
Optical Sensing of Molecular Oxygen (O2) via Metal Oxide Photoluminescence: A Comparative Study of TiO2, SnO2 and ZnO

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Nanoparticles Preparations and Characterizations
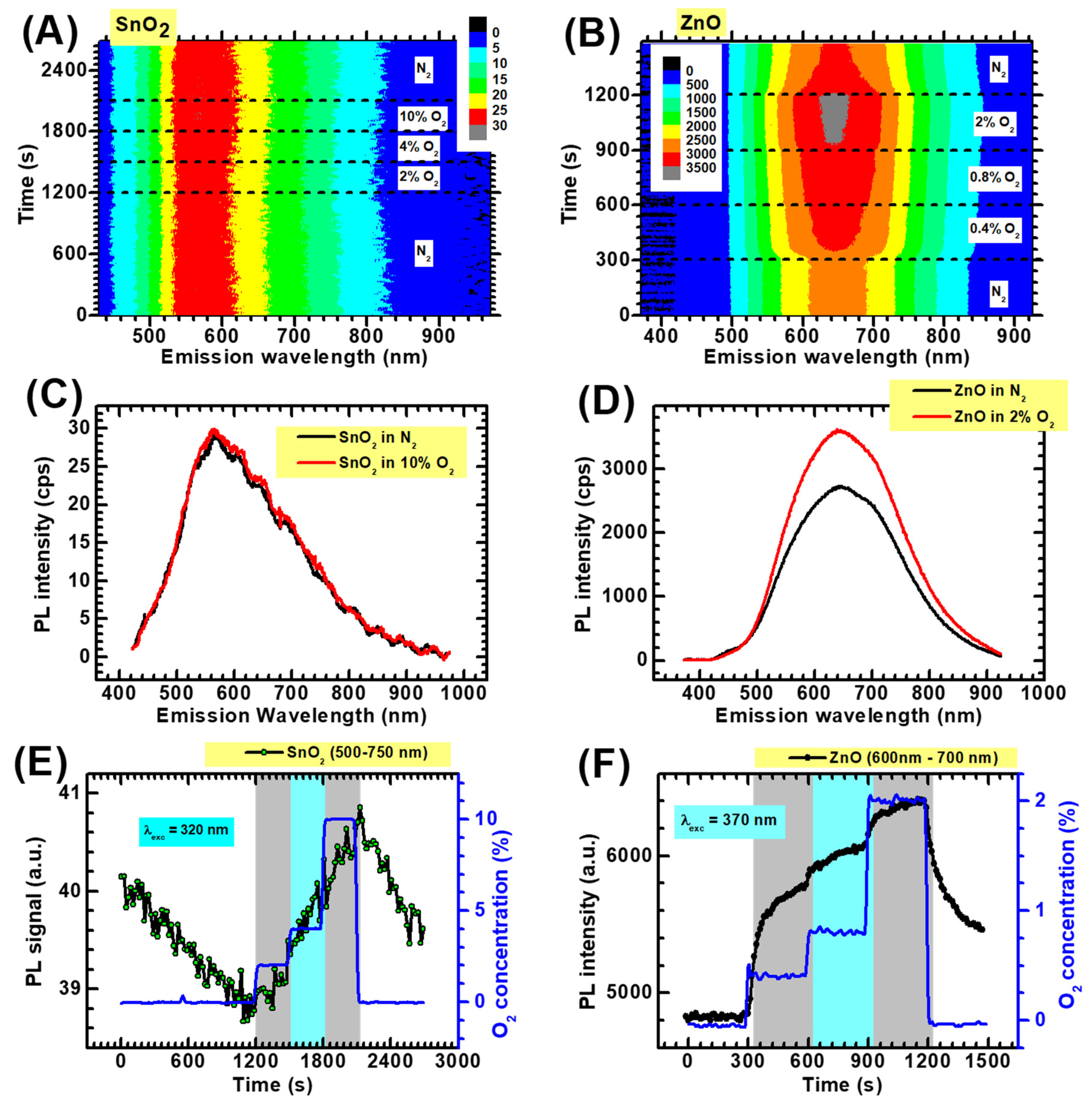
2.2. Photoluminescence Measurements
3. Results
3.1. Structural and Morphological Characterizations
3.2. Photoluminescence Response towards O2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Preininger, C.; Klimant, I.; Wolfbeis, O.S. Optical Fiber Sensor for Biological Oxygen Demand. Anal. Chem. 1994, 66, 1841–1846. [Google Scholar] [CrossRef]
- Crapo, J.D. Morphologic Changes in Pulmonary Oxygen Toxicity. Annu. Rev. Physiol. 1986, 48, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Mantell, L.L.; Lee, P.J. Signal Transduction Pathways in Hyperoxia-Induced Lung Cell Death. Mol. Genet. Metab. 2000, 71, 359–370. [Google Scholar] [CrossRef]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia Promotes Invasive Growth by Transcriptional Activation of the Met Protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.L. Hypoxia--a Key Regulatory Factor in Tumour Growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Ramamoorthy, R.; Dutta, P.K.; Akbar, S.A. Oxygen Sensors: Materials, Methods, Designs and Applications. J. Mater. Sci. 2003, 38, 4271–4282. [Google Scholar] [CrossRef]
- Bittig, H.C.; Körtzinger, A.; Neill, C.; van Ooijen, E.; Plant, J.N.; Hahn, J.; Johnson, K.S.; Yang, B.; Emerson, S.R. Oxygen Optode Sensors: Principle, Characterization, Calibration, and Application in the Ocean. Front. Mar. Sci. 2018, 4. [Google Scholar] [CrossRef]
- Quaranta, M.; Borisov, S.M.; Klimant, I. Indicators for Optical Oxygen Sensors. Bioanal. Rev. 2012, 4, 115–157. [Google Scholar] [CrossRef] [Green Version]
- Amao, Y. Probes and Polymers for Optical Sensing of Oxygen. Microchim. Acta 2003, 143, 1–12. [Google Scholar] [CrossRef]
- Wang, X.; Wolfbeis, O.S. Optical Methods for Sensing and Imaging Oxygen: Materials, Spectroscopies and Applications. Chem. Soc. Rev. 2014, 43, 3666–3761. [Google Scholar] [CrossRef] [Green Version]
- Gehlen, M.H. The Centenary of the Stern-Volmer Equation of Fluorescence Quenching: From the Single Line Plot to the SV Quenching Map. J. Photochem. Photobiol. C: Photochem. Rev. 2020, 42, 100338. [Google Scholar] [CrossRef]
- Xu, K.; Chen, Y.; Okhai, T.A.; Okhai, T.A.; Snyman, L.W. Micro Optical Sensors Based on Avalanching Silicon Light-Emitting Devices Monolithically Integrated on Chips. Opt. Mater. Express OME 2019, 9, 3985–3997. [Google Scholar] [CrossRef]
- Lipka, T.; Moldenhauer, L.; Wahn, L.; Trieu, H.K. Optofluidic Biomolecule Sensors Based on A-Si:H Microrings Embedded in Silicon–Glass Microchannels. Opt. Lett. 2017, 42, 1084–1087. [Google Scholar] [CrossRef]
- Coscia, U.; Ambrosone, G.; Lettieri, S.; Maddalena, P.; Rigato, V.; Restello, S.; Bobeico, E.; Tucci, M. Preparation of Microcrystalline Silicon–Carbon Films. Sol. Energy Mater. Sol. Cells 2005, 87, 433–444. [Google Scholar] [CrossRef]
- Freeman, R.; Willner, I. Optical Molecular Sensing with Semiconductor Quantum Dots (QDs). Chem. Soc. Rev. 2012, 41, 4067–4085. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, I. Porous Silicon Structures as Optical Gas Sensors. Sensors 2015, 15, 19968–19991. [Google Scholar] [CrossRef]
- Kim, H.-J.; Kim, Y.-Y.; Lee, K.-W. Multiparametric Sensor Based on DBR Porous Silicon for Detection of Ethanol Gas. Curr. Appl. Phys. 2010, 10, 181–183. [Google Scholar] [CrossRef]
- Comini, E. Metal Oxide Nano-Crystals for Gas Sensing. Anal. Chim. Acta 2006, 568, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Ambrosone, G.; Coscia, U.; Lettieri, S.; Maddalena, P.; Minarini, C. Optical, Structural and Electrical Properties of Μc-Si:H Films Deposited by SiH4+H2. Mater. Sci. Eng. B 2003, 101, 236–241. [Google Scholar] [CrossRef]
- Baratto, C.; Faglia, G.; Sberveglieri, G.; Gaburro, Z.; Pancheri, L.; Oton, C.; Pavesi, L. Multiparametric Porous Silicon Sensors. Sensors 2002, 2, 121–126. [Google Scholar] [CrossRef]
- Setaro, A.; Bismuto, A.; Lettieri, S.; Maddalena, P.; Comini, E.; Bianchi, S.; Baratto, C.; Sberveglieri, G. Optical Sensing of NO2 in Tin Oxide Nanowires at Sub-Ppm Level. Sens. Actuators B Chem. 2008, 130, 391–395. [Google Scholar] [CrossRef]
- Cretì, A.; Valerini, D.; Taurino, A.; Quaranta, F.; Lomascolo, M.; Rella, R. Photoluminescence Quenching Processes by NO 2 Adsorption in ZnO Nanostructured Films. J. Appl. Phys. 2012, 111, 073520. [Google Scholar] [CrossRef]
- Mercado, C.; Seeley, Z.; Bandyopadhyay, A.; Bose, S.; McHale, J.L. Photoluminescence of Dense Nanocrystalline Titanium Dioxide Thin Films: Effect of Doping and Thickness and Relation to Gas Sensing. ACS Appl. Mater. Interfaces 2011, 3, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Valencia, J.R.; Alcaire, M.; Romero-Gómez, P.; Macias-Montero, M.; Aparicio, F.J.; Borras, A.; Gonzalez-Elipe, A.R.; Barranco, A. Oxygen Optical Sensing in Gas and Liquids with Nanostructured ZnO Thin Films Based on Exciton Emission Detection. J. Phys. Chem. C 2014, 118, 9852–9859. [Google Scholar] [CrossRef]
- Pallotti, D.; Orabona, E.; Amoruso, S.; Maddalena, P.; Lettieri, S. Modulation of Mixed-Phase Titania Photoluminescence by Oxygen Adsorption. Appl. Phys. Lett. 2014, 105, 031903. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, C.; Ballarini, V.; Galli, M.; Liscidini, M.; Andreani, L.C.; Losurdo, M.; Bruno, G.; Lettieri, S.; Gesuele, F.; Maddalena, P.; et al. Amorphous Silicon Nitride: A Suitable Alloy for Optical Multilayered Structures. J. Non-Cryst. Solids 2006, 352, 1294–1297. [Google Scholar] [CrossRef]
- Passoni, L.; Criante, L.; Fumagalli, F.; Scotognella, F.; Lanzani, G.; Di Fonzo, F. Self-Assembled Hierarchical Nanostructures for High-Efficiency Porous Photonic Crystals. ACS Nano 2014, 8, 12167–12174. [Google Scholar] [CrossRef]
- Yu, J.; Lei, J.; Wang, L.; Zhang, J.; Liu, Y. TiO2 Inverse Opal Photonic Crystals: Synthesis, Modification, and Applications—A Review. J. Alloy. Compd. 2018, 769, 740–757. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, W.; Fan, Y.; Song, Q.; Xiao, S. TiO2 Metasurfaces: From Visible Planar Photonics to Photochemistry. Sci. Adv. 2019, 5, eaax0939. [Google Scholar] [CrossRef] [Green Version]
- Terracciano, M.; Galstyan, V.; Rea, I.; Casalino, M.; De Stefano, L.; Sbervegleri, G. Chemical Modification of TiO2 Nanotube Arrays for Label-Free Optical Biosensing Applications. Appl. Surf. Sci. 2017, 419, 235–240. [Google Scholar] [CrossRef]
- Chiarello, G.L.; Zuliani, A.; Ceresoli, D.; Martinazzo, R.; Selli, E. Exploiting the Photonic Crystal Properties of TiO2 Nanotube Arrays To Enhance Photocatalytic Hydrogen Production. ACS Catal. 2016, 6, 1345–1353. [Google Scholar] [CrossRef]
- Likodimos, V. Photonic Crystal-Assisted Visible Light Activated TiO2 Photocatalysis. Appl. Catal. B: Environ. 2018, 230, 269–303. [Google Scholar] [CrossRef]
- Corn, R.M.; Higgins, D.A. Optical Second Harmonic Generation as a Probe of Surface Chemistry. Chem. Rev. 1994, 94, 107–125. [Google Scholar] [CrossRef]
- Borbone, F.; Carella, A.; Caruso, U.; Roviello, G.; Tuzi, A.; Dardano, P.; Lettieri, S.; Maddalena, P.; Barsella, A. Large Second-Order NLO Activity in Poly(4-Vinylpyridine) Grafted with PdII and CuII Chromophoric Complexes with Tridentate Bent Ligands Containing Heterocycles. Eur. J. Inorg. Chem. 2008, 2008, 1846–1853. [Google Scholar] [CrossRef]
- Stevanovic, A.; Ma, S.; Yates, J.T. Effect of Gold Nanoparticles on Photoexcited Charge Carriers in Powdered TiO2 −Long Range Quenching of Photoluminescence. J. Phys. Chem. C 2014, 118, 21275–21280. [Google Scholar] [CrossRef]
- Stevanovic, A.; Yates, J.T. Electron Hopping through TiO2 Powder: A Study by Photoluminescence Spectroscopy. J. Phys. Chem. C 2013, 117, 24189–24195. [Google Scholar] [CrossRef]
- Lettieri, S.; Pavone, M.; Fioravanti, A.; Santamaria Amato, L.; Maddalena, P. Charge Carrier Processes and Optical Properties in TiO2 and TiO2-Based Heterojunction Photocatalysts: A Review. Materials 2021, 14, 1645. [Google Scholar] [CrossRef] [PubMed]
- Vequizo, J.J.M.; Kamimura, S.; Ohno, T.; Yamakata, A. Oxygen Induced Enhancement of NIR Emission in Brookite TiO2 Powders: Comparison with Rutile and Anatase TiO2 Powders. Phys. Chem. Chem. Phys. 2018, 20, 3241–3248. [Google Scholar] [CrossRef]
- Lettieri, S.; Pallotti, D.K.; Gesuele, F.; Maddalena, P. Unconventional Ratiometric-Enhanced Optical Sensing of Oxygen by Mixed-Phase TiO2. Appl. Phys. Lett. 2016, 109, 031905. [Google Scholar] [CrossRef] [Green Version]
- Kaur, N.; Singh, M.; Moumen, A.; Duina, G.; Comini, E. 1D Titanium Dioxide: Achievements in Chemical Sensing. Materials 2020, 13, 2974. [Google Scholar] [CrossRef] [PubMed]
- Preiß, E.M.; Rogge, T.; Krauß, A.; Seidel, H. Tin Oxide-Based Thin Films Prepared by Pulsed Laser Deposition for Gas Sensing. Sens. Actuators B Chem. 2016, 236, 865–873. [Google Scholar] [CrossRef]
- Sanz, M.; López-Arias, M.; Marco, J.F.; de Nalda, R.; Amoruso, S.; Ausanio, G.; Lettieri, S.; Bruzzese, R.; Wang, X.; Castillejo, M. Ultrafast Laser Ablation and Deposition of Wide Band Gap Semiconductors. J. Phys. Chem. C 2011, 115, 3203–3211. [Google Scholar] [CrossRef]
- Rella, R.; Spadavecchia, J.; Manera, M.G.; Capone, S.; Taurino, A.; Martino, M.; Caricato, A.P.; Tunno, T. Acetone and Ethanol Solid-State Gas Sensors Based on TiO2 Nanoparticles Thin Film Deposited by Matrix Assisted Pulsed Laser Evaporation. Sens. Actuators B Chem. 2007, 127, 426–431. [Google Scholar] [CrossRef]
- Morandi, S.; Fioravanti, A.; Cerrato, G.; Lettieri, S.; Sacerdoti, M.; Carotta, M.C. Facile Synthesis of ZnO Nano-Structures: Morphology Influence on Electronic Properties. Sens. Actuators B Chem. 2017, 249, 581–589. [Google Scholar] [CrossRef]
- Fioravanti, A.; Bonanno, A.; Gherardi, S.; Carotta, M.C.; Skouloudis, A.N. A Portable Air-Quality Station Based on Thick Film Gas Sensors for Real Time Detection of Traces of Atmospheric Pollutants. IOP Conf. Ser. Mater. Sci. Eng. 2016, 108, 012005. [Google Scholar] [CrossRef] [Green Version]
- Fioravanti, A.; Marani, P.; Massarotti, G.P.; Lettieri, S.; Morandi, S.; Carotta, M.C. (Ti, Sn) Solid Solution Based Gas Sensors for New Monitoring of Hydraulic Oil Degradation. Materials 2021, 14, 605. [Google Scholar] [CrossRef]
- Carotta, M.C.; Fioravanti, A.; Gherardi, S.; Malagù, C.; Sacerdoti, M.; Ghiotti, G.; Morandi, S. (Ti,Sn) Solid Solutions as Functional Materials for Gas Sensing. Sens. Actuators B Chem. 2014, 194, 195–205. [Google Scholar] [CrossRef]
- Morandi, S.; Amodio, A.; Fioravanti, A.; Giacomino, A.; Mazzocchi, M.; Sacerdoti, M.; Carotta, M.C.; Skouloudis, A.N. Operational Functionalities of Air-Quality W Sn Metal-Oxide Sensors Correlating Semiconductor Defect Levels and Surface Potential Barriers. Sci. Total Environ. 2020, 706, 135731. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, A.; Marani, P.; Morandi, S.; Lettieri, S.; Mazzocchi, M.; Sacerdoti, M.; Carotta, M.C. Growth Mechanisms of ZnO Micro-Nanomorphologies and Their Role in Enhancing Gas Sensing Properties. Sensors 2021, 21, 1331. [Google Scholar] [CrossRef] [PubMed]
- Carotta, M.C.; Gherardi, S.; Malagù, C.; Nagliati, M.; Vendemiati, B.; Martinelli, G.; Sacerdoti, M.; Lesci, I.G. Comparison between Titania Thick Films Obtained through Sol–Gel and Hydrothermal Synthetic Processes. Thin Solid Film. 2007, 515, 8339–8344. [Google Scholar] [CrossRef]
- Pallotti, D.K.; Orabona, E.; Amoruso, S.; Aruta, C.; Bruzzese, R.; Chiarella, F.; Tuzi, S.; Maddalena, P.; Lettieri, S. Multi-Band Photoluminescence in TiO2 Nanoparticles-Assembled Films Produced by Femtosecond Pulsed Laser Deposition. J. Appl. Phys. 2013, 114, 043503. [Google Scholar] [CrossRef]
- Baratto, C.; Comini, E.; Faglia, G.; Sberveglieri, G.; Zha, M.; Zappettini, A. Metal Oxide Nanocrystals for Gas Sensing. Sens. Actuators B Chem. 2005, 109, 2–6. [Google Scholar] [CrossRef]
- Trani, F.; Causà, M.; Lettieri, S.; Setaro, A.; Ninno, D.; Barone, V.; Maddalena, P. Role of Surface Oxygen Vacancies in Photoluminescence of Tin Dioxide Nanobelts. Microelectron. J. 2009, 40, 236–238. [Google Scholar] [CrossRef]
- Valerini, D.; Cretì, A.; Caricato, A.P.; Lomascolo, M.; Rella, R.; Martino, M. Optical Gas Sensing through Nanostructured ZnO Films with Different Morphologies. Sens. Actuators B Chem. 2010, 145, 167–173. [Google Scholar] [CrossRef]
- Orabona, E.; Pallotti, D.; Fioravanti, A.; Gherardi, S.; Sacerdoti, M.; Carotta, M.C.; Maddalena, P.; Lettieri, S. On Mechanism of NO2 Detection by ZnO Excitonic Luminescence. Sens. Actuators B Chem. 2015, 210, 706–711. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, D.; Zhao, Q.; Li, Z.; Ma, Y.; Yang, M. Influence of Adsorbed Oxygen on the Surface Photovoltage and Photoluminescence of ZnO Nanorods. Nanotechnology 2006, 17, 2110–2115. [Google Scholar] [CrossRef]
- Ma, S.; Reish, M.E.; Zhang, Z.; Harrison, I.; Yates, J.T. Anatase-Selective Photoluminescence Spectroscopy of P25 TiO2 Nanoparticles: Different Effects of Oxygen Adsorption on the Band Bending of Anatase. J. Phys. Chem. C 2017, 121, 1263–1271. [Google Scholar] [CrossRef]
- Santara, B.; Giri, P.K.; Imakita, K.; Fujii, M. Evidence for Ti Interstitial Induced Extended Visible Absorption and Near Infrared Photoluminescence from Undoped TiO2 Nanoribbons: An In Situ Photoluminescence Study. J. Phys. Chem. C 2013, 117, 23402–23411. [Google Scholar] [CrossRef]
- Marotti, R.E.; Badán, J.A.; Quagliata, E.; Dalchiele, E.A. Red Photoluminescence and Band Edge Shift from ZnO Thin Films. Phys. B: Condens. Matter 2007, 398, 337–340. [Google Scholar] [CrossRef]
- Choi, S.; Phillips, M.R.; Aharonovich, I.; Pornsuwan, S.; Cowie, B.C.C.; Ton-That, C. Photophysics of Point Defects in ZnO Nanoparticles. Adv. Opt. Mater. 2015, 3, 821–827. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Calcination Temperature (°C) | Crystalline Phase | Composition (at. %) | Crystallite Sizes (nm) | SSA (m2/g) |
|---|---|---|---|---|---|
| TiO2 | 650 | Rutile | 5 | 61 | 29 |
| Anatase | 95 | 34 | |||
| ZnO | 450 | Wurtzite | 100 | 33 | 12 |
| SnO2 | 650 | Cassiterite | 100 | 21 | 28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fioravanti, A.; Marani, P.; Morandi, S.; Giordano, L.; Maddalena, P.; Carotta, M.C.; Lettieri, S. Optical Sensing of Molecular Oxygen (O2) via Metal Oxide Photoluminescence: A Comparative Study of TiO2, SnO2 and ZnO. Chemosensors 2021, 9, 163. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9070163
Fioravanti A, Marani P, Morandi S, Giordano L, Maddalena P, Carotta MC, Lettieri S. Optical Sensing of Molecular Oxygen (O2) via Metal Oxide Photoluminescence: A Comparative Study of TiO2, SnO2 and ZnO. Chemosensors. 2021; 9(7):163. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9070163
Chicago/Turabian StyleFioravanti, Ambra, Pietro Marani, Sara Morandi, Laura Giordano, Pasqualino Maddalena, Maria Cristina Carotta, and Stefano Lettieri. 2021. "Optical Sensing of Molecular Oxygen (O2) via Metal Oxide Photoluminescence: A Comparative Study of TiO2, SnO2 and ZnO" Chemosensors 9, no. 7: 163. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9070163