
Development of an Ultrasound-Assisted Emulsification Microextraction Method for the Determination of Volatile Compounds in Wines

Abstract
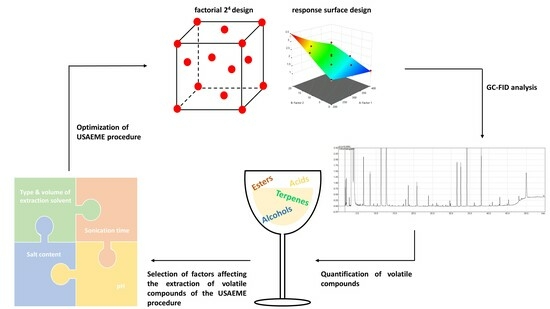
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Standard Solutions
2.2. Sample Preparation
2.2.1. Commercial Wine
2.2.2. Synthetic Wine
2.3. Laboratory-Scale Fermentations
2.3.1. Yeast Strains
2.3.2. Alcoholic Fermentations
2.4. USAEME Procedure
2.5. Determination of Volatile Compounds
2.6. Statistical Analysis
2.6.1. Experimental Design
2.6.2. Statistics of Wine Samples
3. Results and Discussion
3.1. Preliminary Experiments: Selection of Extraction Solvent
3.2. Optimization of USAEME Procedure
3.2.1. Screening Design
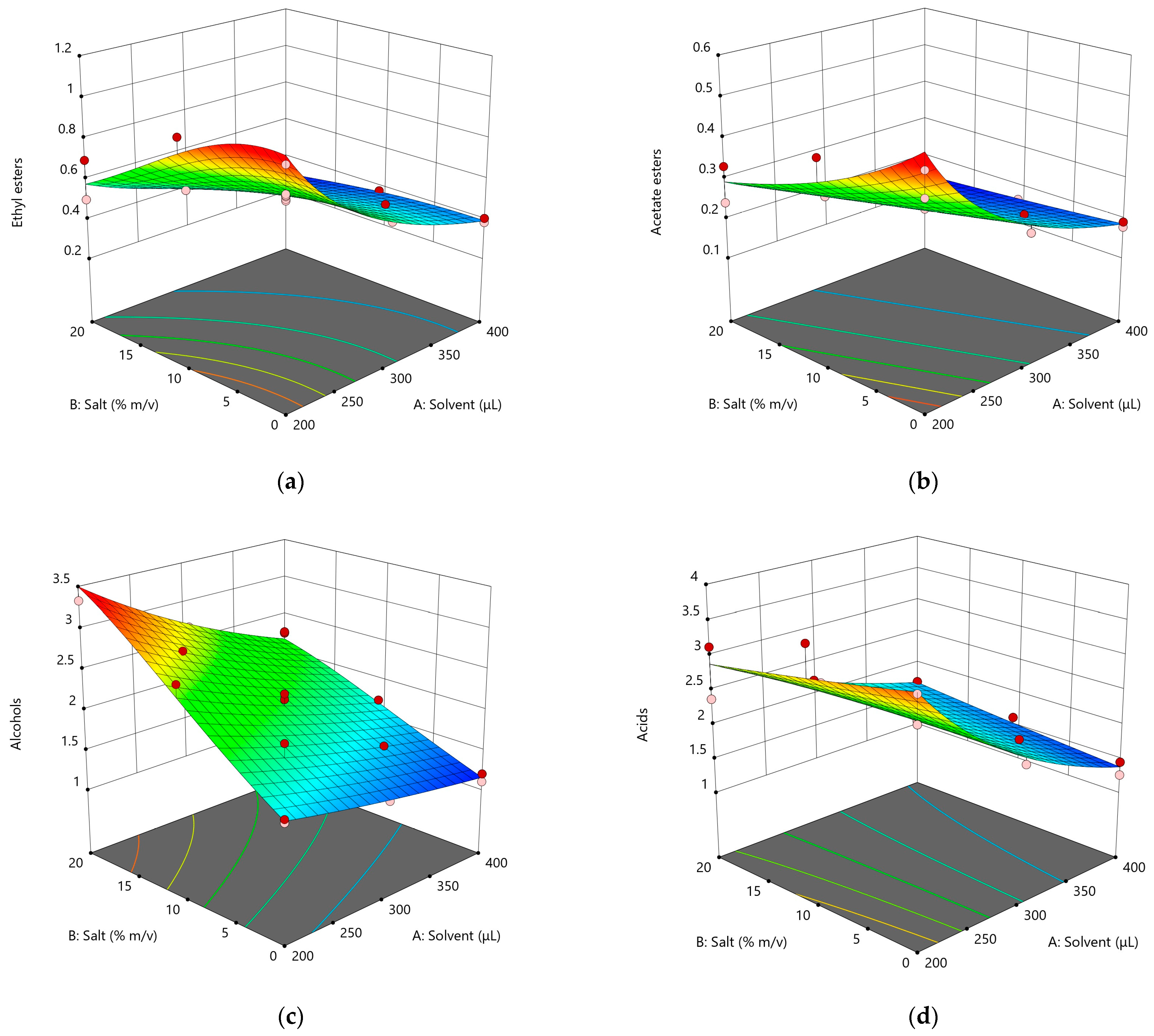
3.2.2. Response Surface Design
3.3. Multiple Response Optimization
3.4. Evaluation of the Methodology
3.4.1. Performance of the Analytical Method
3.4.2. Application of the USAEME Method to White Wines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jofré, V.P.; Assof, M.V.; Fanzone, M.L.; Goicoechea, H.C.; Martínez, L.D.; Silva, M.F. Optimization of Ultrasound Assisted-Emulsification-Dispersive Liquid-Liquid Microextraction by Experimental Design Methodologies for the Determination of Sulfur Compounds in Wines by Gas Chromatography-Mass Spectrometry. Anal. Chim. Acta 2010, 683, 126–135. [Google Scholar] [CrossRef]
- Lambrechts, M.G.; Pretorius, I.S. Yeast and Its Importance to Wine Aroma—A Review. S. Afr. J. Enol. Vitic. 2000, 21, 97–129. [Google Scholar] [CrossRef]
- Andujar-Ortiz, I.; Moreno-Arribas, M.V.; Martín-Álvarez, P.J.; Pozo-Bayón, M.A. Analytical Performance of Three Commonly Used Extraction Methods for the Gas Chromatography-Mass Spectrometry Analysis of Wine Volatile Compounds. J. Chromatogr. A 2009, 1216, 7351–7357. [Google Scholar] [CrossRef]
- Costa Freitas, A.M.; Gomes da Silva, M.D.R.; Cabrita, M.J. Sampling Techniques for the Determination of Volatile Components in Grape Juice, Wine and Alcoholic Beverages. Compr. Sampl. Sample Prep. Anal. Tech. Sci. 2012, 4, 27–41. [Google Scholar] [CrossRef]
- Villamor, R.R.; Ross, C.F. Wine Matrix Compounds Affect Perception of Wine Aromas. Annu. Rev. Food Sci. Technol. 2013, 4, 1–20. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Q.; Li, Y.; Liu, S.; Tu, Q.; Yuan, C. Characterization of Wine Volatile Compounds from Different Regions and Varieties by HS-SPME/GC-MS Coupled with Chemometrics. Curr. Res. Food Sci. 2023, 6, 100418. [Google Scholar] [CrossRef] [PubMed]
- Hernanz, D.; Gallo, V.; Recamales, Á.F.; Meléndez-Martínez, A.J.; Heredia, F.J. Comparison of the Effectiveness of Solid-Phase and Ultrasound-Mediated Liquid-Liquid Extractions to Determine the Volatile Compounds of Wine. Talanta 2008, 76, 929–935. [Google Scholar] [CrossRef]
- Marín-San Román, S.; Rubio-Bretón, P.; Pérez-Álvarez, E.P.; Garde-Cerdán, T. Advancement in Analytical Techniques for the Extraction of Grape and Wine Volatile Compounds. Food Res. Int. 2020, 137, 109712. [Google Scholar] [CrossRef]
- Ortega-Heras, M.; González-SanJosé, M.L.; Beltrán, S. Aroma Composition of Wine Studied by Different Extraction Methods. Anal. Chim. Acta 2002, 458, 85–93. [Google Scholar] [CrossRef]
- Fontana, A.R.; Patil, S.H.; Banerjee, K.; Altamirano, J.C. Ultrasound-Assisted Emulsification Microextraction for Determination of 2,4,6-Trichloroanisole in Wine Samples by Gas Chromatography Tandem Mass Spectrometry. J. Agric. Food Chem. 2010, 58, 4576–4581. [Google Scholar] [CrossRef] [PubMed]
- Demirhan, B.; Kara, H.E.Ş.; Demirhan, B.E. Overview of Green Sample Preparation Techniques in Food Analysis. In Ideas and Applications Toward Sample Preparation for Food and Beverage Analysis; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Fontana, A.R.; Muñoz De Toro, M.; Altamirano, J.C. One-Step Derivatization and Preconcentration Microextraction Technique for Determination of Bisphenol a in Beverage Samples by Gas Chromatography-Mass Spectrometry. J. Agric. Food Chem. 2011, 59, 3559–3565. [Google Scholar] [CrossRef]
- Puertas, B.; Jimenez-Hierro, M.J.; Cantos-Villar, E.; Marrufo-Curtido, A.; Carbú, M.; Cuevas, F.J.; Moreno-Rojas, J.M.; González-Rodríguez, V.E.; Cantoral, J.M.; Ruiz-Moreno, M.J. The Influence of Yeast on Chemical Composition and Sensory Properties of Dry White Wines. Food Chem. 2018, 253, 227–235. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, X.; Huang, Y.; Lu, J.; Zhang, Y. Volatile Aroma Compounds in Wines from Chinese Wild/Hybrid Species. J. Food Biochem. 2019, 43, 12684. [Google Scholar] [CrossRef]
- Knight, S.; Klaere, S.; Fedrizzi, B.; Goddard, M.R. Regional Microbial Signatures Positively Correlate with Differential Wine Phenotypes: Evidence for a Microbial Aspect to Terroir. Sci. Rep. 2015, 5, srep14233. [Google Scholar] [CrossRef]
- Gamero, A.; Wesselink, W.; de Jong, C. Comparison of the Sensitivity of Different Aroma Extraction Techniques in Combination with Gas Chromatography-Mass Spectrometry to Detect Minor Aroma Compounds in Wine. J. Chromatogr. A 2013, 1272, 1–7. [Google Scholar] [CrossRef]
- Cabredo-Pinillos, S.; Cedrón-Fernández, T.; González-Briongos, M.; Puente-Pascual, L.; Sáenz-Barrio, C. Ultrasound-Assisted Extraction of Volatile Compounds from Wine Samples: Optimisation of the Method. Talanta 2006, 69, 1123–1129. [Google Scholar] [CrossRef]
- Ubeda, C.; Callejón, R.M.; Troncoso, A.M.; Peña-Neira, A.; Morales, M.L. Volatile Profile Characterisation of Chilean Sparkling Wines Produced by Traditional and Charmat Methods via Sequential Stir Bar Sorptive Extraction. Food Chem. 2016, 207, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Tredoux, A.; De Villiers, A.; Májek, P.; Lynen, F.; Crouch, A.; Sandra, P. Stir Bar Sorptive Extraction Combined with GC-MS Analysis and Chemometric Methods for the Classification of South African Wines According to the Volatile Composition. J. Agric. Food Chem. 2008, 56, 4286–4296. [Google Scholar] [CrossRef]
- Fang, Y.; Qian, M.C. Quantification of Selected Aroma-Active Compounds in Pinot Noir Wines from Different Grape Maturities. J. Agric. Food Chem. 2006, 54, 8567–8573. [Google Scholar] [CrossRef] [PubMed]
- Marín-San Román, S.; Carot-Sierra, J.M.; Sáenz de Urturi, I.; Rubio-Bretón, P.; Pérez-Álvarez, E.P.; Garde-Cerdán, T. Optimization of Stir Bar Sorptive Extraction (SBSE) and Multi-Stir Bar Sorptive Extraction (MSBSE) to Improve Must Volatile Compounds Extraction. Lwt 2022, 172, 114182. [Google Scholar] [CrossRef]
- Pizarro, C.; Sáenz-González, C.; Pérez-del-Notario, N.; González-Sáiz, J.M. Ultrasound-Assisted Emulsification-Microextraction for the Sensitive Determination of Brett Character Responsible Compounds in Wines. J. Chromatogr. A 2011, 1218, 8975–8981. [Google Scholar] [CrossRef] [PubMed]
- Reboredo-Rodríguez, P.; Rey-Salgueiro, L.; Regueiro, J.; González-Barreiro, C.; Cancho-Grande, B.; Simal-Gándara, J. Ultrasound-Assisted Emulsification-Microextraction for the Determination of Phenolic Compounds in Olive Oils. Food Chem. 2014, 150, 128–136. [Google Scholar] [CrossRef]
- Kiejza, D.; Kotowska, U.; Polińska, W.; Karpińska, J. USAEME-GC/MS Method for Easy and Sensitive Determination of Nine Bisphenol Analogues in Water and Wastewater. Molecules 2022, 27, 4977. [Google Scholar] [CrossRef]
- Vila, M.; Pablo Lamas, J.; Garcia-Jares, C.; Dagnac, T.; Llompart, M. Ultrasound-Assisted Emulsification Microextraction Followed by Gas Chromatography-Mass Spectrometry and Gas Chromatography-Tandem Mass Spectrometry for the Analysis of UV Filters in Water. Microchem. J. 2016, 124, 530–539. [Google Scholar] [CrossRef]
- Regueiro, J.; Llompart, M.; Garcia-Jares, C.; Garcia-Monteagudo, J.C.; Cela, R. Ultrasound-Assisted Emulsification-Microextraction of Emergent Contaminants and Pesticides in Environmental Waters. J. Chromatogr. A 2008, 1190, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Becerril-Bravo, E.; Pablo Lamas, J.; Sanchez-Prado, L.; Lores, M.; Garcia-Jares, C.; Jimenez, B.; Llompart, M. Ultrasound-Assisted Emulsification-Microextraction of Fragrance Allergens in Water. Chemosphere 2010, 81, 1378–1385. [Google Scholar] [CrossRef]
- Pérez-Outeiral, J.; Millán, E.; Garcia-Arrona, R. Ultrasound-Assisted Emulsification Microextraction Coupled with High-Performance Liquid Chromatography for the Simultaneous Determination of Fragrance Allergens in Cosmetics and Water. J. Sep. Sci. 2015, 38, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Najafi, N.M.; Tavakoli, H.; Abdollahzadeh, Y.; Alizadeh, R. Comparison of Ultrasound-Assisted Emulsification and Dispersive Liquid-Liquid Microextraction Methods for the Speciation of Inorganic Selenium in Environmental Water Samples Using Low Density Extraction Solvents. Anal. Chim. Acta 2012, 714, 82–88. [Google Scholar] [CrossRef]
- Ma, J.J.; Du, X.; Zhang, J.W.; Li, J.C.; Wang, L.Z. Ultrasound-Assisted Emulsification-Microextraction Combined with Flame Atomic Absorption Spectrometry for Determination of Trace Cadmium in Water Samples. Talanta 2009, 80, 980–984. [Google Scholar] [CrossRef]
- Ferreira, V.J.; De Jesus, M.S.; Dos Santos, M.C.; Guedes, W.N.; Lemos, V.A.; Novaes, C.G.; Costa, F.S.; Pacheco, C.S.V.; Da Silva, E.G.P.; Amorim, F.A.C. Multivariate Optimization of Ultrasound-Assisted Liquid-Liquid Microextraction Based on Two Solvents for Cadmium Preconcentration Prior to Determination by Flame Atomic Absorption Spectrometry. Anal. Methods 2021, 13, 267–273. [Google Scholar] [CrossRef]
- Nunes, L.S.; Lemos, V.A. Ultrasound-Assisted Emulsification Microextraction in an Online System for Determination of Cadmium in Water and Tea Samples. J. AOAC Int. 2018, 101, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Hryniewicka, M.; Starczewska, B.; Tkaczuk, N. Simple Approach Based On Ultrasound-Assisted Emulsification Microextraction For Determination Of β-Sitosterol In Dietary Supplements And Selected Food Products. Microchem. J. 2020, 155, 104775. [Google Scholar] [CrossRef]
- Liao, Q.G.; Li, W.H.; Luo, L.G. Ultrasound-Assisted Emulsification-Microextraction for the Sensitive Determination of Ethyl Carbamate in Alcoholic Beverages. Anal. Bioanal. Chem. 2013, 405, 6791–6797. [Google Scholar] [CrossRef] [PubMed]
- Cortada, C.; Vidal, L.; Canals, A. Determination of Geosmin and 2-Methylisoborneol in Water and Wine Samples by Ultrasound-Assisted Dispersive Liquid-Liquid Microextraction Coupled to Gas Chromatography-Mass Spectrometry. J. Chromatogr. A 2011, 1218, 17–22. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Wang, S.; Liu, F.; Shi, K. Ultrasound-Assisted Surfactant-Enhanced Emulsification Microextraction Based on the Solidification of a Floating Organic Droplet Used for the Simultaneous Determination of Six Fungicide Residues in Juices and Red Wine. J. Chromatogr. A 2013, 1300, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jares, C.; Celeiro, M.; Lamas, J.P.; Iglesias, M.; Lores, M.; Llompart, M. Rapid Analysis of Fungicides in White Wines from Northwest Spain by Ultrasound-Assisted Emulsification-Microextraction and Gas Chromatography-Mass Spectrometry. Anal. Methods 2014, 6, 3108–3116. [Google Scholar] [CrossRef]
- Peña, R.M.; Barciela, J.; Herrero, C.; García-Martín, S. Comparison of Ultrasound-Assisted Extraction and Direct Immersion Solid-Phase Microextraction Methods for the Analysis of Monoterpenoids in Wine. Talanta 2005, 67, 129–135. [Google Scholar] [CrossRef]
- Pizarro, C.; Sáenz-González, C.; Pérez-del-Notario, N.; González-Sáiz, J.M. Development of an Ultrasound-Assisted Emulsification-Microextraction Method for the Determination of the Main Compounds Causing Cork Taint in Wines. J. Chromatogr. A 2012, 1229, 63–71. [Google Scholar] [CrossRef]
- Pizarro, C.; Sáenz-González, C.; Pérez-del-Notario, N.; González-Sáiz, J.M. Optimisation of a Sensitive Method Based on Ultrasound-Assisted Emulsification-Microextraction for the Simultaneous Determination of Haloanisoles and Volatile Phenols in Wine. J. Chromatogr. A 2012, 1244, 37–45. [Google Scholar] [CrossRef]
- Chalvantzi, I.; Banilas, G.; Tassou, C.; Nisiotou, A. Biogeographical Regionalization of Wine Yeast Communities in Greece and Environmental Drivers of Species Distribution at a Local Scale. Front. Microbiol. 2021, 12, 705001. [Google Scholar] [CrossRef]
- Esteve-Zarzoso, B.; Belloch, C.; Uruburu, F.; Querol, A. Identification of Yeasts by RFLP Analysis of the 5.8S RRNA Gene and the Two Ribosomal Internal Transcribed Spacers. Int. J. Syst. Bacteriol. 1999, 49, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Legras, J.L.; Karst, F. Optimisation of Interdelta Analysis for Saccharomyces Cerevisiae Strain Characterisation. FEMS Microbiol. Lett. 2003, 221, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Hammer, D.A.T.; Ryan, P.D.; Hammer, Ø.; Harper, D.A.T. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 178. [Google Scholar]
- Wu, C.; Liu, N.; Wu, Q.; Wang, C.; Wang, Z. Application of Ultrasound-Assisted Surfactant-Enhanced Emulsification Microextraction for the Determination of Some Organophosphorus Pesticides in Water Samples. Anal. Chim. Acta 2010, 679, 56–62. [Google Scholar] [CrossRef]
- Viñas, P.; Campillo, N.; López-García, I.; Hernández-Córdoba, M. Dispersive Liquid-Liquid Microextraction in Food Analysis. A Critical Review Microextraction Techniques. Anal. Bioanal. Chem. 2014, 406, 2067–2099. [Google Scholar] [CrossRef]
- Priser, C.; Etiévant, P.X.; Nicklaus, S.; Brun, O. Representative Champagne Wine Extracts for Gas Chromatography Olfactometry Analysis. J. Agric. Food Chem. 1997, 45, 3511–3514. [Google Scholar] [CrossRef]
- Ortega, C.; López, R.; Cacho, J.; Ferreira, V. Fast Analysis of Important Wine Volatile Compounds—Development and Validation of a New Method Based on Gas Chromatographic-Flame Ionisation Detection Analysis of Dichloromethane Microextracts. J. Chromatogr. A 2001, 923, 205–214. [Google Scholar] [CrossRef]
- Ribereau-Gayon, P.; Glories, Y.; Dubourdieu, D.; Maujean, A. Handbook of Enology, Volume 2: The Chemistry of Wine Stabilization and Treatments, 2nd ed.; Wiley: Hoboken, NJ, USA, 2006; ISBN 9780470010389. [Google Scholar]
- Jackson, R.S. Wine Science. In Principles and Applications, 3rd ed.; Academic Press: San Diego, CA, USA, 2008; ISBN 9780123736468. [Google Scholar]
- Smucker, B.; Krzywinski, M.; Altman, N. Optimal Experimental Design. Nat. Methods 2018, 15, 559–560. [Google Scholar] [CrossRef]
- Vera Candioti, L.; De Zan, M.M.; Cámara, M.S.; Goicoechea, H.C. Experimental Design and Multiple Response Optimization. Using the Desirability Function in Analytical Methods Development. Talanta 2014, 124, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Callejon, R.M.; Clavijo, A.; Ortigueira, P.; Troncoso, A.M.; Paneque, P.; Morales, M.L. Volatile and Sensory Profile of Organic Red Wines Produced by Different Selected Autochthonous and Commercial Saccharomyces Cerevisiae Strains. Anal. Chim. Acta 2010, 660, 68–75. [Google Scholar] [CrossRef]
- Çelik, Z.D.; Erten, H.; Cabaroglu, T. The Influence of Selected Autochthonous Saccharomyces Cerevisiae Strains on the Physicochemical and Sensory Properties of Narince Wines. Fermentation 2019, 5, 70. [Google Scholar] [CrossRef]
- Liu, N.; Qin, Y.; Song, Y.Y.; Tao, Y.S.; Sun, Y.; Liu, Y.L. Aroma Composition and Sensory Quality of Cabernet Sauvignon Wines Fermented by Indigenous Saccharomyces Cerevisiae Strains in the Eastern Base of the Helan Mountain, China. Int. J. Food Prop. 2016, 19, 2417–2431. [Google Scholar] [CrossRef]
- Torrens, J.; Urpí, P.; Riu-Aumatell, M.; Vichi, S.; López-Tamames, E.; Buxaderas, S. Different Commercial Yeast Strains Affecting the Volatile and Sensory Profile of Cava Base Wine. Int. J. Food Microbiol. 2008, 124, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Swiegers, J.H.; Bartowsky, E.J.; Henschke, P.A.; Pretorius, I.S. Yeast and Bacterial Modulation of Wine Aroma and Flavour. Aust. J. Grape Wine Res. 2005, 11, 139–173. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Factors 1 | Response (× 1000) 2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Block | Run No. | A | B | C | D | EtE | AcE | Alc | Acd | Trp | Ethyl Lactate | Acetic Acid |
| 1 | 1 | 150 | 5 | 0 | 3 | 1345 | 791 | 9280 | 4722 | 1576 | 414 | 150 |
| 1 | 2 | 450 | 0 | 20 | 4 | 90 | 68 | 6307 | 803 | 204 | 381 | 129 |
| 1 | 3 | 450 | 0 | 0 | 4 | 363 | 206 | 6377 | 1876 | 490 | 351 | 58 |
| 1 | 4 | 150 | 0 | 20 | 3 | 426 | 296 | 16,880 | 4289 | 803 | 723 | 233 |
| 1 | 5 | 450 | 5 | 0 | 3 | 275 | 159 | 4448 | 1310 | 333 | 242 | 69 |
| 1 | 6 | 150 | 0 | 0 | 3 | 773 | 492 | 6099 | 3157 | 927 | 275 | 42 |
| 1 | 7 | 450 | 5 | 20 | 4 | 102 | 78 | 6784 | 1246 | 262 | 400 | 168 |
| 1 | 8 | 150 | 5 | 20 | 4 | 352 | 221 | 11,739 | 2449 | 634 | 487 | 227 |
| 2 | 9 | 450 | 5 | 20 | 3 | 98 | 82 | 6497 | 828 | 240 | 389 | 174 |
| 2 | 10 | 450 | 0 | 0 | 3 | 236 | 146 | 4340 | 1038 | 308 | 234 | 23 |
| 2 | 11 | 150 | 5 | 20 | 3 | 238 | 182 | 10,419 | 2112 | 558 | 451 | 192 |
| 2 | 12 | 150 | 0 | 20 | 4 | 146 | 78 | 8091 | 1473 | 334 | 374 | 75 |
| 2 | 13 | 450 | 0 | 20 | 3 | 66 | 60 | 5531 | 760 | 179 | 340 | 109 |
| 2 | 14 | 450 | 5 | 0 | 4 | 200 | 109 | 4001 | 862 | 259 | 208 | 22 |
| 2 | 15 | 150 | 0 | 0 | 4 | 634 | 371 | 5663 | 2534 | 801 | 240 | 17 |
| 2 | 16 | 150 | 5 | 0 | 4 | 646 | 371 | 5417 | 2515 | 787 | 239 | 83 |
| Response | Model | F-Value | p-Value 1 | Adjusted R2 | Predicted R2 | CV% | ||
|---|---|---|---|---|---|---|---|---|
| Model | Lack of Fit | Model | Lack of Fit | |||||
| Ethyl esters | Quadratic | 133.66 | 0.6095 | <0.0001 | 0.7622 | 0.9593 | 0.9292 | 7.14 |
| Acetate esters | Linear | 166.42 | 1.07 | <0.0001 | 0.4732 | 0.9430 | 0.9183 | 7.67 |
| Alcohols | Quadratic | 169.35 | 1.47 | <0.0001 | 0.3128 | 0.9637 | 0.9503 | 2.71 |
| Acids | 2FI | 89.98 | 0.6174 | <0.0001 | 0.7565 | 0.9303 | 0.8726 | 3.71 |
| Terpenes | Linear | 241.95 | 1.46 | <0.0001 | 0.3037 | 0.9602 | 0.9422 | 5.88 |
| 3-Methyl-1-butanol 2 | Quadratic | 123.81 | 0.7940 | <0.0001 | 0.6330 | 0.9485 | 0.9250 | 7.45 |
| Ethyl lactate 2 | Linear | 131.71 | 1.35 | <0.0001 | 0.3429 | 0.9289 | 0.9005 | 7.43 |
| Acetic acid 2 | Linear | 156.22 | 0.7268 | <0.0001 | 0.6878 | 0.9395 | 0.9103 | 7.14 |
| Compounds | HL a | R2 | LOD (μg L−1) | LOQ (μg L−1) | RSD% b | Recovery % c |
|---|---|---|---|---|---|---|
| Ethyl esters | ||||||
| Ethyl butanoate | 960 | 0.9998 | 9.7 | 29 | 5.3 | 82.9 |
| Ethyl hexanoate | 1020 | 0.9994 | 5.4 | 16 | 7.0 | 92.8 |
| Ethyl octanoate | 990 | 0.9997 | 1.1 | 3.2 | 8.4 | 107.8 |
| Ethyl decanoate | 990 | 0.9997 | 2.3 | 7.0 | 6.2 | 93.2 |
| Diethyl succinate | 920 | 0.9999 | 3.7 | 11 | 4.3 | 69.0 |
| Acetate esters | ||||||
| Isobutyl acetate | 980 | 0.9995 | 16.3 | 49 | 5.6 | 86.5 |
| Butyl acetate | 1010 | 0.9999 | 7.9 | 24 | 3.1 | 87.1 |
| 3-Methylbutyl acetate | 10,550 | 0.9999 | 31 | 93 | 6.4 | 88.5 |
| Hexyl acetate | 1030 | 0.9998 | 9.8 | 30 | 7.8 | 88.1 |
| 2-Phenylethyl acetate | 1180 | 0.9999 | 8.3 | 25 | 6.7 | 82.2 |
| Alcohols | ||||||
| 1-Propanol | 221,950 | 0.9993 | 837 | 2537 | 4.3 | 86.8 |
| 2-Methyl-1-propanol | 220,850 | 0.9992 | 96 | 292 | 3.2 | 86.6 |
| 1-Butanol | 1150 | 0.9996 | 18 | 54 | 3.7 | 88.3 |
| 3-Methyl-1-butanol | 408,350 | 0.9998 | 264 | 800 | 2.0 | 96.3 |
| 3-Methyl-1-pentanol | 950 | 0.9999 | 5.2 | 16 | 2.3 | 99.1 |
| 1-Hexanol | 1060 | 0.9999 | 3.1 | 9.3 | 0.5 | 93.2 |
| (E)-3-Hexen-1-ol | 930 | 0.9999 | 5.2 | 16 | 1.8 | 101.4 |
| (Z)-3-Hexen-1-ol | 990 | 0.9998 | 9.8 | 30 | 1.2 | 99.3 |
| 1-Octanol | 1450 | 0.9999 | 11 | 34 | 4.4 | 94.9 |
| 2-Phenylethanol | 8450 | 0.9999 | 46 | 138 | 2.3 | 95.3 |
| Acids | ||||||
| Acetic acid | 500,000 | 0.9951 | 2475 | 7500 | 5.1 | 71.5 |
| Butanoic acid | 4950 | 0.9987 | 135 | 411 | 3.5 | 72.8 |
| Hexanoic acid | 5400 | 0.9997 | 66 | 201 | 1.5 | 88.9 |
| Octanoic acid | 3550 | 0.9999 | 10 | 31 | 3.8 | 70.5 |
| Decanoic acid | 5600 | 0.9999 | 19 | 57 | 4.7 | 72.3 |
| γ-Butyrolactone | 1240 | 0.9999 | 17 | 53 | 7.2 | 96.1 |
| Terpenes | ||||||
| d-Limonene | 950 | 0.9999 | 6.5 | 20 | 12.0 | 34.7 |
| γ-Terpinene | 1020 | 0.9999 | 5.7 | 17 | 7.7 | 33.9 |
| p-Cymene | 900 | 0.9998 | 8.6 | 26 | 10.1 | 38.9 |
| Linalool | 1020 | 0.9999 | 2.9 | 8.6 | 4.7 | 90.0 |
| α-Terpineol | 1040 | 0.9998 | 9.6 | 29 | 2.8 | 89.1 |
| Compounds | Sc1 | Sc2 | Sc3 | Sc4 |
|---|---|---|---|---|
| Concentration (μg L−1) | ||||
| Ethyl esters | ||||
| Ethyl butanoate | 81 (10) b | 68 (4) b | 142 (15) a | 87 (4) b |
| Ethyl hexanoate | 48 (13) b | 20 (5) b | 195 (7) a | 31 (9) b |
| Ethyl octanoate | 22 (3) b | 11 (3) b | 47 (6) a | 19 (7) b |
| Ethyl decanoate | 3.6 (0.4) a (<LOQ) | nd 2 | 3.6 (1.5) a (<LOQ) | nd 2 |
| Diethyl succinate | nd 2 | 10 (0) a (<LOQ) | 7.6 (2.1) a (<LOQ) | 7.6 (0.6) a (<LOQ) |
| Total | 155 | 109 | 395 | 145 |
| Acetate esters | ||||
| Isobutyl acetate | nd 2 | nd 2 | 22 (1) (<LOQ) | nd 2 |
| 3-Methylbutyl acetate | 80 (14) b (<LOQ) | 86 (0) b (<LOQ) | 546 (55) a | 115 (5) b |
| 2-Phenylethyl acetate | 81 (4) b | 79 (7) b | 260 (24) a | 83 (5) b |
| Total | 161 | 165 | 828 | 198 |
| Alcohols | ||||
| 1-Propanol | 64,815 (1508) a | 19,096 (170) b | 22,506 (689) b | 23,878 (3108) b |
| 2-Methyl-1-propanol | 13,943 (434) b | 19,946 (204) a | 20,693 (33) a | 19,613 (2135) a |
| 1-Butanol | 739 (7) a | 217 (9) d | 268 (3) c | 507 (12) b |
| 3-Methyl-1-butanol | 71,388 (2152) b | 87,859 (1274) a | 90,862 (1229) a | 96,345 (5676) a |
| 1-Hexanol | 329 (17) a | 322 (1) a | 299 (9) a | 308 (6) a |
| (E)-3-Hexen-1-ol | 5.5 (0.4) a (<LOQ) | 5.7 (0.1) a (<LOQ) | nd | 6.5 (0.7) a (<LOQ) |
| (Z)-3-Hexen-1-ol | 141 (1) a | 140 (1) a | 143 (1) a | 142 (1) a |
| 2-Phenylethanol | 13,346 (491) b | 15,888 (363) a | 12,721 (10) b | 13,665 (199) b |
| Total | 164,707 | 143,474 | 147,492 | 154,465 |
| Carboxylic acids | ||||
| Acetic acid | 516,147 (7071) b | 593,867 (7366)a | 415,929 (11,986) c | 417,221 (8066) c |
| Butanoic acid | 737 (31) a | 595 (23) a | 582 (73) a | 614 (24) a |
| Hexanoic acid | 2724 (35) a | 1444 (78) b | 1565 (14) b | 1454 (112) b |
| Octanoic acid | 1425 (25) a | 738 (24) b | 1400 (98) a | 841 (35) b |
| Decanoic acid | 244 (11) b | 102 (16) c | 738 (32) a | 135 (4) c |
| Total 1 | 5130 | 2879 | 4285 | 3044 |
| Terpenes | ||||
| Linalool | 20 (0) b | 20 (1) b | 43 (10) a | 19 (1) b |
| α-Terpineol | 49 (2) b | 57 (4) b | 76 (7) a | 55 (4) b |
| Total | 69 | 77 | 119 | 74 |
| Lactones | ||||
| γ-Butyrolactone | 1967 (38) c | 3119 (47) a | 3021 (57) a | 2434 (36) b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chalvantzi, I.; Nisiotou, A.; Banilas, G.; Mallouchos, A. Development of an Ultrasound-Assisted Emulsification Microextraction Method for the Determination of Volatile Compounds in Wines. Separations 2023, 10, 525. https://0-doi-org.brum.beds.ac.uk/10.3390/separations10100525
Chalvantzi I, Nisiotou A, Banilas G, Mallouchos A. Development of an Ultrasound-Assisted Emulsification Microextraction Method for the Determination of Volatile Compounds in Wines. Separations. 2023; 10(10):525. https://0-doi-org.brum.beds.ac.uk/10.3390/separations10100525
Chicago/Turabian StyleChalvantzi, Ioanna, Aspasia Nisiotou, Georgios Banilas, and Athanasios Mallouchos. 2023. "Development of an Ultrasound-Assisted Emulsification Microextraction Method for the Determination of Volatile Compounds in Wines" Separations 10, no. 10: 525. https://0-doi-org.brum.beds.ac.uk/10.3390/separations10100525