
Evolutionary History and Functional Diversification of the JmjC Domain-Containing Histone Demethylase Gene Family in Plants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
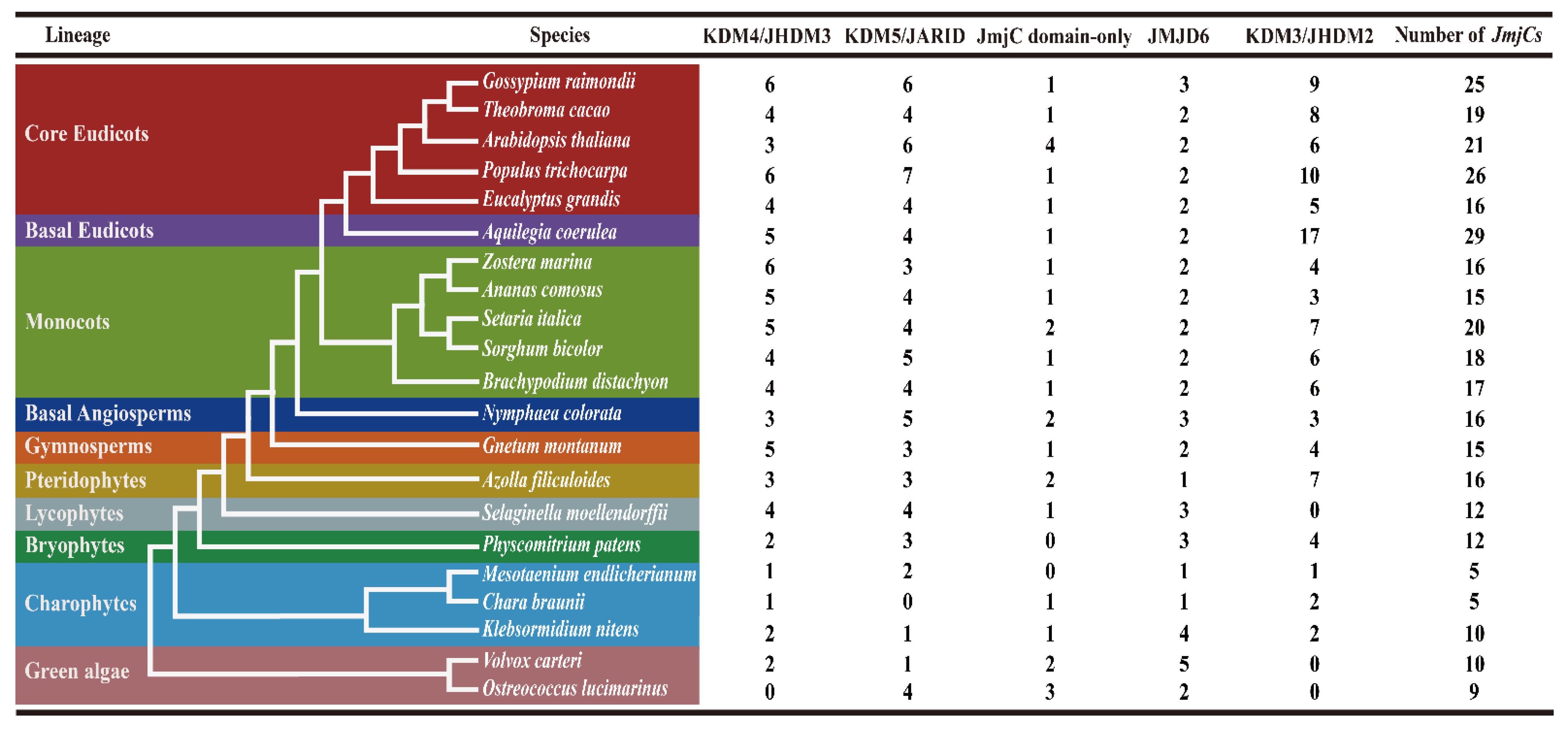
2.1. Identification of JmjC Genes in Green Plants
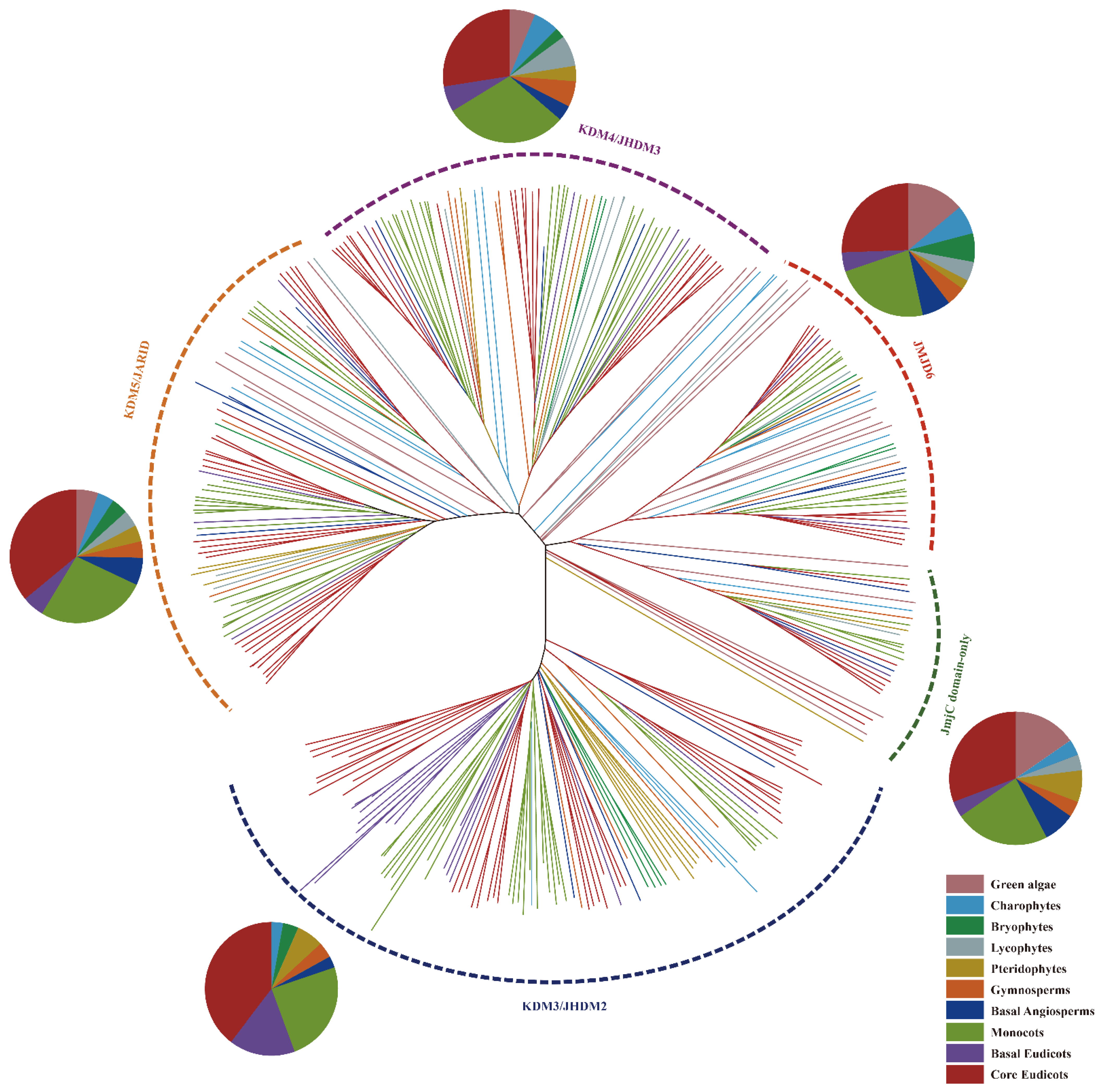
2.2. Phylogenetic Evolution Analysis of Plant JmjC Genes
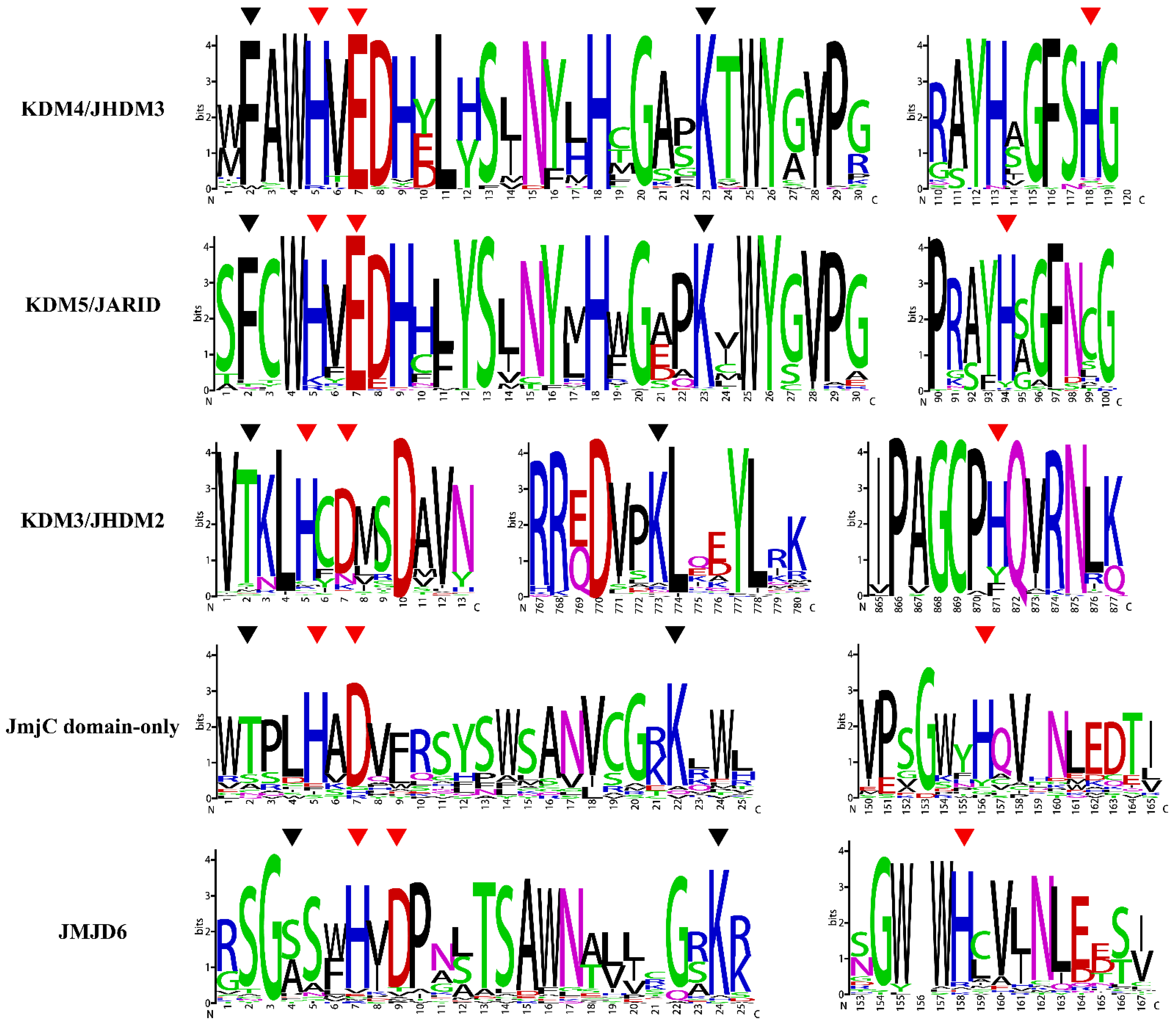
2.3. Analysis of Conserved Amino Acid Residues of JmjC
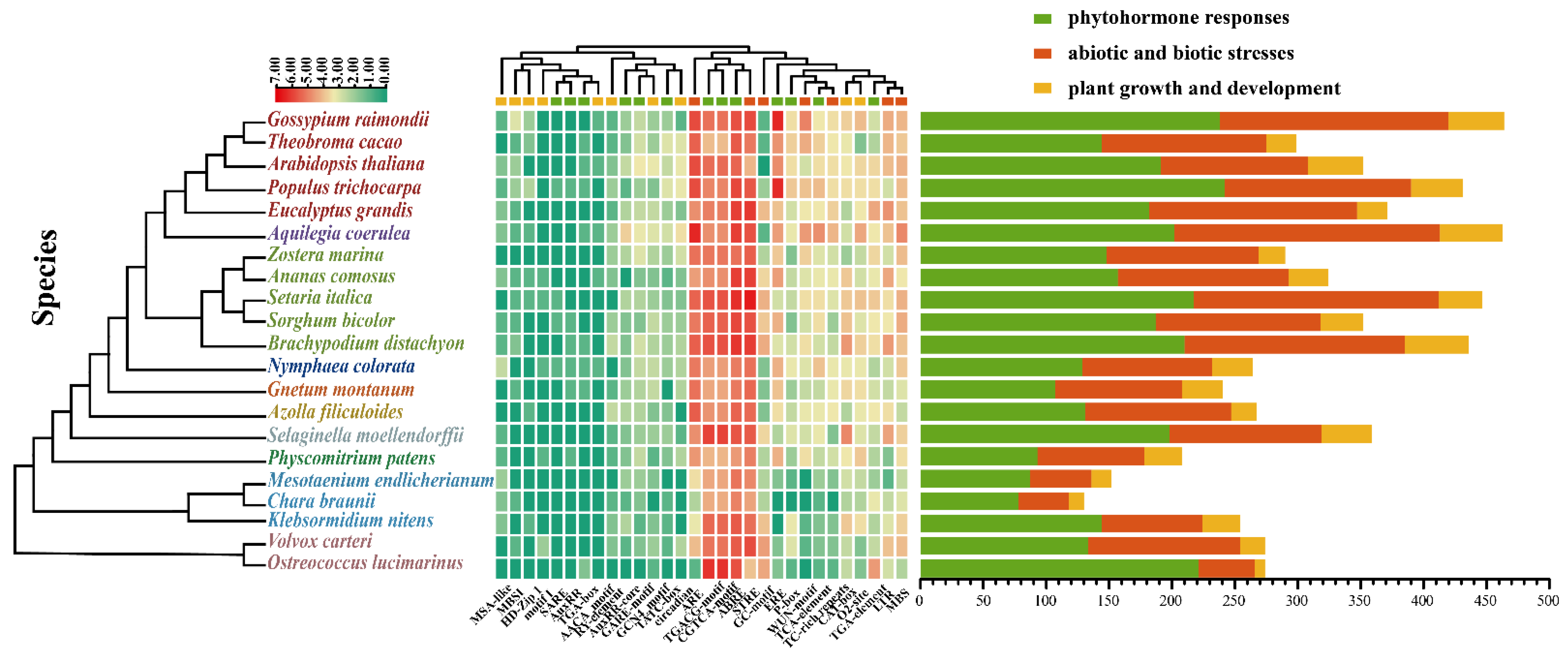
2.4. Analysis of Cis-Acting Elements in the Promoter Regions of JmjC Genes
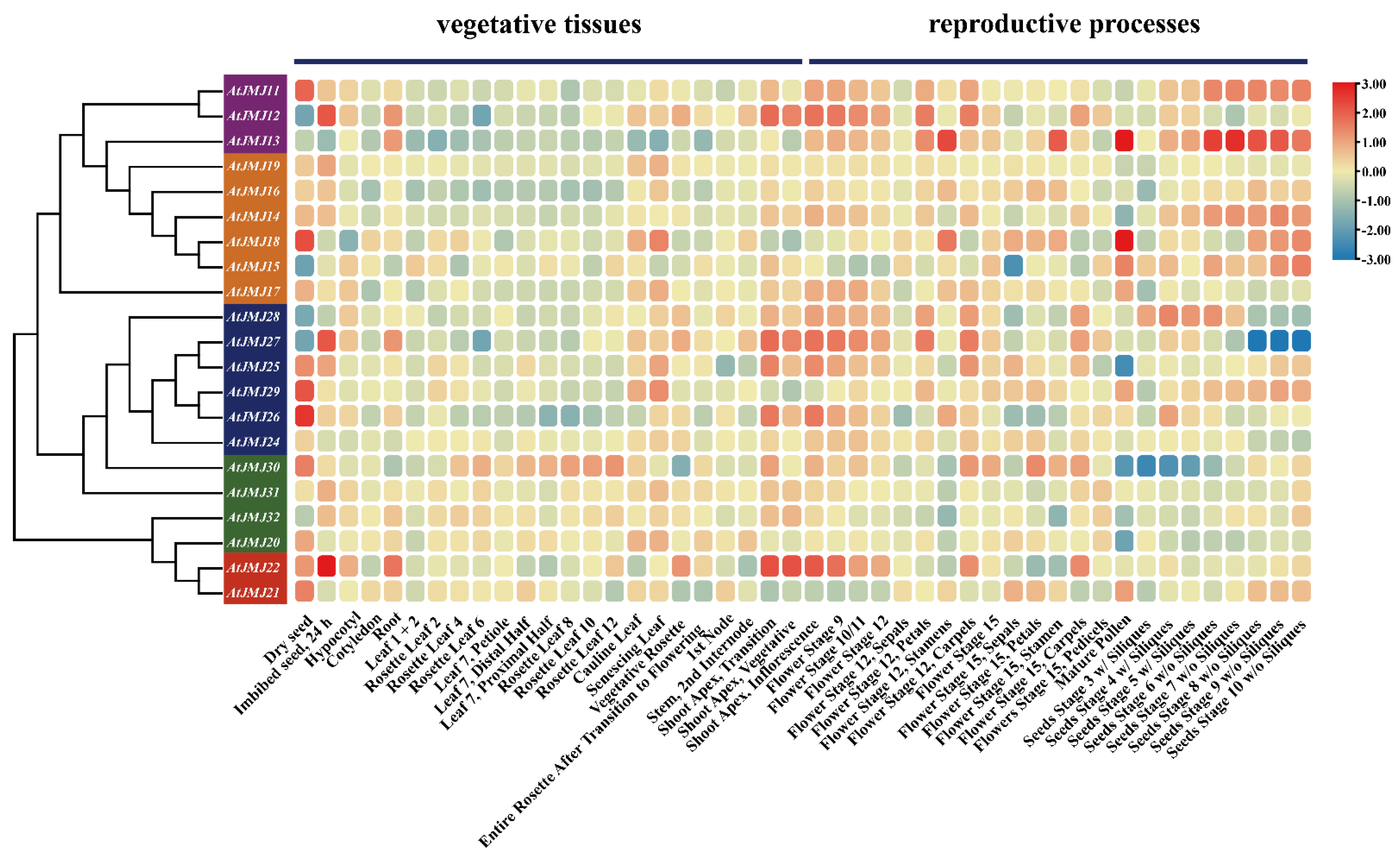
2.5. Expression Profiles of JmjC Genes in Different Tissues
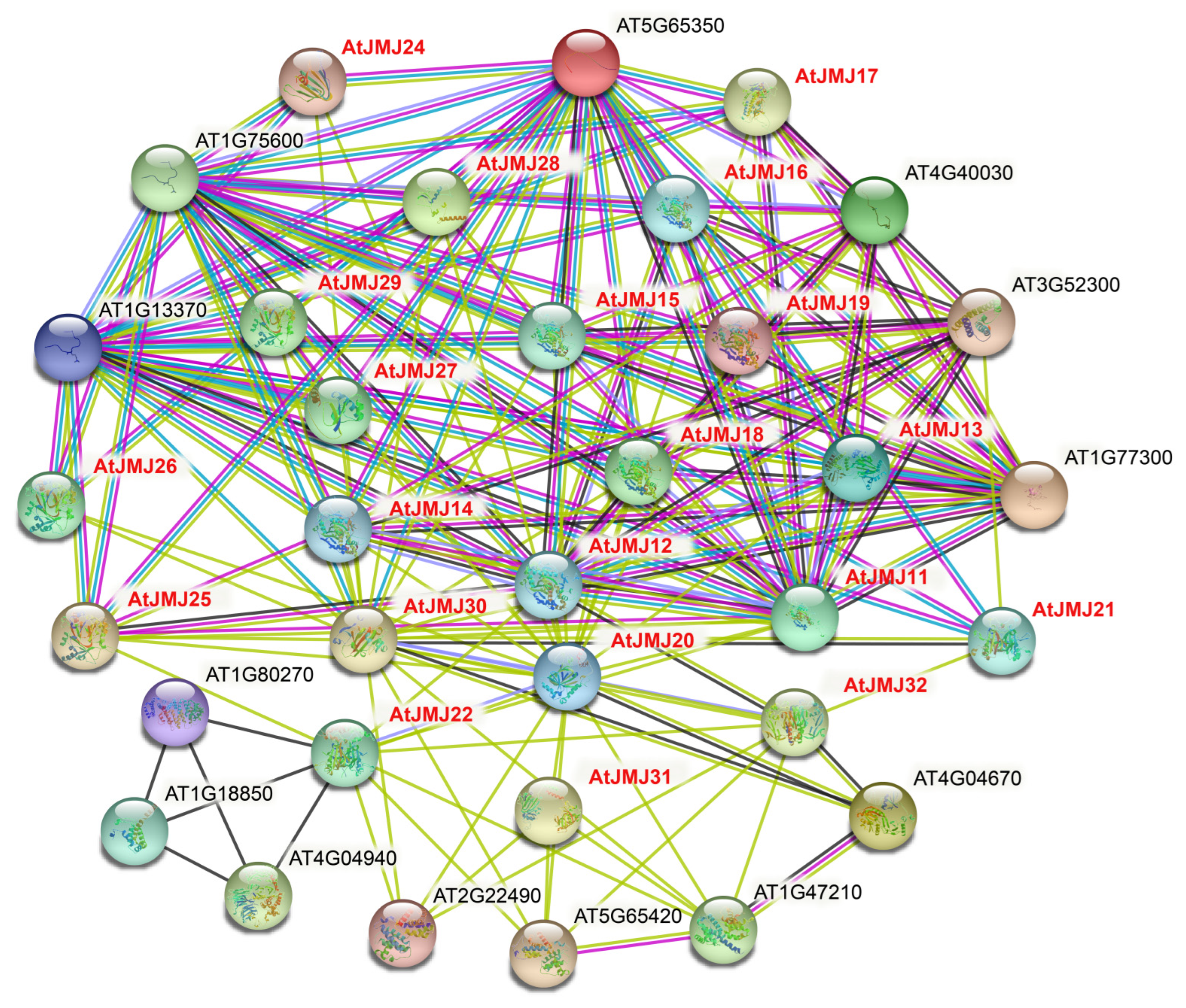
2.6. Interaction of the JmjC Protein in Plants
3. Discussion
4. Materials and Methods
4.1. Genome-Wide Identification of JmjC Family Genes in Plants
4.2. Evolutionary Analysis
4.3. Analysis of Conserved Amino Acid Residues of JmjC
4.4. Analysis of Cis-Acting Elements in the Promoter Regions of JmjC Genes
4.5. Expression Pattern of Plant JmjC Genes in Different Tissues
4.6. Prediction of JmjC Protein Interactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Venegas, R.; Abdallat, A.A.; Guo, M.; Alfano, J.R.; Avramova, Z. Epigenetic control of a transcription factor at the cross section of two antagonistic pathways. Epigenetics 2007, 2, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, B.R. The logic of chromatin architecture and remodelling at promoters. Nature 2009, 461, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Xiao, J.; Lee, U.S.; Wagner, D. Tug of war: Adding and removing histone lysine methylation in Arabidopsis. Curr. Opin. Plant Biol. 2016, 34, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone methylation in higher plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Mosammaparast, N.; Shi, Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Bioch. 2010, 79, 155–179. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Chen, C.; Jiang, L.; Zhang, J.; Ren, Q. Genome-wide identification, classification and expression analysis of the JmjC domain-containing histone demethylase gene family in maize. BMC Genom. 2019, 20, 256. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, X.; Cheng, L.; Liu, Y.; Wang, H.; Ke, D.; Yuan, H.; Zhang, L.; Wang, L. Genome-wide analysis of soybean JmjC domain-containing proteins suggests evolutionary conservation following whole-genome duplication. Front. Plant Sci. 2016, 7, 1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowrasia, S.; Panda, A.K.; Rawal, H.C.; Kaur, H.; Mondal, T.K. Identification of jumonjiC domain containing gene family among the Oryza species and their expression analysis in FL478, a salt tolerant rice genotype. Plant Physiol. Biochem. 2018, 130, 43–53. [Google Scholar] [CrossRef]
- Lu, F.; Li, G.; Cui, X.; Liu, C.; Wang, X.J.; Cao, X. Comparative analysis of JmjC domain-containing proteins reveals the potential histone demethylases in Arabidopsis and rice. J. Integr. Plant Biol. 2008, 50, 886–896. [Google Scholar] [CrossRef]
- Luo, M.; Hung, F.-Y.; Yang, S.; Liu, X.; Wu, K. Histone lysine demethylases and their functions in plants. Plant Mol. Biol. Rep. 2013, 32, 558–565. [Google Scholar] [CrossRef]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 2007, 447, 601–605. [Google Scholar] [CrossRef]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.A.; Morohashi, K.; Grotewold, E.; Harmer, S.L. Arabidopsis JMJD5/JMJ30 acts independently of LUX ARRHYTHMO within the plant circadian clock to enable temperature compensation. Fron. Plant Sci. 2019, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Hu, Y.; Zhou, D.X. Epigenetic gene regulation by plant Jumonji group of histone demethylase. Biochim. Biophys. Acta 2011, 1809, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Noh, B.; Lee, S.H.; Kim, H.J.; Yi, G.; Shin, E.A.; Lee, M.; Jung, K.J.; Doyle, M.R.; Amasino, R.M.; Noh, Y.S. Divergent roles of a pair of homologous jumonji/zinc-finger-class transcription factor proteins in the regulation of Arabidopsis flowering time. Plant Cell 2004, 16, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Han, Z.; Cao, Y.; Fan, D.; Li, H.; Mo, H.; Feng, Y.; Liu, L.; Wang, Z.; Yue, Y.; et al. A companion cell-dominant and developmentally regulated H3K4 demethylase controls flowering time in Arabidopsis via the repression of FLC expression. PLoS Genet. 2012, 8, e1002664. [Google Scholar] [CrossRef]
- Yang, H.; Mo, H.; Fan, D.; Cao, Y.; Cui, S.; Ma, L. Overexpression of a histone H3K4 demethylase, JMJ15, accelerates flowering time in Arabidopsis. Plant Cell Rep. 2012, 31, 1297–1308. [Google Scholar] [CrossRef]
- Park, J.; Lee, N.; Kim, W.; Lim, S.; Choi, G. ABI3 and PIL5 collaboratively activate the expression of SOMNUS by directly binding to its promoter in imbibed Arabidopsis seeds. Plant Cell 2011, 23, 1404–1415. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.N.; Ryu, J.Y.; Jeong, Y.M.; Park, J.; Song, J.J.; Amasino, R.M.; Noh, B.; Noh, Y.S. Control of seed germination by light-induced histone arginine demethylation activity. Dev. Cell 2012, 22, 736–748. [Google Scholar] [CrossRef] [Green Version]
- Saze, H.; Shiraishi, A.; Miura, A.; Kakutani, T. Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science 2008, 319, 462–465. [Google Scholar] [CrossRef]
- Sun, Q.; Zhou, D.X. Rice jmjC domain-containing gene JMJ706 encodes H3K9 demethylase required for floral organ development. Proc. Nat. Acad. Sci. USA 2008, 105, 13679–13684. [Google Scholar] [CrossRef] [Green Version]
- Audonnet, L.; Shen, Y.; Zhou, D.X. JMJ24 antagonizes histone H3K9 demethylase IBM1/JMJ25 function and interacts with RNAi pathways for gene silencing. Gene Expr. Patterns 2017, 25–26, 1–7. [Google Scholar] [CrossRef]
- Lu, S.X.; Knowles, S.M.; Webb, C.J.; Celaya, R.B.; Cha, C.; Siu, J.P.; Tobin, E.M. The Jumonji C domain-containing protein JMJ30 regulates period length in the Arabidopsis circadian clock. Plant Physiol. 2011, 155, 906–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Conde, E.S.N.; Audonnet, L.; Servet, C.; Wei, W.; Zhou, D.X. Over-expression of histone H3K4 demethylase gene JMJ15 enhances salt tolerance in Arabidopsis. Front. Plant Sci. 2014, 5, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Wang, L.; Wang, L.; Liu, L.; Li, L.; Sun, L.; Rao, Q.; Zhang, J.; Huang, S. JMJ704 positively regulates rice defense response against Xanthomonas oryzae pv. oryzae infection via reducing H3K4me2/3 associated with negative disease resistance regulators. BMC Plant Biol. 2015, 15, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, X.; Zhong, X.; Zhao, Y.; Liu, X.; Zhou, S.; Cheng, S.; Zhou, D.X. Jumonji C domain protein JMJ705-mediated removal of histone H3 lysine 27 trimethylation is involved in defense-related gene activation in rice. Plant Cell 2013, 25, 4725–4736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Guo, S.; Xu, Y.; Li, C.; Zhang, Z.; Zhang, D.; Xu, S.; Zhang, C.; Chong, K. OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiol. 2014, 165, 160–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Feng, J.; Liu, W.; Ren, Z.; Zhao, J.; Pei, X.; Liu, Y.; Yang, D.; Ma, X. Characterization and stress response of the JmjC domain-containing histone demethylase gene family in the allotetraploid cotton species Gossypium hirsutum. Plants 2020, 9, 1617. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Qiao, K.; Fan, S.; Ma, Q. Genome-wide analysis of JMJ-C histone demethylase family involved in salt-tolerance in Gossypium hirsutum L. Plant Physiol. Biochem. 2021, 158, 420–433. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Xu, B.; Tian, R.; Zuo, Y. Modular arrangements of sequence motifs determine the functional diversity of KDM proteins. Brief. Bioinform. 2021, 22, bbaa215. [Google Scholar] [CrossRef]
- Yu, T.F.; Zhao, W.Y.; Fu, J.D.; Liu, Y.W.; Chen, M.; Zhou, Y.B.; Ma, Y.Z.; Xu, Z.S.; Xi, Y.J. Genome-Wide Analysis of CDPK Family in Foxtail Millet and Determination of SiCDPK24 Functions in Drought Stress. Front. Plant Sci. 2018, 9, 651. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217–218, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.X. Regulatory mechanism of plant gene transcription by GT-elements and GT-factors. Trends Plant Sci. 1999, 4, 210–214. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Huang, S.N.; Wang, G.; Xuan, J.P.; Guo, Z.R. Overexpression of Actinidia deliciosa pyruvate decarboxylase 1 gene enhances waterlogging stress in transgenic Arabidopsis thaliana. Plant Physiol. Biochem. 2016, 106, 244–252. [Google Scholar] [CrossRef]
- Wasternack, C.; Hause, B. Jasmonates and octadecanoids: Signals in plant stress responses and development. Prog. Nucleic Acid Res. Mol. Biol. 2002, 72, 165–221. [Google Scholar]
- Yu, X.; Li, L.; Li, L.; Guo, M.; Chory, J.; Yin, Y. Modulation of brassinosteroid-regulated gene expression by Jumonji domain-containing proteins ELF6 and REF6 in Arabidopsis. Proc. Nat. Acad. Sci. USA 2008, 105, 7618–7623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Cui, X.; Zhang, S.; Jenuwein, T.; Cao, X. Arabidopsis REF6 is a histone H3 lysine 27 demethylase. Nat. Genet. 2011, 43, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Hu, H.; Ren, H.; Yang, Z.; Qiu, Q.; Qi, W.; Liu, X.; Chen, X.; Cui, X.; Li, S.; et al. The Arabidopsis H3K27me3 demethylase JUMONJI 13 is a temperature and photoperiod dependent flowering repressor. Nat. Commun. 2019, 10, 1303. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Shafiq, S.; Berr, A.; Shen, W.H. Genome-wide gene expression profiling to investigate molecular phenotypes of Arabidopsis mutants deprived in distinct histone methyltransferases and demethylases. Genom. Data 2015, 4, 143–145. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, W.; Ma, Z.; Zhu, W.; Jia, L. Transcriptional characterization and response to defense elicitors of mevalonate pathway genes in cotton (Gossypium arboreum L.). PeerJ 2019, 7, e8123. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Letunic, I.; Copley, R.R.; Schmidt, S.; Ciccarelli, F.D.; Doerks, T.; Schultz, J.; Ponting, C.P.; Bork, P. SMART 4.0: Towards genomic data integration. Nucleic Acids Res. 2004, 32, D142–D144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.B. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics 2003, 19, 1585–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhang, Z.; Zhu, W.; Ren, Z.; Jia, L.; Li, W.; Ma, Z. Evolutionary conservation and divergence of genes encoding 3-Hydroxy-3-methylglutaryl coenzyme A synthase in the allotetraploid cotton species Gossypium hirsutum. Cells 2019, 8, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Li, W.; Sun, K.; Ren, Z.; Song, C.; Pei, X.; Liu, Y.; Wang, Z.; He, K.; Zhang, F.; Zhou, X.; et al. Molecular evolution and stress and phytohormone responsiveness of SUT genes in Gossypium hirsutum. Front Genet. 2018, 9, 494. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N.J. An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2007, 2, e718. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Palenik, B.; Grimwood, J.; Aerts, A.; Rouzé, P.; Salamov, A.; Putnam, N.; Dupont, C.; Jorgensen, R.; Derelle, E.; Rombauts, S.; et al. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc. Natl. Acad. Sci. USA 2007, 104, 7705–7710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochnik, S.E.; Umen, J.; Nedelcu, A.M.; Hallmann, A.; Miller, S.M.; Nishii, I.; Ferris, P.; Kuo, A.; Mitros, T.; Fritz-Laylin, L.K.; et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 2010, 329, 223–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Xian, W.; Fu, Y.; Marin, B.; Keller, J.; Wu, T.; Sun, W.; Li, X.; Xu, Y.; Zhang, Y.; et al. Genomes of subaerial zygnematophyceae provide insights into land plant evolution. Cell 2019, 179, 1057–1067.e14. [Google Scholar] [CrossRef]
- Nishiyama, T.; Sakayama, H.; de Vries, J.; Buschmann, H.; Saint-Marcoux, D.; Ullrich, K.K.; Haas, F.B.; Vanderstraeten, L.; Becker, D.; Lang, D.; et al. The chara genome: Secondary complexity and implications for plant terrestrialization. Cell 2018, 174, 448–464.e424. [Google Scholar] [CrossRef] [Green Version]
- Hori, K.; Maruyama, F.; Fujisawa, T.; Togashi, T.; Yamamoto, N.; Seo, M.; Sato, S.; Yamada, T.; Mori, H.; Tajima, N.; et al. Klebsormidium flaccidum genome reveals primary factors for plant terrestrial adaptation. Nat. Commun. 2014, 5, 3978. [Google Scholar] [CrossRef]
- Lang, D.; Ullrich, K.K.; Murat, F.; Fuchs, J.; Jenkins, J.; Haas, F.B.; Piednoel, M.; Gundlach, H.; Van Bel, M.; Meyberg, R.; et al. The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J. 2018, 93, 515–533. [Google Scholar] [CrossRef] [Green Version]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; de Pamphilis, C.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef] [Green Version]
- Li, F.W.; Brouwer, P.; Carretero-Paulet, L.; Cheng, S.; de Vries, J.; Delaux, P.M.; Eily, A.; Koppers, N.; Kuo, L.Y.; Li, Z.; et al. Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nat. Plants 2018, 4, 460–472. [Google Scholar] [CrossRef] [Green Version]
- Wan, T.; Liu, Z.M.; Li, L.F.; Leitch, A.R.; Leitch, I.J.; Lohaus, R.; Liu, Z.J.; Xin, H.P.; Gong, Y.B.; Liu, Y.; et al. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 2018, 4, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, F.; Zhang, X.; Li, Z.; Zhao, Y.; Lohaus, R.; Chang, X.; Dong, W.; Ho, S.Y.W.; Liu, X.; et al. The water lily genome and the early evolution of flowering plants. Nature 2020, 577, 79–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef] [PubMed]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B.; et al. The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, J.L.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.C.; Estep, M.; Feng, L.; Vaughn, J.N.; Grimwood, J.; et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, R.; VanBuren, R.; Wai, C.M.; Tang, H.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.L.; Chen, J.; Biggers, E.; et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.L.; Rouzé, P.; Verhelst, B.; Lin, Y.C.; Bayer, T.; Collen, J.; Dattolo, E.; De Paoli, E.; Dittami, S.; Maumus, F.; et al. The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 2016, 530, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Filiault, D.L.; Ballerini, E.S.; Mandáková, T.; Aköz, G.; Derieg, N.J.; Schmutz, J.; Jenkins, J.; Grimwood, J.; Shu, S.; Hayes, R.D.; et al. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. Elife 2018, 7, e36426. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar]
- Argout, X.; Martin, G.; Droc, G.; Fouet, O.; Labadie, K.; Rivals, E.; Aury, J.M.; Lanaud, C. The cacao Criollo genome v2.0: An improved version of the genome for genetic and functional genomic studies. BMC Genom. 2017, 18, 730. [Google Scholar] [CrossRef]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Zhang, Z.; Long, Y.; Huo, W.; Zhang, Y.; Yang, X.; Zhang, J.; Li, X.; Du, Q.; Liu, W.; et al. Evolutionary History and Functional Diversification of the JmjC Domain-Containing Histone Demethylase Gene Family in Plants. Plants 2022, 11, 1041. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11081041
Ma S, Zhang Z, Long Y, Huo W, Zhang Y, Yang X, Zhang J, Li X, Du Q, Liu W, et al. Evolutionary History and Functional Diversification of the JmjC Domain-Containing Histone Demethylase Gene Family in Plants. Plants. 2022; 11(8):1041. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11081041
Chicago/Turabian StyleMa, Shifeng, Zhiqiang Zhang, Yingqiang Long, Wenqi Huo, Yuzhi Zhang, Xiaoqing Yang, Jie Zhang, Xinyang Li, Qiying Du, Wei Liu, and et al. 2022. "Evolutionary History and Functional Diversification of the JmjC Domain-Containing Histone Demethylase Gene Family in Plants" Plants 11, no. 8: 1041. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11081041