
A Systematic Evaluation of Semispecific Peptide Search Parameter Enables Identification of Previously Undescribed N-Terminal Peptides and Conserved Proteolytic Processing in Cancer Cell Lines

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Proteomic Sample Preparation of the Different Specimens
2.1.1. Murine Formalin-Fixed, Paraffin-Embedded FFPE Kidney Tissue
2.1.2. Human FFPE Liver Metastasis Sample
2.1.3. Human Embryonic Kidney (HEK293T) Whole Proteome Samples
2.2. LC-MS/MS Analysis
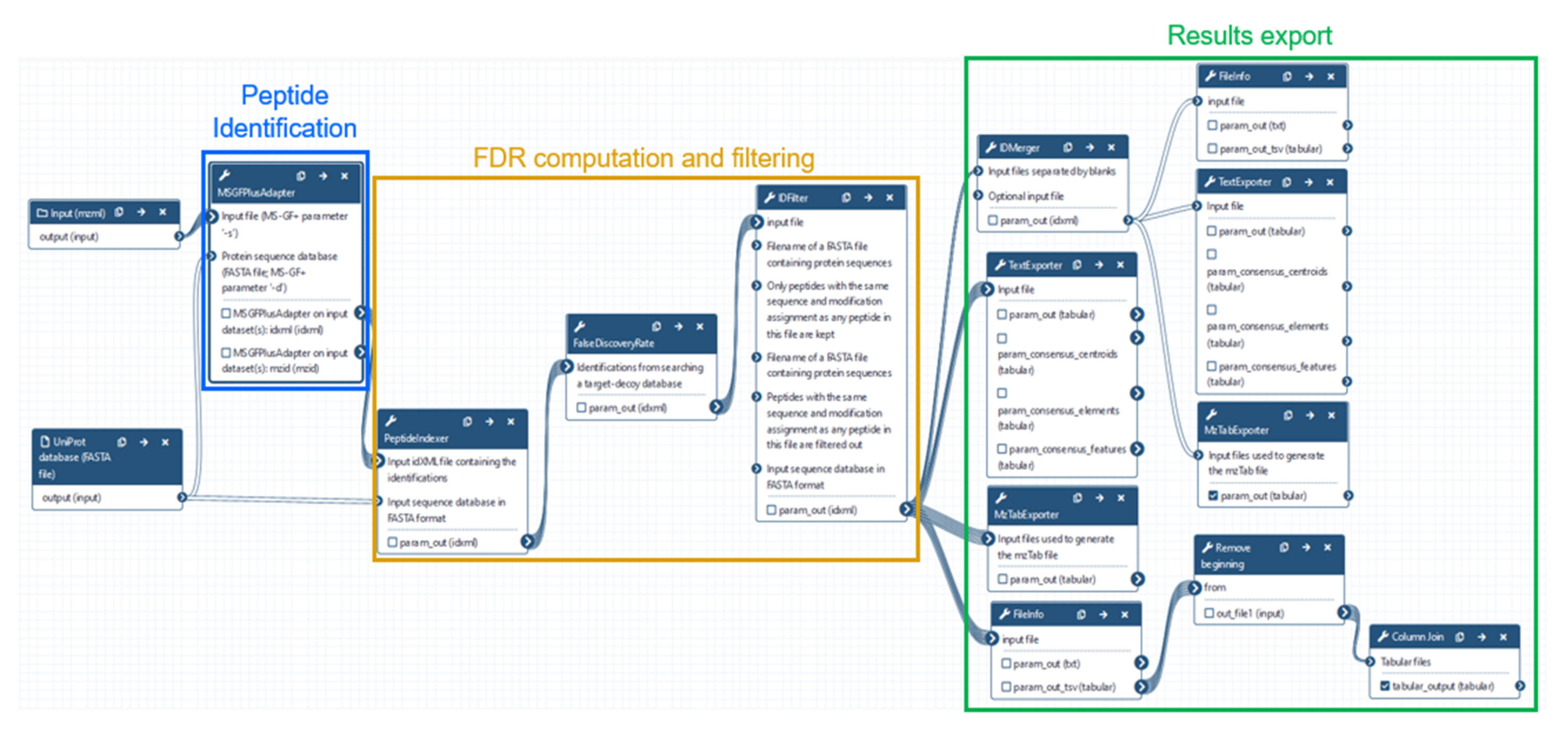
2.3. Data Analysis Using OpenMS
2.4. Reanalysis of Published NCI-60 Data
3. Results
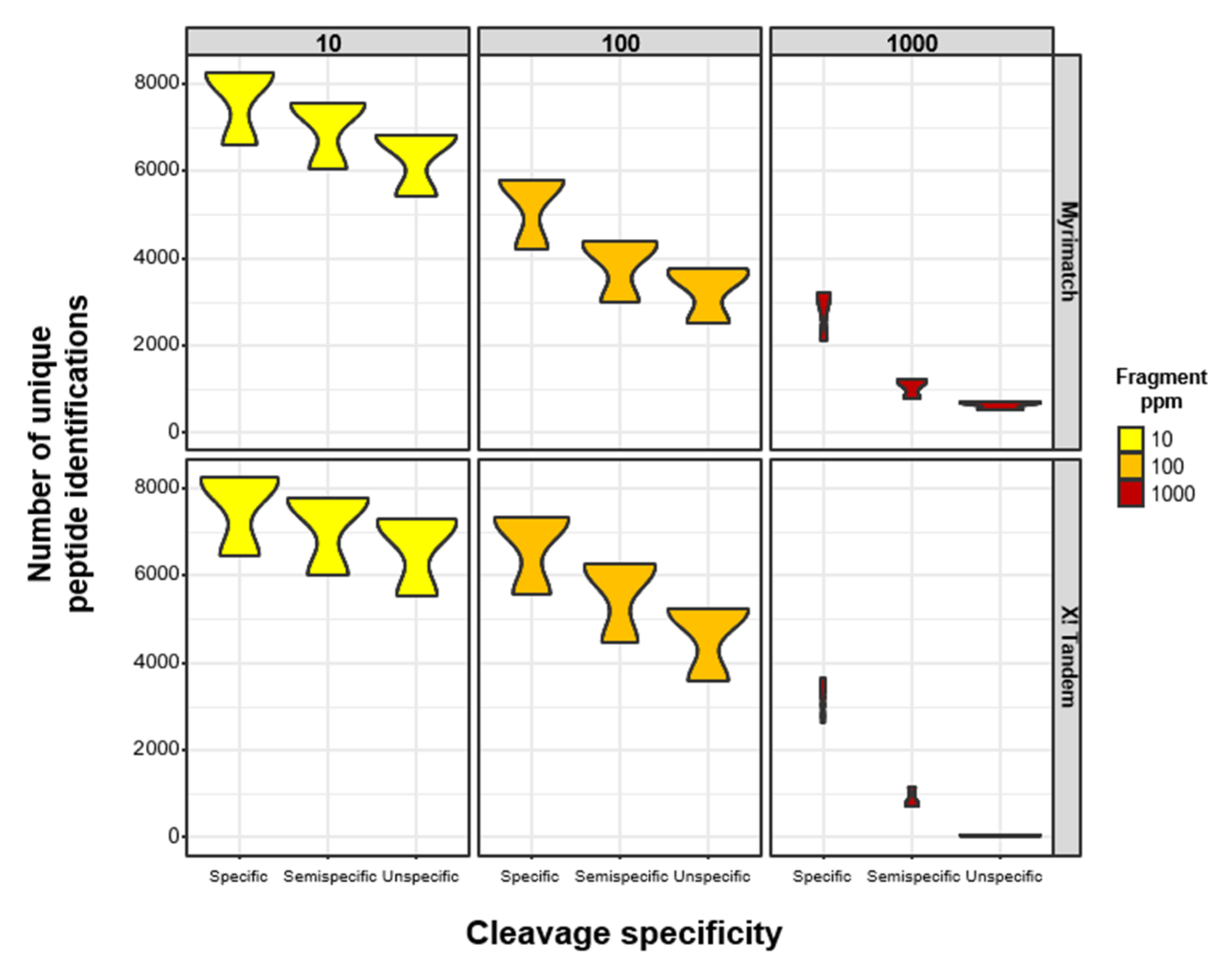
3.1. Reduced Enzymatic Constraints Combined with Increased Fragment Mass Tolerances Lead to Fewer Peptide Identifications in High-Resolution LC-MS/MS (MS) Data
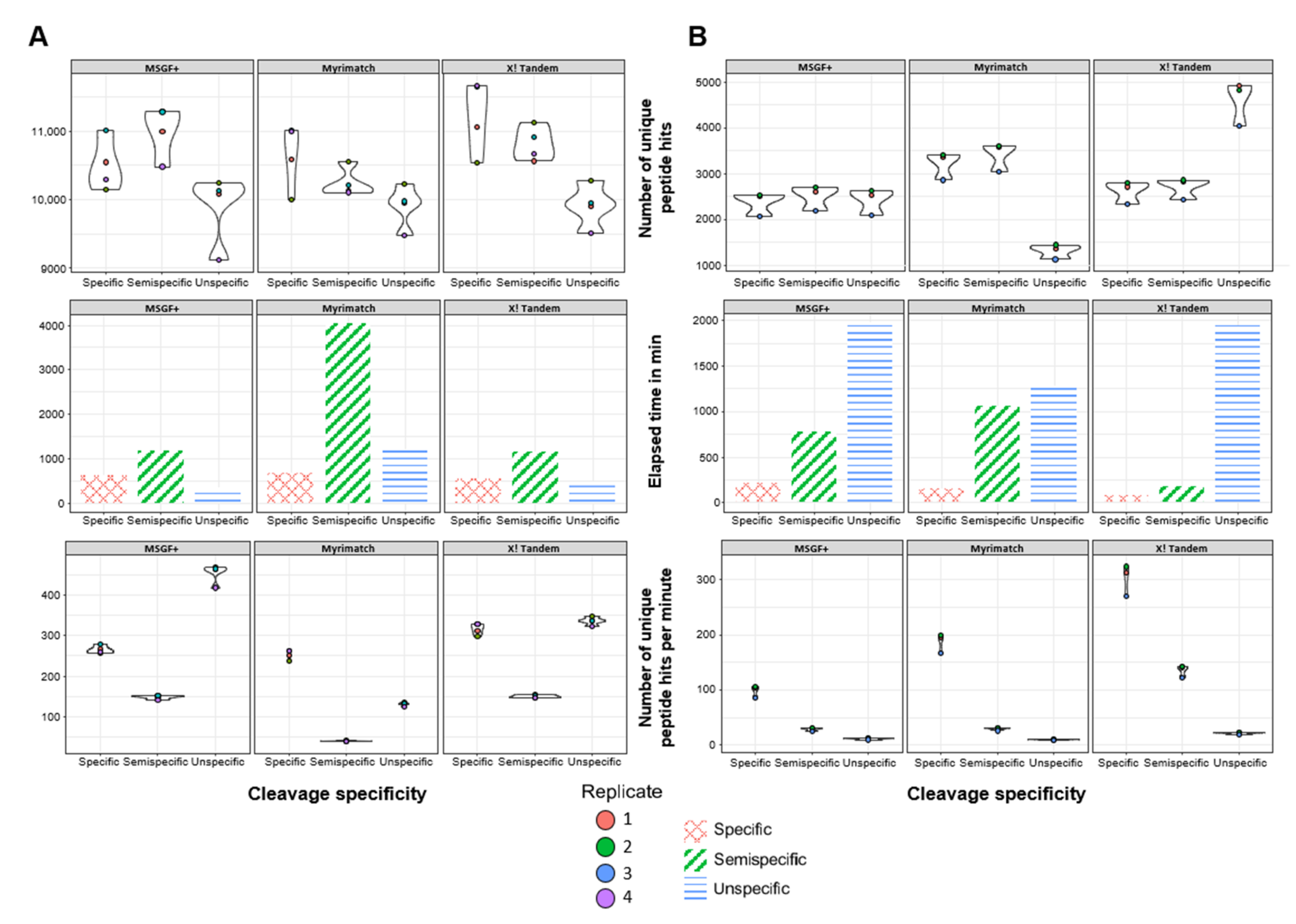
3.2. Semispecific PSMs Yield Comparable Numbers of Peptide Identifications Using Different Open-Source Search Engines
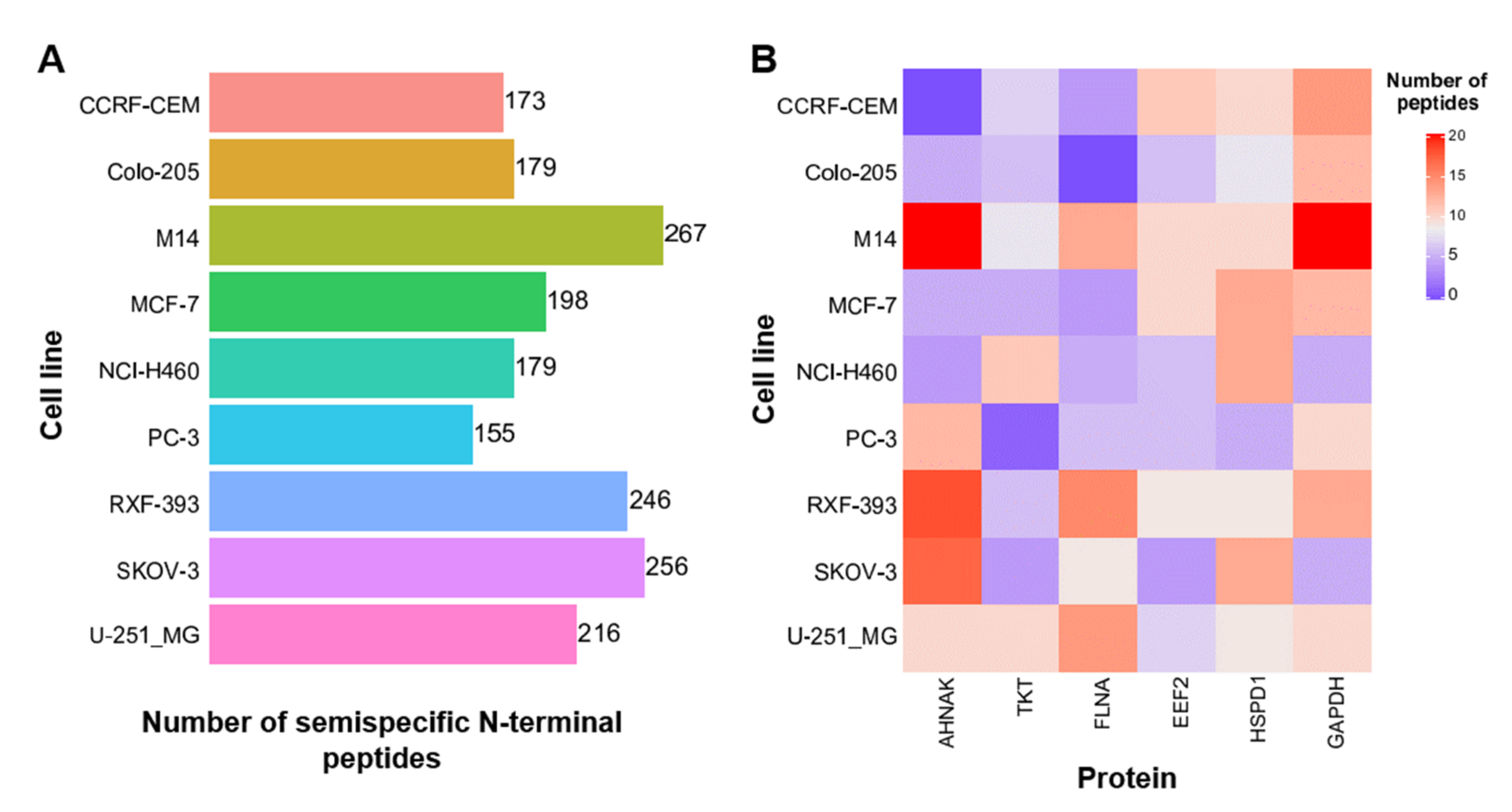
3.3. Semispecific Reanalysis of Published Data Enables the Identification of Previously Non-Described N-Terminal Peptides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nat. Cell Biol. 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Föll, M.C.; Fahrner, M.; Gretzmeier, C.; Thoma, K.; Biniossek, M.L.; Kiritsi, D.; Meiss, F.; Schilling, O.; Nyström, A.; Kern, J.S. Identification of tissue damage, extracellular matrix remodeling and bacterial challenge as common mechanisms associated with high-risk cutaneous squamous cell carcinomas. Matrix Biol. 2018, 66, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Oria, V.; Bronsert, P.; Thomsen, A.; Föll, M.; Zamboglou, C.; Hannibal, L.; Behringer, S.; Biniossek, M.; Schreiber, C.; Grosu, A.; et al. Proteome Profiling of Primary Pancreatic Ductal Adenocarcinomas Undergoing Additive Chemoradiation Link ALDH1A1 to Early Local Recurrence and Chemoradiation Resistance. Transl. Oncol. 2018, 11, 1307–1322. [Google Scholar] [CrossRef]
- Müller, A.-K.; Föll, M.; Heckelmann, B.; Kiefer, S.; Werner, M.; Schilling, O.; Biniossek, M.L.; Jilg, C.A.; Drendel, V. Proteomic Characterization of Prostate Cancer to Distinguish Nonmetastasizing and Metastasizing Primary Tumors and Lymph Node Metastases. Neoplasia 2018, 20, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Brosch, M.; Swamy, S.; Hubbard, T.; Choudhary, J. Comparison of Mascot and X!Tandem Performance for Low and High Accuracy Mass Spectrometry and the Development of an Adjusted Mascot Threshold. Mol. Cell. Proteom. 2008, 7, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Ma, J.; Wang, P.; Zhu, Y.; Zhou, B.B.; Yang, Y.H. Improving X!Tandem on Peptide Identification from Mass Spectrometry by Self-Boosted Percolator. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1273–1280. [Google Scholar] [CrossRef]
- Hsieh, E.J.; Hoopmann, M.R.; MacLean, B.; MacCoss, M.J. Comparison of Database Search Strategies for High Precursor Mass Accuracy MS/MS Data. J. Proteome Res. 2009, 9, 1138–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabb, D.L.; Fernando, C.G.; Chambers, M.C. MyriMatch: Highly Accurate Tandem Mass Spectral Peptide Identification by Multivariate Hypergeometric Analysis. J. Proteome Res. 2007, 6, 654–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, P.; Arnold, R.J.; Clemmer, D.E.; Li, Y.; Reilly, J.P.; Sheng, Q.; Tang, H.; Xun, Z.; Zeng, R.; Radivojac, P. Fast and accurate identification of semi-tryptic peptides in shotgun proteomics. Bioinformatics 2007, 24, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Murphy, G.; Murthy, A.; Khokha, R. Clipping, shedding and RIPping keep immunity on cue. Trends Immunol. 2008, 29, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kleifeld, O.; Doucet, A.; Keller, U.A.D.; Prudova, A.; Schilling, O.; Kainthan, R.K.; Starr, A.E.; Foster, L.J.; Kizhakkedathu, J.N.; Overall, C.M. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 2010, 28, 281–288. [Google Scholar] [CrossRef]
- Schilling, O.; Overall, C.M. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 2008, 26, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Schilling, O.; Huesgen, P.F.; Barré, O.; Keller, U.A.D.; Overall, C.M. Characterization of the prime and non-prime active site specificities of proteases by proteome-derived peptide libraries and tandem mass spectrometry. Nat. Protoc. 2011, 6, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Coradin, M.; Karch, K.R.; Garcia, B.A. Monitoring proteolytic processing events by quantitative mass spectrometry. Expert Rev. Proteom. 2017, 14, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uliana, F.; Vizovišek, M.; Acquasaliente, L.; Ciuffa, R.; Fossati, A.; Frommelt, F.; Goetze, S.; Wollscheid, B.; Gstaiger, M.; De Filippis, V.; et al. Mapping specificity, cleavage entropy, allosteric changes and substrates of blood proteases in a high-throughput screen. Nat. Commun. 2021, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Eckhard, U.; Dufour, A.; Solis, N.; Overall, C.M. Proteolytic Cleavage—Mechanisms, Function, and “Omic” Approaches for a Near-Ubiquitous Posttranslational Modification. Chem. Rev. 2018, 118, 1137–1168. [Google Scholar] [CrossRef] [PubMed]
- Gholami, A.M.; Hahne, H.; Wu, Z.; Auer, F.J.; Meng, C.; Wilhelm, M.; Kuster, B. Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Rep. 2013, 4, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Föll, M.C.; Fahrner, M.; Oria, V.O.; Kühs, M.; Biniossek, M.L.; Werner, M.; Bronsert, P.; Schilling, O. Reproducible proteomics sample preparation for single FFPE tissue slices using acid-labile surfactant and direct trypsinization. Clin. Proteom. 2018, 15, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef]
- Röst, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Junker, J.; Bielow, C.; Bertsch, A.; Sturm, M.; Reinert, K.; Kohlbacher, O. TOPPAS: A Graphical Workflow Editor for the Analysis of High-Throughput Proteomics Data. J. Proteome Res. 2012, 11, 3914–3920. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, J.; Arentz, G.; Hoffmann, P. Proteomic developments in the analysis of formalin-fixed tissue. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2015, 1854, 559–580. [Google Scholar] [CrossRef] [PubMed]
- Shahinian, J.H.; Mayer, B.; Tholen, S.; Brehm, K.; Biniossek, M.L.; Füllgraf, H.; Kiefer, S.; Heizmann, U.; Heilmann, C.; Rüter, F.; et al. Proteomics highlights decrease of matricellular proteins in left ventricular assist device therapy†. Eur. J. Cardio Thorac. Surg. 2017, 51, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Föll, M.C.; Moritz, L.; Wollmann, T.; Stillger, M.N.; Vockert, N.; Werner, M.; Bronsert, P.; Rohr, K.; Grüning, B.A.; Schilling, O. Accessible and reproducible mass spectrometry imaging data analysis in Galaxy. GigaScience 2019, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Na, C.H.; Barbhuiya, M.; Kim, M.-S.; Verbruggen, S.; Eacker, S.M.; Pletnikova, O.; Troncoso, J.C.; Halushka, M.K.; Menschaert, G.; Overall, C.M.; et al. Discovery of noncanonical translation initiation sites through mass spectrometric analysis of protein N termini. Genome Res. 2017, 28, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Tholen, S.; Biniossek, M.L.; Gansz, M.; Gomez-Auli, A.; Bengsch, F.; Noel, A.; Kizhakkedathu, J.N.; Boerries, M.; Busch, H.; Reinheckel, T.; et al. Deletion of Cysteine Cathepsins B or L Yields Differential Impacts on Murine Skin Proteome and Degradome. Mol. Cell. Proteom. 2013, 12, 611–625. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.L.; Kristie, T.M. Autocatalytic proteolysis of the transcription factor-coactivator C1 (HCF): A potential role for proteolytic regulation of coactivator function. Proc. Natl. Acad. Sci. USA 2000, 97, 9425–9430. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accessions | Sequence | Function | AA Before | Position | Expected Gel Slice | Observed Gel Slice |
|---|---|---|---|---|---|---|
| sp|Q9C0E2|XPO4_HUMAN | .(Acetyl)AAALGPPEVIAQLENAAK | Double clipping ** | M | 3 | 5 | 6 |
| sp|Q9BWU0|NADAP_HUMAN | .(Acetyl)ADILSQSETLASQDLSGDFKKPALPVSPAAR | Potential ATIS * | M | 56 | 7 | 7 |
| sp|O14980|XPO1_HUMAN | .(Acetyl)TM(Oxidation)LADHAAR | ATIS * | M | 6 | 6 | 7 |
| sp|Q9Y4L1|HYOU1_HUMAN | LAVM(Oxidation)SVDLGSESM(Oxidation)K | Removal of signal peptide | T | 33 | 6 | 6 |
| sp|P51610|HCFC1_HUMAN | THETGTTNTATTSNAGSAQR | Cleavage by autolysis/HCF C-terminal chain 4 | E | 1296 | 7 | 8 |
| sp|Q8N766|EMC1_HUMAN | VYEDQVGK | Removal of signal peptide | A | 22 | 6 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahrner, M.; Kook, L.; Fröhlich, K.; Biniossek, M.L.; Schilling, O. A Systematic Evaluation of Semispecific Peptide Search Parameter Enables Identification of Previously Undescribed N-Terminal Peptides and Conserved Proteolytic Processing in Cancer Cell Lines. Proteomes 2021, 9, 26. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes9020026
Fahrner M, Kook L, Fröhlich K, Biniossek ML, Schilling O. A Systematic Evaluation of Semispecific Peptide Search Parameter Enables Identification of Previously Undescribed N-Terminal Peptides and Conserved Proteolytic Processing in Cancer Cell Lines. Proteomes. 2021; 9(2):26. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes9020026
Chicago/Turabian StyleFahrner, Matthias, Lucas Kook, Klemens Fröhlich, Martin L. Biniossek, and Oliver Schilling. 2021. "A Systematic Evaluation of Semispecific Peptide Search Parameter Enables Identification of Previously Undescribed N-Terminal Peptides and Conserved Proteolytic Processing in Cancer Cell Lines" Proteomes 9, no. 2: 26. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes9020026