

New Challenges in (Bio)Analytical Sample Treatment Procedures for Clinical Applications

 ,
,  , ,
, , 
 ,
,  , ,
, ,
Abstract
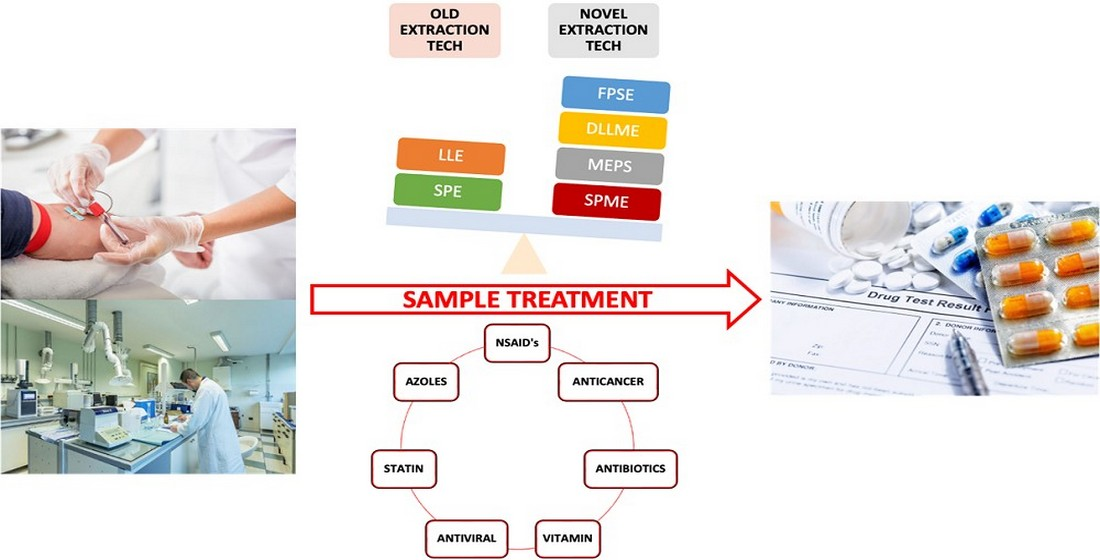
:
1. Introduction
2. Clinical Applications
2.1. Anticancer Drugs
2.2. Antibiotics
2.3. Vitamin
2.4. Antiviral
2.5. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
2.6. Statin
2.7. Imidazoles and Triazoles
3. Sample Preparation Techniques for Diagnostic Purposes
3.1. Solidphase Microextraction (SPME)
3.2. Microextraction by Packed Sorbent (MEPS)
3.3. Dispersive Liquid–Liquid Microextraction (DLLME)
3.4. Fabric Phase Sorptive Extraction (FPSE)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Ovidio, C.; Bonelli, M.; Rosato, E.; Tartaglia, A.; Ulusoy, H.I.; Samanidou, V.; Furton, K.G.; Kabir, A.; Ali, I.; Savini, F.; et al. Novel Applications of Microextraction Techniques Focused on Biological and Forensic Analyses. Separations 2022, 9, 18. [Google Scholar] [CrossRef]
- Ingle, R.G.; Zeng, S.; Jiang, H.; Fang, W.J. Current developments of bioanalytical sample preparation techniques in pharmaceuticals. J. Pharm. Anal. 2022, 12, 517–529. [Google Scholar] [CrossRef]
- Pandey, K.; Mishra, O.P. Advancement in Analytical and Bioanalytical Techniques as a Boon to Medical Sciences. In Biochemical Testing; Bobbarala, V., Zaman, G.S., Desa, M.N.M., Akim, A.M., Eds.; IntechOpen: London, UK, 2020; Volume 12, pp. 29–46. [Google Scholar]
- Pourshamsi, T.; Amri, F.; Abniki, M. A comprehensive review on application of the syringe in liquidand solid phase microextraction methods. J. Iran. Chem. Soc. 2021, 18, 245–264. [Google Scholar] [CrossRef]
- Hansen, F.A.; Pedersen-Bjergaard, S. Emerging Extraction Strategies in Analytical Chemistry. Anal. Chem. 2020, 92, 2–15. [Google Scholar] [CrossRef]
- Knikma, J.E.; Rosing, H.; Guchelaar, H.J.; Cats, A.; Beijnen, J.H. A review of the bioanalytical methods for the quantitative determination of capecitabine and its metabolites in biological matrices. Biomed. Chromatogr. 2020, 34, e4732. [Google Scholar]
- Zufia, L.; Aldaz, A.; Giraldez, J. Simple determination of capecitabine and its metabolites by liquid chromatography with ultraviolet detection in a single injection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 809, 51–58. [Google Scholar] [CrossRef]
- Piórkowska, E.; Kaza, M.; Fitatiuk, J.; Szlaska, I.; Pawinski, T.; Rudzki, P.J. Rapid and simplified HPLC-UV method with on-line wavelengths switching for determination of capecitabine in human plasma. Pharmazie 2014, 69, 500–505. [Google Scholar]
- Licea-Perez, H.; Wang, S.; Bowen, C. Development of a sensitive and selective LC-MS/MS method for the determination of á-fluoroâ-alanine, 5-fluorouracil and capecitabine in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1040–1046. [Google Scholar] [CrossRef]
- Salvador, A.; Millerioux, L.; Renou, A. Simultaneous LC-MS-MS Analysis of Capecitabine and its Metabolites (50 -deoxy5-fluorocytidine, 50 -deoxy-5-fluorouridine, 5-fluorouracil) After OffLine SPE from Human Plasma. Chroma 2006, 63, 609–615. [Google Scholar] [CrossRef]
- Buchner, P.; Mihola, E.; Sahmanovic, A.; Steininger, T.; Dittrich, C.; Czejka, M. Validation of a Simple Assay for the Quantification of the Capecitabine Metabolites 5′–DFCR and 5′–DFUR for Drug Monitoring in Patients Receiving Outpatient Chemotherapy. Anticancer Res. 2013, 33, 881–886. [Google Scholar]
- Farkouh, A.; Ettlinger, D.; Schueller, J.; Georgopoulos, A.; Scheithauer, W.; Czejka, M. A rapid and simple HPLC assay for quantification of capecitabine for drug monitoring purposes. Anticancer Res. 2010, 30, 5207–5211. [Google Scholar]
- Xu, Y.; Grem, J.L. Liquid chromatography-mass spectrometry method for the analysis of the anti-cancer agent capecitabine and its nucleoside metabolites in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2003, 783, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, P.; Ji, C.; Dai, X.; Zhong, D.; Ding, L.; Chen, X. Simultaneous determination of capecitabine and its three nucleoside metabolites in human plasma by high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 989, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Thorat, S.G.; Chikhale, R.V.; Tajne, M.R. A rapid and simple HPTLC assay for therapeutic drug monitoring of capecitabine in colorectal cancer patients. Biomed. Chromatogr. 2018, 32, e4100. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Mayer, I.; Jodrell, D.I. Simultaneous determination of capecitabine and its metabolites by HPLC and mass spectrometry for preclinical and clinical studies. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 826, 232–237. [Google Scholar] [CrossRef]
- Vainchtein, L.D.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. A new, validated HPLC-MS/MS method for the simultaneous determination of the anti-cancer agent capecitabine and its metabolites: 50 -deoxy-5- fluorocytidine, 50-deoxy-5-fluorouridine, 5-fluorouracil and 5-fluorodihydrouracil, in human plasma. Biomed. Chromatogr. 2010, 24, 374–386. [Google Scholar] [CrossRef]
- Wang, Z.; Li, X.; Yang, Y.; Zhang, F.; Li, M.; Chen, W.; Gao, S.; Chen, W. A Sensitive and Efficient Method for Determination of Capecitabine and Its Five Metabolites in Human Plasma Based on One-Step LiquidLiquid Extraction. J. Anal. Methods Chem. 2019, 2019, 9371790. [Google Scholar] [CrossRef]
- Deenen, M.J.; Rosing, H.; Hillebrand, M.J.; Schellens, J.H.M.; Beijnen, J.H. Quantitative determination of capecitabine and its six metabolites in human plasma using liquid chromatography coupled to electrospray tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 913–914, 30–40. [Google Scholar] [CrossRef]
- Fresnai, M.; Roth, A.; Foerster, K.I.; Jäger, D.; Pfister, S.M.; Haefeli, W.E.; Burhenne, J.; Longuespée, R. Rapid and Sensitive Quantification of Osimertinib in Human Plasma Using a Fully Validated MALDI–IM–MS/MS Assay. Cancers 2020, 17, 1897. [Google Scholar] [CrossRef]
- Llopis, B.; Robidou, P.; Tissot, N.; Pinna, B.; Gougis, P.; Aubart, F.C.; Campedel, L.; Abbar, B.; Weil, D.R.; Uzunov, M.; et al. Development and clinical validation of a simple and fast UPLC-ESI-MS/MS method for simultaneous quantification of nine kinase inhibitors and two antiandrogen drugs in human plasma: Interest for their therapeutic drug monitoring. J. Pharm. Biomed. Anal. 2021, 197, 113968. [Google Scholar] [CrossRef]
- Locatelli, M.; Tinari, N.; Grassadonia, A.; Tartaglia, A.; Macerola, D.; Piccolantonio, S.; Sperandio, E.; D’Ovidio, C.; Carradori, S.; Ulusoy, H.I.; et al. FPSE-HPLC-DAD method for the quantification of anticancer drugs in human whole blood, plasma, and urine. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1095, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Ripa, M.; Premaschi, S.; Banfi, G.; Castagna, A.; Locatelli, M. LC-MS/MS method for simultaneous determination of linezolid, meropenem, piperacillin and teicoplanin in human plasma samples. Journal of Pharmaceutical and biomedical analysis. J Pharm Biomed Anal. 2019, 169, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wongchang, T.; Winterberg, M.; Tarning, J.; Sriboonvorakul, N.; Muangnoicharoen, S.; Blessborn, D. Determination of ceftriaxone in human plasma using liquid chromatography–tandem mass spectrometry. Wellcome Open Res. 2021, 4, 47. [Google Scholar] [CrossRef]
- Krnac, D.; Reiffova, K.; Rolinski, B. A new HPLC-MS/MS analytical method for quantification of tazobactam, piperacillin, and meropenem in human plasma. J. Sep. Sci. 2021, 44, 2744–2753. [Google Scholar] [CrossRef] [PubMed]
- Rola, R.; Kowalski, K.; Bienkowski, T.; Studzinska, S. Improved sample preparation method for fast LC-MS/MS analysis of vitamin D metabolites in serum. J. Pharm. Biomed. Anal. 2020, 190, 113529. [Google Scholar] [CrossRef]
- Hotta, K.; Wang, Y.; Mano, Y. A sensitive bioanalytical assay for methylcobalamin, an endogenous and light-labile substance, in human plasma by liquid chromatography with tandem mass spectrometry and its application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2020, 191, 113621. [Google Scholar] [CrossRef] [PubMed]
- Bollen, P.D.J.; de Graaff-Teulen, M.J.A.; Schalkwijk, S.; van Erp, N.P.; Burger, D.M. Development and validation of an UPLC-MS/MS bioanalytical method for simultaneous quantification of the antiretroviral drugs dolutegravir, elvitegravir, raltegravir, nevirapine and etravirine in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1105, 76–84. [Google Scholar] [CrossRef]
- Elkadya, E.F.; Aboelwaf, A.A. Rapid bioanalytical LC-MS/MS method for the simultaneous determination of sofosbuvir and velpatasvir in human plasma-application to a pharmacokinetic study in Egyptian volunteers. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1102–1103, 116–124. [Google Scholar] [CrossRef]
- Raabovà, H.; Havlíková, L.C.; Erben, J.; Chvojka, J.; Švec, F.; Šatínský, D. Polycaprolactone Composite Micro/Nanofibrous Material as an Alternative to Restricted Access Media for Direct Extraction and Separation of Non-Steroidal Anti-Inflammatory Drugs from Human Serum Using Column-Switching Chromatography. Nanomaterials 2021, 11, 2669. [Google Scholar] [CrossRef]
- Kabir, A.; Furton, K.G.; Tinari, N.; Grossi, L.; Innosad, D.; Macerola, D.; Tartaglia, A.; Di Donato, V.; D’Ovidio, C.; Locatelli, M. Fabric phase sorptive extraction-high performance liquid chromatography photo diode array detection method for simultaneous monitoring of three inflammatory bowel disease treatment drugs in whole blood, plasma and urine. J. Chromatogr. B 2018, 1084, 53–63. [Google Scholar] [CrossRef]
- Tartaglia, A.; Kabir, A.; D’Ambrosio, F.; Ramundo, P.; Ulusoy, S.; Ulusoy, H.I.; Merone, G.M.; Savini, F.; D’Ovidio, C.; De Grazia, U.; et al. Fast off-line FPSE-HPLC-PDA determination of six NSAIDs in saliva samples. J. Chromatogr. B 2020, 1144, 9. [Google Scholar] [CrossRef] [PubMed]
- D’angelo, V.; Tessari, F.; Bellagamba, G.; De Luca, E.; Cifelli, R.; Celia, C.; Primavera, R.; Di Francesco, M.; Paolino, D.; Di Marzio, L.; et al. Microextraction by packed sorbent and HPLC–PDA quantification of multiple anti-inflammatory drugs and fluoroquinolones in human plasma and urine. J. Enzym. Inhib. Med. Chem. 2016, 31 (Suppl. S3), 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courlet, P.; Spaggiari, D.; Desfontaine, V.; Cavassini, M.; Saldanha, S.A.; Buclin, T.; Marzolini, C.; Csajka, C.; Decoster, L.A. UHPLC-MS/MS assay for simultaneous determination of amlodipine, metoprolol, pravastatin, rosuvastatin, atorvastatin with its active metabolites in human plasma, for population-scale drug-drug interactions studies in people living with HIV. J. Chromatogr. B. 2019, 1125, 11. [Google Scholar] [CrossRef] [PubMed]
- Campestre, C.; Locatelli, M.; Guglielmi, P.; De Luca, E.; Bellagamba, G.; Menta, S.; Zengin, G.; Celia, C.; Di Marzio, L.; Carradori, S. Analysis of imidazoles and triazoles in biological samples after MicroExtraction by packed sorbent. J. Enzym. Inhib. Med. Chem. 2017, 32, 1053–1063. [Google Scholar] [CrossRef]
- Locatelli, M.; Kabir, A.; Innosa, D.; Lopatriello, T.; Furton, K.G. A fabric phase sorptive extraction-High performance liquid chromatography-Photo diode array detection method for the determination of twelve azole antimicrobial drug residues in human plasma and urine. J. Chromatogr. B 2017, 1040, 192–198. [Google Scholar] [CrossRef]
- Kabir, A.; Locatelli, M.; Ulusoy, H.I. Recent Trends in Microextraction Techniques Employed in Analytical and Bioanalytical Sample Preparation. Separations 2017, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, M.; Tartaglia, A.; Piccolantonio, S.; Di Iorio, L.A.; Sperandio, E.; Ulusoy, H.I.; Furton, K.G.; Kabir, A. Innovative configurations of sample preparation Techniques Applied in bioanalytical chemistry: A review. Curr. Anal. Chem. 2019, 15, 731–744. [Google Scholar] [CrossRef]
- Matys, J.; Gieroba, B.; Jó’zwiak, K. Recent developments of bioanalytical methods in determination of neurotransmitters in vivo. J. Pharm. Biomed. Anal. 2020, 180, 12. [Google Scholar] [CrossRef]
- Naccarato, A.; Gionfriddo, E.; Sindona, G.; Tagarelli, A. Development of a simple and rapid solid phase microextraction-gas chromatography–triple quadrupole mass spectrometry method for the analysis of dopamine, serotonin and norepinephrine in human urine. Anal. Chim. Acta 2014, 810, 17–24. [Google Scholar] [CrossRef]
- Monteiro, M.; Carvalho, M.; Henrique, R.; Jeronimo, C.; Moreira, N.; de Lourdes Bastos, M.; de Pinho, P.G. Analysis of volatile human urinary metabolome by solid-phase microextraction in combination with gas chromatography–mass spectrometry for biomarker discovery: Application in a pilot study to discriminate patients with renal cell carcinoma. Eur. J. Cancer 2014, 50, 1993–2002. [Google Scholar] [CrossRef]
- Deev, V.; Solovieva, S.; Andreev, E.; Protoshchak, V.; Karpushchenko, E.; Sleptsov, A.; Kartsova, L.; Bessonova, E.; Legin, A.; Kirsanov, D. Prostate cancer screening using chemometric processing of GC–MS profiles obtained in the headspace above urine samples. J. Chromatogr. B 2020, 1155, 8. [Google Scholar] [CrossRef] [PubMed]
- Bessonneau, V.; Boyaci, E.; Maciazek-Jurczyk, M.; Pawliszyn, J. In vivo solid phase microextraction sampling of human saliva for non-invasive and on-site monitoring. Anal. Chim. Acta 2015, 856, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Costa Queiroz, M.E.; Donizeti de Souza, I.; de Oliveira, I.G.; Grecco, C.F. In vivo solid phase microextraction for bioanalysis. Trends Anal. Chem. 2022, 153, 18. [Google Scholar]
- Ghimenti, S.; Lomonaco, T.; Bellagambi, F.G.; Biagini, D.; Salvo, P.; Trivella, M.G.; Scali, M.C.; Barletta, V.; Marzilli, M.; Di Francesco, F.; et al. Salivary lactate and 8-isoprostaglandin F2á as potential non-invasive biomarkers for monitoring heart failure: A pilot study. Sci. Rep. 2020, 10, 7441. [Google Scholar] [CrossRef]
- Berenguer, P.H.; Camacho, I.C.; Câmara, R.; Oliveira, S.; Câmara, J.S. Determination of potential childhood asthma biomarkers using a powerful methodology based on microextraction by packed sorbent combined with ultra-high pressure liquid chromatography. Eicosanoids as case study. J. Chromatogr. A 2019, 1584, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-S.; Yuan, Y.-C.; Yin, Y.; Tang, Y.P.; Xu, R.-J.; Liu, Y.; Chen, P.-D.; Yin, L.; Duan, J.-A. Hydrophilic interaction chromatography combined with ultrasound-assisted ionic liquid dispersive liquid-liquid microextraction for determination of underivatized neurotransmitters in dementia patients’ urine samples. Anal. Chim. Acta 2020, 1107, 74–84. [Google Scholar] [CrossRef]
- Monteleone, M.; Naccarato, A.; Sindona, G.; Tagarelli, A. A reliable and simple method for the assay of neuroendocrine tumor markers in human urine by solid-phase microextraction–gas chromatography-triple quadrupole mass spectrometry. Anal. Chim. Acta 2013, 759, 66–73. [Google Scholar] [CrossRef]
- Lord, H.; Yu, Y.; Segal, A.; Pawliszyn, J. Breath analysis and monitoring by membrane extraction with sorbent interface. Anal. Chem. 2002, 74, 5650–5657. [Google Scholar] [CrossRef]
- Lee, C.Y.J.; Jenner, A.M.; Halliwell, B. Rapid preparation of human urine and plasma samples for analysis of F 2-isoprostanes bygas chromatography-mass spectrometry. Biochem. Biophys. Res. Commun. 2004, 320, 696–702. [Google Scholar] [CrossRef]
- Perestrelo, R.; Silva, C.L.; Câmara, J.S. Determination of urinary levels of leukotriene B4 using ad highly specific and sensitive methodology based on automatic MEPS combined with UHPLC-PDA analysis. Talanta 2015, 144, 382–389. [Google Scholar] [CrossRef]
- Xiong, X.; Zhang, Y.; Zhao, R. Quantitative Measurement of Plasma Free Metanephrines by a Simple and Cost-Effective Microextraction Packed Sorbent with Porous Graphitic Carbon and Liquid Chromatography-Tandem Mass Spectrometr. J. Anal. Met. Chem. 2021, 2021, 8821276. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Lew, B.-L.; Sim, W.-Y.; Hong, J.; Chung, B.-C. Serial Hydrolysis for the Simultaneous Analysis of Catecholamines and Steroids in the Urine of Patients with Alopecia Areata. Molecules 2021, 26, 2734. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguirre, A.; Maldonado, M. Preparation of Methacrylate-Based Polymers Modified with Chiral Resorcinarenes and Their Evaluation as Sorbents in Norepinephrine Microextraction. Polymers 2019, 11, 1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Zhang, Y. Simple, rapid, and cost-effective microextraction by the packed sorbent method for quantifying of urinary free catecholamines and metanephrines using liquid chromatography-tandem mass spectrometry and its application in clinical analysis. Anal. Bioanal. Chem. 2020, 412, 2763–2775. [Google Scholar] [CrossRef] [PubMed]
- Kabir, A.; Furton, K.G. Fabric Phase Sorptive Extractors. U.S. Patents 9557252, 31 January 2017. [Google Scholar]
- Samanidou, V.; Kaltzi, I.; Kabir, A.; Furton, K.G. Simplifying sample preparation using fabric phase sorptive extraction technique for the determination of benzodiazepines in blood serum by high-performance liquid chromatography. Biomed. Chromatogr. 2016, 30, 829–836. [Google Scholar] [CrossRef]
- Tartaglia, A.; Locatelli, M.; Kabir, A.; Furton, K.G.; Macerola, D.; Sperandio, E.; Piccolantonio, S.; Ulusoy, H.I.; Maroni, F.; Bruni, P.; et al. Comparison between Exhaustive and Equilibrium Extraction Using Different SPE Sorbents and Sol-Gel Carbowax 20M Coated FPSE Media. Molecules 2019, 24, 382. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, A.; Covone, S.; Rosato, E.; Bonelli, M.; Savini, F.; Furton, K.G.; Gazioglu, I.; D’Ovidio, C.; Kabir, A.; Locatelli, M. Fabric phase sorptive extraction (FPSE) as an efficient sample preparation platform for the extraction of antidepressant drugs from biological fluids. Adv. Sample Prep. 2022, 3, 100022. [Google Scholar] [CrossRef]
- Gazioglu, I.; Tekkeli, S.E.K.; Tartaglia, A.; Aslan, C.; Locatelli, M.; Kabir, A. Simultaneous determination of febuxostat and montelukast in human plasma using fabric phase sorptive extraction and high-performance liquid chromatography-fluorimetric detection. J. Chromatogr. B 2022, 1188, 123070. [Google Scholar] [CrossRef]
- Tiris, G.; Gazioglu, I.; Furton, K.G.; Kabir, A.; Locatelli, M. Fabric phase sorptive extraction combined with high performance liquid chromatography for the determination of favipiravir in human plasma and breast milk. J. Pharm. Biomed. Anal. 2023, 223, 115131. [Google Scholar] [CrossRef]
- Locatelli, M.; Tartaglia, A.; Ulusoy, H.I.; Ulusoy, S.; Savini, F.; Rossi, S.; Santavenere, F.; Merone, G.M.; Bassotti, E.; D’Ovidio, C.; et al. Fabric-Phase sorptive membrane array as a noninvasive in vivo sampling device for human exposure to different compounds. Anal. Chem. 2021, 93, 1957–1961. [Google Scholar] [CrossRef]
- Tartaglia, A.; Kabir, A.; Ulusoy, S.; Sperandio, E.; Piccolantonio, S.; Ulusoy, H.I.; Furton, K.G.; Locatelli, M. FPSE-HPLC-PDA analysis of seven paraben residues in human whole blood, plasma, and urine. J. Chromatogr. B 2019, 1125, 10. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Furton, K.G.; Tartaglia, A.; Sperandio, E.; Ulusoy, H.I.; Kabir, A. An FPSE-HPLC-PDA method for rapid determination of solar UV filters in human whole blood, plasma, and urine. J. Chromatogr. B 2019, 1118, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Sidiropoulou, G.; Kabir, A.; Furton, K.G.; Kika, F.S.; Fytianos, K.; Tzanavaras, P.D.; Zacharis, C.K. Combination of fabric phase sorptive extraction with UHPLC-ESI-MS/MS for the determination of adamantine analogues in human urine. Microchem. J. 2022, 176, 107250. [Google Scholar] [CrossRef]
- Mazaraki, K.; Kabir, A.; Furton, K.; Fytianos, K.G.; Samanidou, V.F.; Zacharis, C.K. Fast fabric phase sorptive extraction of selected β-blockers from human serum and urine followed by UHPLC-ESI-MS/MS analysis. J. Pharm. Biomed. Anal. 2021, 199, 114053. [Google Scholar] [CrossRef]
- Locatelli, M.; Covone, S.; Rosato, E.; Bonelli, M.; Savini, F.; Furton, K.G.; Gazioglu, I.; D’Ovidio, C.; Kabir, A.; Tartaglia, A. Analysis of seven selected antidepressant drugs in post–mortem samples using fabric phase sorptive extraction followed by high performance liquid chromatography-photodiode array detection. Forensic. Chem. 2022, 31, 100460. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Analyte | Sample | Treatment | LOD/LOQ | Ref. |
|---|---|---|---|---|
| Capecitabine 5′-DFUR 5-FU 5-FUH2 | Human plasma | LLE | LOQ: 0.025 ug/mL LOD: 0.01 ug/mL | [7] |
| Capecitabine | Human plasma | LLE | CC: 0.05/10 ug/mL | [8] |
| Capecitabine 5-FU FBAL | Human plasma | LLE LLE LLE + SPE | LLQ: 1.00/10.00 ng/mL | [9] |
| Capecitabine 5′-DFCR 5′-DFUR 5-FU | Human plasma | SPE | LOD: 0.007(Capecitabine) 0.08 (5-DFUR) 0.6 (5-DFCR) 0.08 (5-FU) ng/mL LOQ: 0.02 (Capecitabine) 0.3 (5-DFUR) 2 (5-DFCR) 0.3 (5-FU) ng/mL | [10] |
| Capecitabine | Human plasma | SPE | LOD: 39 ng LOQ: 156 ng | [11] |
| Capecitabine | Human plasma | SPE | LOQ: 156 ng/mL LOD: 78 ng/mL | [12] |
| Capecitabine 5′-DFCR 5′-DFUR | Human plasma | Online SPE | LOQ 1.4 ng/mL (Capecitabine) 17.6 ng/mL (5-DFCR) 8.4 ng/mL (5-DFUR) | [13] |
| Capecitabine 5′-DFCR 5′-DFUR 5-FU | Human plasma | PP | LOQ: 10.0 (Capecitabine) 10.0 (DFCR) 10.0 (DFUR) 2.0 (5-FU) | [14] |
| Capecitabine | Human serum | PP | LOQ: 1 ug/mL | [15] |
| Capecitabine 5′-DFCR 5′-DFUR 5-FU | Plasma Tumor tissue liver | PP | LOQ ng/mL Plasma: 4.0(Capecitabine) 1.4 (DFCR) 3.3 (DFUR) 45.8 (5-FU) Tumor tissue: 1.3 Capecitabine 1.7 DFCR 0.5 DFUR 50.0 5-FU Liver: 13.0 Capecitabine 3.0 DFCR 92.0 5-FU No interference DFUR | [16] |
| Capecitabine 5′-DFCR 5′-DFUR 5-FU 5-FUH2 | Human plasma | PP | LOQ: 1 ug/mL | [17] |
| Capecitabine 5′-DFCR 5′-DFUR 5-FU 5-FUH2 FBAL | Human plasma | LLE | LOQ: 20.0 ng/mL | [18] |
| 5′-DFCR 5′-DFUR 5-FU 5-FUH2 FUPA FBAL | Human plasma | PP | LOQ: 50.0 ng/mL | [19] |
| Target Analytes | Matrix | Extraction Technique | Analytical Characteristics | Ref. |
|---|---|---|---|---|
| Dopamine (DA), Serotonin (5-HT), Norepinephrine (NE) | Human urine | SPME-GC-QqQ-MS | DA: LOD 0.59 μg L−1, LOQ 0.81 μg L−1 5-HT: LOD 0.38 μg L−1, LOQ 0.74 μg L−1 NE: LOD 13.5 μg L−1, LOQ 21.3 μg L−1 | [40] |
| Homovanillic acid (HVA), vanylmandelic acid (VMA), 5-hydroxyindolacetic acid (5-HIAA) | Human urine | SPME-GC-QqQ-MS | HVA: LOD 1.3 μg L−1, LOQ 2.7 μg L−1 VMA: LOD 0.046 μg L−1, LOQ 0.063 μg L−1 5-HIAA: LOD 24.3 μg L−1, LOQ 49.6 μg L−1 | [48] |
| VOCs | Human Breathe | MESI-GC | LOD Acetone 0.4 μg/L LOD Ethanol 0.5 μg/L | [49] |
| F2-isoprostanes | Human plasma Urine | SPE-GC-MS | LOD (plasma) 0.037 ng/mL LOD (urine) 0.007 ng/mg | [50] |
| LTB4 | Human urine | MEPS-UHPLC-PDA | LOD 0.37 ng/mL LOQ 1.22 ng/mL | [51] |
| Metanephrine (MN) Normetanephrine (NMN) | Human plasma | MEPS-HILIC-MS/MS | NM LOD 12.4 pg/mL NMN LOD 12.3 pg/mL | [52] |
| Dopamine (DA), Norepinephrine (NE), Metanephrine (MN), Normetanephrine (NMN), L-3,4-dihydroxyphenylalanine (L-DOPA), Epinephrine (E), Epinephrine-d6 (E-d6), Metanephrine-d3 (MN-d3), Normetanephrine-d3 (NMN-d3), Serotonin (5-HT), Testosterone (T), Epitestosterone (EpiT), Dihydrotestosterone (DHT), 17-hydroxyprogesterone (17-OHP); Androstenedione (A), Progesterone (P4), Epitestosterone-d3 (EpiT-d3) | Human urine | LLE-LC/MS | DA: LOD 1 ng/mL NE: LOD 1 ng/mL MN: LOD 1 ng/mL NMN: LOD 1 ng/mL L-DOPA: LOD 20 ng/mL E: LOD 1 ng/mL E-d6: LOD 1 ng/mL MN-d3: LOD 1 ng/mL NMN-d3: LOD 1 ng/mL 5-HT: LOD 1 ng/mL T: LOD 1 ng/mL EpiT: LOD 1 ng/mL DHT: LOD 1 ng/mL 17-OHP: LOD 1 ng/mL A: LOD 1 ng/mL P4: LOD 1 ng/mL EpiT-d3: LOD 1 ng/mL | [53] |
| Norepinephrine (NE) | Artificial urine | Rotating-disk sorptive extraction technique (RDSE) | NE: LOD 11.3 μg L−1, LLOQ 34.0 μg L−1 | [54] |
| Epinephrine (EPI), Norepinephrine (NE), Dopamine (DA), Metanephrine (MN), Normetanephrine (NMN), 3-methoxytyramine (3-MT) | Human urine | MEPS-LC-MS/MS | EPI: LOQ 0.167 ng/mL, LOD 0.0800 ng/mL NE: LOQ 0.650 ng/mL, LOD 0.300 ng/mL DA: LOQ 1.53 ng/mL, LOD 0.530 ng/mL MN: LOQ 0.440 ng/mL, LOD 0.176 ng/mL NMN: LOQ 1.10 ng/mL, LOD 0.440 ng/mL 3-MT: LOQ 0.880 ng/mL, LOD 0.176 ng/mL | [55] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, V.; Locatelli, M.; Savini, F.; Grazia, U.d.; Montanaro, O.; Rosato, E.; Perrucci, M.; Ciriolo, L.; Kabir, A.; Ulusoy, H.I.; et al. New Challenges in (Bio)Analytical Sample Treatment Procedures for Clinical Applications. Separations 2023, 10, 62. https://0-doi-org.brum.beds.ac.uk/10.3390/separations10010062
Greco V, Locatelli M, Savini F, Grazia Ud, Montanaro O, Rosato E, Perrucci M, Ciriolo L, Kabir A, Ulusoy HI, et al. New Challenges in (Bio)Analytical Sample Treatment Procedures for Clinical Applications. Separations. 2023; 10(1):62. https://0-doi-org.brum.beds.ac.uk/10.3390/separations10010062
Chicago/Turabian StyleGreco, Valentina, Marcello Locatelli, Fabio Savini, Ugo de Grazia, Ottavia Montanaro, Enrica Rosato, Miryam Perrucci, Luigi Ciriolo, Abuzar Kabir, Halil Ibrahim Ulusoy, and et al. 2023. "New Challenges in (Bio)Analytical Sample Treatment Procedures for Clinical Applications" Separations 10, no. 1: 62. https://0-doi-org.brum.beds.ac.uk/10.3390/separations10010062