84.0–84.9 | −2.8 | 31.9 |
| 2015 | iMT2-1 | 2 | 11.0 | 35.2 | 34.2–35.4 | 0.9 | 36.3 | |
| 2015 | iMT2-2 | 2 | 15.7 | 44.9 | 44.4–45.0 | 0.9 | 36.3 | |
| iSL | 2014 | iSL3-1 | 3 | 11.9 | 96.3 | 95.8–97 | 0.1 | 30.1 |
| 2015 | iSL2-1 | 2 | 12.1 | 44.9 | 44.4–45.1 | 0.1 | 31.2 | |
| iML | 2014 | iML3-1 | 3 | 6.9 | 96.3 | 95.8–97.5 | 0.1 | 16.5 |
| iTL | 2015 | iTL2-1 | 2 | 8.2 | 37.9 | 36.7–39.0 | 0.0 | 27.6 |
| 2015 | iTL2-2 | 2 | 7.3 | 45.8 | 45.4–46.4 | 0.1 | 22.8 | |
| 2015 | iTL2-3 | 2 | 5.0 | 76.1 | 74.7–79.0 | −0.0 | 12.3 | |
| iLR | 2014 | iLR3-1 | 3 | 5.7 | 96.7 | 96.3–100.1 | 0.2 | 15.7 |
| 2015 | iLR6-1 | 6 | 6.3 | 25.1 | 24.1–26.9 | 0.1 | 16.0 | |
| 2015 | iLR6-2 | 6 | 6.2 | 87.1 | 85.9–89.3 | −0.2 | 18.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ou, C.; Sun, T.; Liu, X.; Li, C.; Li, M.; Wang, X.; Ren, H.; Zhao, Z.; Zhuang, F. Detection of Chromosomal Segments Introgressed from Wild Species of Carrot into Cultivars: Quantitative Trait Loci Mapping for Morphological Features in Backcross Inbred Lines. Plants 2022, 11, 391. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11030391
Ou C, Sun T, Liu X, Li C, Li M, Wang X, Ren H, Zhao Z, Zhuang F. Detection of Chromosomal Segments Introgressed from Wild Species of Carrot into Cultivars: Quantitative Trait Loci Mapping for Morphological Features in Backcross Inbred Lines. Plants. 2022; 11(3):391. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11030391
Chicago/Turabian StyleOu, Chenggang, Tingting Sun, Xing Liu, Chengjiang Li, Min Li, Xuewei Wang, Huaifu Ren, Zhiwei Zhao, and Feiyun Zhuang. 2022. "Detection of Chromosomal Segments Introgressed from Wild Species of Carrot into Cultivars: Quantitative Trait Loci Mapping for Morphological Features in Backcross Inbred Lines" Plants 11, no. 3: 391. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11030391