
Stochastic Neural Networks for Automatic Cell Tracking in Microscopy Image Sequences of Bacterial Colonies


 , , , and
, , , and
Abstract
:1. Introduction
1.1. Related Work
1.2. Contributions
1.3. Outline
2. Datasets
2.1. Synthetic Videos of Simulated Cell Colonies
2.2. Laboratory Image Sequences (Real Biological Data)
2.3. Cell Characteristics
3. Methodology
3.1. Registration Mappings
- Case 1:
- Cell did not divide in the interframe , and has become a cell ; that is, has grown and moved during the interframe time interval.
- Case 2:
- Cell divided between J and , and generated two children cells ; we then denote .
- Case 3:
- Cell disappeared in the interframe , so that is not defined.
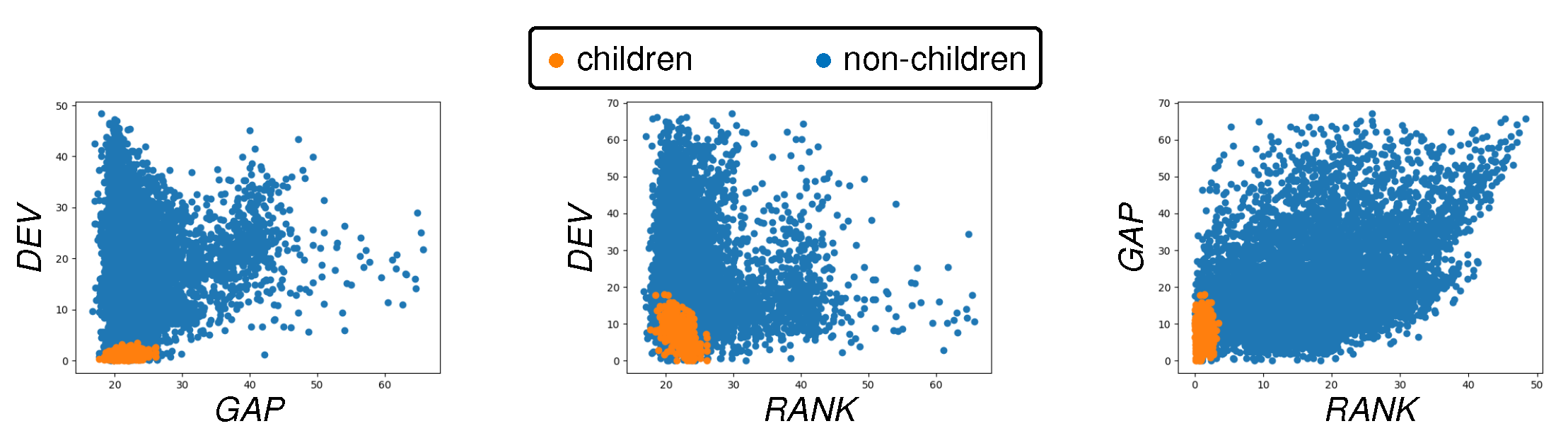
3.2. Calibration of Cost Function Weights
3.3. Cell Divisions and Parent–Children Short Lineages
3.3.1. Cell Divisions
3.3.2. Most Likely Parent Cell for a Given Children Pair
3.3.3. Penalties to Enforce Adequate Parent–Children Links
3.4. Generic Boltzmann Machines (BMs)
3.5. Optimized Set of Parent–Children Triplets
3.6. Performance of Automatic Children Pairing on Synthetic Videos
3.6.1. Children Pairing: Fast BM Simulations
3.6.2. Children Pairing: Implementation on Synthetic Videos
3.6.3. Parent–Children Matching: Accuracy on Synthetic Videos
3.7. Reduction to Registrations with No Cell Division
3.8. Automatic Cell Registration after Reduction to Cases with No Cell Division
3.8.1. The Set of Many-to-One Cell Registrations
3.8.2. Registration Cost Functional
3.9. BM Minimization of Registration Cost Function
3.9.1. BM Minimization of over
3.9.2. BM Energy Function
3.9.3. Cliques of Interactive Neurons
3.9.4. Test Set of 100 Synthetic Image Pairs
3.9.5. Implementation of BM Minimization for
3.9.6. Weight Calibration
3.9.7. BM Simulations
4. Results
4.1. Tests of Cell Registration Algorithms on Synthetic Data
4.2. Tests of Cell Registration Algorithms on Laboratory Image Sequences

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task | Accuracy | |||||
|---|---|---|---|---|---|---|
| correctly detected parents | 15/19 | 79% | 20/21 | 95% | 7/10 | 70% |
| correctly detected children | 35/38 | 92% | 32/42 | 76% | 14/20 | 70% |
| correct parent–children triplets | 15/19 | 78% | 16/21 | 76% | 7/10 | 70% |
| correctly registered cell pairs | 36/36 | 100% | 44/49 | 90% | 76/80 | 95% |
5. Conclusions and Future Work
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Stochastic Dynamics of BMs
Appendix A.1. Asynchronous BM Dynamics
Appendix A.2. Synchronous BM Dynamics
Appendix A.3. Comparing Asynchronous and Synchronous BM Dynamics
Appendix B. Computer Hardware
Appendix C. Parameters for Simulation Software
Appendix D. Cell Segmentation
Appendix D.1. Watershed Algorithm
Appendix D.2. Segmentation Errors: Correction Steps
| Characteristic | 5% Quantile | Min | Max | Mean |
|---|---|---|---|---|
| Watershed area | 56.00 | 43.00 | 984.00 | 211.00 ± 138.00 |
| Mean intensity of area | 0.34 | 0.00 | 0.57 | 0.41 ± 0.06 |
| Mean intensity of boundary segment | 0.46 | 0.30 | 0.99 | 0.74 ± 0.14 |
| Height of boundary segment | 0.05 | −0.09 | 0.62 | 0.33 ± 0.14 |
Appendix D.3. Cell Boundary Detection
Appendix D.4. Convolutional Neural Networks (CNNs)
| 0 | 1 | |
| 0 | 0.97 | 0.03 |
| 1 | 0.11 | 0.89 |
References
- Butts-Wilmsmeyer, C.J.; Rapp, S.; Guthrie, B. The technological advancements that enabled the age of big data in the environmental sciences: A history and future directions. Curr. Opin. Environ. Sci. Health 2020, 18, 63–69. [Google Scholar] [CrossRef]
- Sivarajah, U.; Kamal, M.M.; Irani, Z.; Weerakkody, V. Critical analysis of Big Data challenges and analytical methods. J. Bus. Res. 2017, 70, 263–286. [Google Scholar] [CrossRef] [Green Version]
- Balomenos, A.D.; Tsakanikas, P.; Aspridou, Z.; Tampakaki, A.P.; Koutsoumanis, K.P.; Manolakos, E.S. Image analysis driven single-cell analytics for systems microbiology. BMC Syst. Biol. 2017, 11, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Klein, J.; Leupold, S.; Biegler, I.; Biedendieck, R.; Münch, R.; Jahn, D. TLM-Tracker: Software for cell segmentation, tracking and lineage analysis in time-lapse microscopy movies. Bioinformatics 2012, 28, 2276–2277. [Google Scholar] [CrossRef] [Green Version]
- Stylianidou, S.; Brennan, C.; Nissen, S.B.; Kuwada, N.J.; Wiggins, P.A. SuperSegger: Robust image segmentation, analysis and lineage tracking of bacterial cells. Mol. Microbiol. 2016, 102, 690–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.R.; Hasty, J. Microfluidic devices for measuring gene network dynamics in single cells. Nat. Rev. Genet. 2009, 10, 628–638. [Google Scholar] [CrossRef]
- Danino, T.; Mondragón-Palomino, O.; Tsimring, L.; Hasty, J. A synchronized quorum of genetic clocks. Nature 2010, 463, 326–330. Available online: http://xxx.lanl.gov/abs/15334406 (accessed on 15 December 2021). [CrossRef] [Green Version]
- Mather, W.; Mondragon-Palomino, O.; Danino, T.; Hasty, J.; Tsimring, L.S. Streaming instability in growing cell populations. Phys. Rev. Lett. 2010, 104, 208101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Najjar, N.; Van Teeseling, M.C.; Mayer, B.; Hermann, S.; Thanbichler, M.; Graumann, P.L. Bacterial cell growth is arrested by violet and blue, but not yellow light excitation during fluorescence microscopy. BMC Mol. Cell Biol. 2020, 21, 35. [Google Scholar] [CrossRef]
- Icha, J.; Weber, M.; Waters, J.C.; Norden, C. Phototoxicity in live fluorescence microscopy, and how to avoid it. BioEssays 2017, 39, 1700003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.K.; Chen, Y.; Hirning, A.J.; Alnahhas, R.N.; Josić, K.; Bennett, M.R. Long-range spatio-temporal coordination of gene expression in synthetic microbial consortia. Nat. Chem. Biol. 2019, 15, 1102–1109. [Google Scholar] [CrossRef]
- Winkle, J.; Igoshin, O.A.; Bennett, M.R.; Josic, K.; Ott, W. Modeling mechanical interactions in growing populations of rod-shaped bacteria. Phys. Biol. 2017, 14, 055001. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.; Logan, D.; Madden, K.; Ljosa, V.; Rueden, C.; Harris, G.B.; Eliceiri, K.; et al. Improved structure, function, and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [Green Version]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next,-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Alnahhas, R.N.; Sadeghpour, M.; Chen, Y.; Frey, A.A.; Ott, W.; Josić, K.; Bennett, M.R. Majority sensing in synthetic microbial consortia. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.C.W.; Elowitz, M.B. Using movies to analyse gene circuit dynamics in single cells. Nat. Rev. Microbiol. 2009, 7, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Alnahhas, R.N.; Winkle, J.J.; Hirning, A.J.; Karamched, B.; Ott, W.; Josić, K.; Bennett, M.R. Spatiotemporal Dynamics of Synthetic Microbial Consortia in Microfluidic Devices. ACS Synth. Biol. 2019, 8, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Hand, A.J.; Sun, T.; Barber, D.C.; Hose, D.R.; MacNeil, S. Automated tracking of migrating cells in phase-contrast video microscopy sequences using image registration. J. Microsc. 2009, 234, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Ulman, V.; Maška, M.; Magnusson, K.E.G.; Ronneberger, O.; Haubold, C.; Harder, N.; Matula, P.; Matula, P.; Svoboda, D.; Radojevic, M.; et al. An objective comparison of cell-tracking algorithms. Nat. Methods 2017, 14, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Marvasti-Zadeh, S.M.; Cheng, L.; Ghanei-Yakhdan, H.; Kasaei, S. Deep learning for visual tracking: A comprehensive survey. IEEE Trans. Intell. Transp. Syst. 2021, 1–26. [Google Scholar] [CrossRef]
- Yilmaz, A.; Javed, O.; Shah, M. Object tracking: A survey. ACM Comput. Surv. (CSUR) 2006, 38, 13-es. [Google Scholar] [CrossRef]
- Lucas, B.D.; Kanade, T. An iterative image registration technique with an application to stereo vision. In Proceedings of the International Conference on Artificial Intelligence, Vancouver, BC, Canada, 24–28 August 1981; pp. 674–679. [Google Scholar]
- Mang, A.; Biros, G. An inexact Newton–Krylov algorithm for constrained diffeomorphic image registration. SIAM J. Imaging Sci. 2015, 8, 1030–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mang, A.; Ruthotto, L. A Lagrangian Gauss–Newton–Krylov solver for mass- and intensity-preserving diffeomorphic image registration. SIAM J. Sci. Comput. 2017, 39, B860–B885. [Google Scholar] [CrossRef] [PubMed]
- Mang, A.; Gholami, A.; Davatzikos, C.; Biros, G. CLAIRE: A distributed-memory solver for constrained large deformation diffeomorphic image registration. SIAM J. Sci. Comput. 2019, 41, C548–C584. [Google Scholar] [PubMed]
- Borzi, A.; Ito, K.; Kunisch, K. An optimal control approach to optical flow computation. Int. J. Numer. Methods Fluids 2002, 40, 231–240. [Google Scholar] [CrossRef]
- Horn, B.K.P.; Shunck, B.G. Determining optical flow. Artif. Intell. 1981, 17, 185–203. [Google Scholar] [CrossRef] [Green Version]
- Delpiano, J.; Jara, J.; Scheer, J.; Ramírez, O.A.; Ruiz-del Solar, J.; Härtel, S. Performance of optical flow techniques for motion analysis of fluorescent point signals in confocal microscopy. Mach. Vis. Appl. 2012, 23, 675–689. [Google Scholar]
- Madrigal, F.; Hayet, J.B.; Rivera, M. Motion priors for multiple target visual tracking. Mach. Vis. Appl. 2015, 26, 141–160. [Google Scholar] [CrossRef]
- Banerjee, D.S.; Stephenson, G.; Das, S.G. Segmentation and analysis of mother machine data: SAM. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jug, F.; Pietzsch, T.; Kainmüller, D.; Funke, J.; Kaiser, M.; van Nimwegen, E.; Rother, C.; Myers, G. Optimal Joint Segmentation and Tracking of Escherichia Coli in the Mother Machine. In Bayesian and Graphical Models for Biomedical Imaging; Springer: Cham, Switzerland, 2014; Volume LNCS 8677, pp. 25–36. [Google Scholar]
- Lugagne, J.B.; Lin, H.; Dunlop, M.J. DeLTA: Automated cell segmentation, tracking, and lineage reconstruction using deep learning. PLoS Comput. Biol. 2020, 16, e1007673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollion, J.; Elez, M.; Robert, L. High-throughput detection and tracking of cells and intracellular spots in mother machine experiments. Nat. Protoc. 2019, 14, 3144–3161. [Google Scholar] [CrossRef]
- Sauls, J.T.; Schroeder, J.W.; Brown, S.D.; Le Treut, G.; Si, F.; Li, D.; Wang, J.D.; Jun, S. Mother machine image analysis with MM3. bioRxiv 2019, 810036. [Google Scholar] [CrossRef]
- Smith, A.; Metz, J.; Pagliara, S. MMHelper: An automated framework for the analysis of microscopy images acquired with the mother machine. Sci. Rep. 2019, 9, 10123. [Google Scholar]
- Arbelle, A.; Reyes, J.; Chen, J.Y.; Lahav, G.; Raviv, T.R. A probabilistic approach to joint cell tracking and segmentation in high-throughput microscopy videos. Med. Image Anal. 2018, 47, 140–152. [Google Scholar]
- Okuma, K.; Taleghani, A.; De Freitas, N.; Little, J.J.; Lowe, D.G. A boosted particle filter: Multitarget detection and tracking. In Proceedings of the European Conference on Computer Vision, Prague, Czech Republic, 11–14 May 2004; Springer: Berlin/Heidelberg, Germany, 2004; pp. 28–39. [Google Scholar]
- Smal, I.; Niessen, W.; Meijering, E. Bayesian tracking for fluorescence microscopic imaging. In Proceedings of the 3rd IEEE International Symposium on Biomedical Imaging: Nano to Macro, Arlington, WA, USA, 6–9 April 2006; pp. 550–553. [Google Scholar]
- Kervrann, C.; Trubuil, A. Optimal level curves and global minimizers of cost functionals in image segmentation. J. Math. Imaging Vis. 2002, 17, 153–174. [Google Scholar] [CrossRef]
- Li, K.; Miller, E.D.; Chen, M.; Kanade, T.; Weiss, L.E.; Campbell, P.G. Cell population tracking and lineage construction with spatiotemporal context. Med. Image Anal. 2008, 12, 546–566. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, W.; Metaxas, D.; Mathew, R.; White, E. Cell segmentation and tracking using texture-adaptive snakes. In Proceedings of the IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Arlington, VA, USA, 12–15 April 2007; pp. 101–104. [Google Scholar]
- Yang, F.; Mackey, M.A.; Ianzini, F.; Gallardo, G.; Sonka, M. Cell segmentation, tracking, and mitosis detection using temporal context. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Palm Springs, CA, USA, 26–29 October 2005; pp. 302–309. [Google Scholar]
- Sethuraman, V.; French, A.; Wells, D.; Kenobi, K.; Pridmore, T. Tissue-level segmentation and tracking of cells in growing plant roots. Mach. Vis. Appl. 2012, 23, 639–658. [Google Scholar] [CrossRef]
- Balomenos, A.D.; Tsakanikas, P.; Manolakos, E.S. Tracking single-cells in overcrowded bacterial colonies. In Proceedings of the Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Milano, Italy, 25–29 August 2015; IEEE: Piscataway, NJ, USA, 2015; pp. 6473–6476. [Google Scholar]
- Bise, R.; Yin, Z.; Kanade, T. Reliable cell tracking by global data association. In Proceedings of the IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Chicago, IL, USA, 30 March–2 April 2011; pp. 1004–1010. [Google Scholar]
- Bise, R.; Li, K.; Eom, S.; Kanade, T. Reliably tracking partially overlapping neural stem cells in DIC microscopy image sequences. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention Workshop, London, UK, 20–24 September 2009; pp. 67–77. [Google Scholar]
- Kanade, T.; Yin, Z.; Bise, R.; Huh, S.; Eom, S.; Sandbothe, M.F.; Chen, M. Cell image analysis: Algorithms, system and applications. In Proceedings of the 2011 IEEE Workshop on Applications of Computer Vision (WACV), Kona, HI, USA, 5–7 January 2011; IEEE: Piscataway, NJ, USA, 2011; pp. 374–381. [Google Scholar]
- Primet, M.; Demarez, A.; Taddei, F.; Lindner, A.; Moisan, L. Tracking of cells in a sequence of images using a low-dimensional image representation. In Proceedings of the IEEE International Symposium on Biomedical Imaging, Paris, France, 14–17 May 2008; pp. 995–998. [Google Scholar]
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation. In Proceedings of the Medical Image Computing and Computer Assisted Intervention, Munich, Germany, 5–9 October 2015; Volume LNCS 9351, pp. 234–241. [Google Scholar]
- Su, H.; Yin, Z.; Huh, S.; Kanade, T. Cell segmentation in phase contrast microscopy images via semi-supervised classification over optics-related features. Med. Image Anal. 2013, 17, 746–765. [Google Scholar] [CrossRef]
- Wang, Q.; Niemi, J.; Tan, C.M.; You, L.; West, M. Image segmentation and dynamic lineage analysis in single-cell fluorescence microscopy. Cytom. Part A J. Int. Soc. Adv. Cytom. 2010, 77, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiuqing, W.; Xu, C.; Xianhang, Z. Cell tracking via structured prediction and learning. Mach. Vis. Appl. 2017, 28, 859–874. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, F.; Xi, W.; Chen, H.; Gao, P.; He, C. Joint multi-frame detection and segmentation for multi-cell tracking. In Proceedings of the International Conference on Image and Graphics, Beijing, China, 23–25 August 2019; Volume LNCS 11902, pp. 435–446. [Google Scholar]
- Sixta, T.; Cao, J.; Seebach, J.; Schnittler, H.; Flach, B. Coupling cell detection and tracking by temporal feedback. Mach. Vis. Appl. 2020, 31, 1–18. [Google Scholar]
- Hayashida, J.; Nishimura, K.; Bise, R. MPM: Joint representation of motion and position map for cell tracking. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Seattle, WA, USA, 14–19 June 2020; pp. 3823–3832. [Google Scholar]
- Payer, C.; Stern, D.; Neff, T.; Bishof, H.; Urschler, M. Instance segmentation and tracking with cosine embeddings and recurrent hourglass networks. In Proceedings of the Medical Image Computing and Computer Assisted Intervention, Granada, Spain, 16–20 September 2018; Volume LNCS 11071, pp. 3–11. [Google Scholar]
- Payer, C.; Štern, D.; Feiner, M.; Bischof, H.; Urschler, M. Segmenting and tracking cell instances with cosine embeddings and recurrent hourglass networks. Med. Image Anal. 2019, 57, 106–119. [Google Scholar] [PubMed]
- Vicar, T.; Balvan, J.; Jaros, J.; Jug, F.; Kolar, R.; Masarik, M.; Gumulec, J. Cell segmentation methods for label-free contrast microscopy: Review and comprehensive comparison. BMC Bioinform. 2019, 20, 1–25. [Google Scholar]
- Al-Kofahi, Y.; Zaltsman, A.; Graves, R.; Marshall, W.; Rusu, M. A deep learning-based algorithm for 2D cell segmentation in microscopy images. BMC Bioinform. 2018, 19, 1–11. [Google Scholar]
- Falk, T.; Mai, D.; Bensch, R.; Çiçek, Ö.; Abdulkadir, A.; Marrakchi, Y.; Böhm, A.; Deubner, J.; Jäckel, Z.; Seiwald, K.; et al. U-Net: Deep learning for cell counting, detection, and morphometry. Nat. Methods 2019, 16, 67–70. [Google Scholar] [CrossRef]
- Lux, F.; Matula, P. DIC image segmentation of dense cell populations by combining deep learning and watershed. In Proceedings of the 2019 IEEE 16th International Symposium on Biomedical Imaging (ISBI 2019), Venice, Italy, 8–11 April 2019; pp. 236–239. [Google Scholar]
- Moen, E.; Bannon, D.; Kudo, T.; Graf, W.; Covert, M.; Van Valen, D. Deep learning for cellular image analysis. Nat. Methods 2019, 16, 1233–1246. [Google Scholar] [PubMed]
- Rempfler, M.; Stierle, V.; Ditzel, K.; Kumar, S.; Paulitschke, P.; Andres, B.; Menze, B.H. Tracing cell lineages in videos of lens-free microscopy. Med. Image Anal. 2018, 48, 147–161. [Google Scholar]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: A generalist algorithm for cellular segmentation. Nat. Methods 2021, 18, 100–106. [Google Scholar] [CrossRef]
- Akram, S.U.; Kannala, J.; Eklund, L.; Heikkilä, J. Joint cell segmentation and tracking using cell proposals. In Proceedings of the IEEE 13th International Symposium on Biomedical Imaging (ISBI), Prague, Czech Republic, 13–16 April 2016; pp. 920–924. [Google Scholar]
- Nishimura, K.; Hayashida, J.; Wang, C.; Bise, R. Weakly-Supervised Cell Tracking via Backward-and-Forward Propagation. In Proceedings of the European Conference on Computer Vision, Seoul, Korea, 27 October–2 November 2019; pp. 104–121. [Google Scholar]
- Rempfler, M.; Kumar, S.; Stierle, V.; Paulitschke, P.; Andres, B.; Menze, B.H. Cell lineage tracing in lens-free microscopy videos. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Quebec City, QC, Canada, 11–13 September 2017; pp. 3–11. [Google Scholar]
- Maska, M.; Ulman, V.; Svoboda, D.; Matula, P.; Matula, P.; Ederra, C.; Urbiola, A.; Espana, T.; Venkatesan, S.; Balak, D.M.W.; et al. A benchmark for comparison of cell tracking algorithms. Bioinformatics 2014, 30, 1609–1617. [Google Scholar]
- Löffler, K.; Scherr, T.; Mikut, R. A graph-based cell tracking algorithm with few manually tunable parameters and automated segmentation error correction. bioRxiv 2021, 16, e0249257. [Google Scholar]
- Vo, B.T.; Vo, B.N.; Cantoni, A. The cardinality balanced multi-target multi-Bernoulli filter and its implementations. IEEE Trans. Signal Process. 2008, 57, 409–423. [Google Scholar]
- Pierskalla, W.P. The multidimensional assignment problem. Oper. Res. 1968, 16, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, K.C.; Hofstra, R.B. Multidimensional assignment problems. Decis. Sci. 1988, 19, 306–321. [Google Scholar] [CrossRef]
- Chakraborty, A.; Roy-Chowdhury, A.K. Context aware spatio-temporal cell tracking in densely packed multilayer tissues. Med. Image Anal. 2015, 19, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yadav, R.K.; Roy-Chowdhury, A.; Reddy, G.V. Automated tracking of stem cell lineages of Arabidopsis shoot apex using local graph matching. Plant J. 2010, 62, 135–147. [Google Scholar] [CrossRef]
- Liu, M.; Chakraborty, A.; Singh, D.; Yadav, R.K.; Meenakshisundaram, G.; Reddy, G.V.; Roy-Chowdhury, A. Adaptive cell segmentation and tracking for volumetric confocal microscopy images of a developing plant meristem. Mol. Plant 2011, 4, 922–931. [Google Scholar] [CrossRef]
- Liu, M.; Li, J.; Qian, W. A multi-seed dynamic local graph matching model for tracking of densely packed cells across unregistered microscopy image sequences. Mach. Vis. Appl. 2018, 29, 1237–1247. [Google Scholar] [CrossRef]
- Vo, B.N.; Vo, B.T. A multi-scan labeled random finite set model for multi-object state estimation. IEEE Trans. Signal Process. 2019, 67, 4948–4963. [Google Scholar] [CrossRef] [Green Version]
- Punchihewa, Y.G.; Vo, B.T.; Vo, B.N.; Kim, D.Y. Multiple object tracking in unknown backgrounds with labeled random finite sets. IEEE Trans. Signal Process. 2018, 66, 3040–3055. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Vo, B.N.; Thian, A.; Choi, Y.S. A generalized labeled multi-Bernoulli tracker for time lapse cell migration. In Proceedings of the 2017 International Conference on Control, Automation and Information Sciences, Jeju, Korea, 18–21 October 2017; pp. 20–25. [Google Scholar]
- Winkle, J.J.; Karamched, B.R.; Bennett, M.R.; Ott, W.; Josić, K. Emergent spatiotemporal population dynamics with cell-length control of synthetic microbial consortia. PLoS Comput. Biol. 2021, 17, e1009381. [Google Scholar]
- Bise, R.; Sato, Y. Cell detection from redundant candidate regions under non-overlapping constraints. IEEE Trans. Med Imaging 2015, 34, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Matula, P.; Maška, M.; Sorokin, D.V.; Matula, P.; Ortiz-de Solórzano, C.; Kozubek, M. Cell tracking accuracy measurement based on comparison of acyclic oriented graphs. PLoS ONE 2015, 10, e0144959. [Google Scholar]
- Agrawal, A.; Verschueren, R.; Diamond, S.; Boyd, S. A rewriting system for convex optimization problems. J. Control Decis. 2018, 5, 42–60. [Google Scholar] [CrossRef]
- Diamond, S.; Boyd, S. CVXPY: A Python-embedded modeling language for convex optimization. J. Mach. Learn. Res. 2016, 17, 1–5. [Google Scholar]
- Shen, X.; Diamond, S.; Gu, Y.; Boyd, S. Disciplined convex-concave programming. In Proceedings of the 2016 IEEE 55th Conference on Decision and Control (CDC), Las Vegas, NV, USA, 12–14 December 2016; pp. 1009–1014. [Google Scholar]
- Stricker, J.; Cookson, S.; Bennett, M.R.; Mather, W.H.; Tsimring, L.S.; Hasty, J. A fast, robust and tunable synthetic gene oscillator. Nature 2008, 456, 516–519. [Google Scholar] [CrossRef]
- Chen, Y.; Kim, J.K.; Hirning, A.J.; Josić, K.; Bennett, M.R. Emergent genetic oscillations in a synthetic microbial consortium. Science 2015, 349, 986–989. [Google Scholar] [CrossRef] [Green Version]
- Sloan, S.W. A fast algorithm for constructing Delauny triangulations in the plane. Adv. Eng. Softw. 1987, 9, 34–55. [Google Scholar] [CrossRef]
- Azencott, R. Simulated Annealing: Parallelization Techniques; Wiley-Interscience: Hoboken, NJ, USA, 1992; Volume 27. [Google Scholar]
- Azencott, R.; Chalmond, B.; Coldefy, F. Markov Image Fusion to Detect Intensity Valleys. Int. J. Comput. Vis. 1994, 16, 135–145. [Google Scholar] [CrossRef]
- Boyd, S.; Vandenberghe, L. Convex Optimization; Campridge University Press: Campridge, UK, 2004. [Google Scholar]
- Ackley, D.H.; Hinton, G.E.; Sejnowski, T.J. A learning algorithm for Boltzmann machines. Cogn. Sci. 1985, 9, 147–169. [Google Scholar] [CrossRef]
- Hinton, G.E.; Sejnowski, T.J. Chapter Learning and Relearning in Boltzmann Machines. In Parallel Distributed Processing: Explorations in the Microstructure of Cognition; MIT Press: Cambridge, MA, USA, 1986; pp. 282–317. [Google Scholar]
- Azencott, R. Synchronous Boltzmann machines and Gibbs fields: Learning algorithms. In Neurocomputing; Springer: Berlin/Heidelberg, Germany, 1990; pp. 51–63. [Google Scholar]
- Azencott, R. Synchronous Boltzmann machines and artificial vision. Neural Netw. 1990, 135–143. Available online: https://www.math.uh.edu/~razencot/MyWeb/Research/Selected_Reprints/1990SynchronousBoltzmanMachinesArtificialVision.pdf (accessed on 15 December 2021).
- Azencott, R.; Graffigne, C.; Labourdette, C. Edge Detection and Segmentation of Textured Plane Images. In Stochastic Models, Statistical Methods, and Algorithms in Image Analysis; Springer-Verlag: New York, NY, USA, 1992; Volume 74, pp. 75–88. [Google Scholar]
- Kong, A.; Azencott, R. Binary Markov Random Fields and Interpretable Mass Spectra Discrimination. Stat. Appl. Genet. Mol. Biol. 2017, 16, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Azencott, R.; Doutriaux, A.; Younes, L. Synchronous Boltzmann Machines and Curve Identification Tasks. Netw. Comput. Neural Syst. 1993, 4, 461–480. [Google Scholar]
- Garda, P.; Belhaire, E. An Analog Circuit with Digital I/O for Synchronous Boltzmann Machines. In VLSI for Artificial Intelligence and Neural Networks; Springer: Berlin, Germany, 1991; pp. 245–254. [Google Scholar]
- Lafargue, V.; Belhaire, E.; Pujol, H.; Berechet, I.; Garda, P. Programmable Mixed Implementation of the Boltzmann Machine. In International Conference on Artificial Neural Networks; Springer: Berlin, Germany, 1994; pp. 409–412. [Google Scholar]
- Pujol, H.; Klein, J.-O.; Belhaire, E.; Garda, P. RA: An analog neurocomputer for the synchronous Boltzmann machine. In Proceedings of the Fourth International Conference on Microelectronics for Neural Networks and Fuzzy Systems, Turin, Italy, 26–28 September 1994; IEEE: Piscataway, NJ, USA, 1994; pp. 449–455. [Google Scholar]
- Beucher, S.; Lantuejoul, C. Use of watersheds in contour detection. In Workshop on Image Processing; CCETT/IRISA: Rennes, France, 1979. [Google Scholar]
- Mumford, D.B.; Shah, J. Optimal approximations by piecewise smooth functions and associated variational problems. Commun. Pure Appl. Math. 1989, 42, 577–685. [Google Scholar] [CrossRef] [Green Version]
- Mézard, M.; Parisi, G.; Virasoro, M.A. Spin Glass Theory and Beyond: An Introduction to the Replica Method and Its Applications; World Scientific Publishing Company: Singapore, 1987; Volume 9. [Google Scholar]
- Kirkpatrick, S.; Gelatt, C.D., Jr.; Vecchi, M.P. Optimization by simulated annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Roussel-Ragot, P.; Dreyfus, G. A problem independent parallel implementation of simulated annealing: Models and experiments. IEEE Trans. Comput.-Aided Des. Integr. Circuits Syst. 1990, 9, 827–835. [Google Scholar] [CrossRef]
- Burda, Z.; Krzywicki, A.; Martin, O.C.; Tabor, Z. From simple to complex networks: Inherent structures, barriers, and valleys in the context of spin glasses. Phys. Rev. E 2006, 73, 036110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, P.J. The 1972 Wald Lecture Robust Statistics: A Review. Ann. Math. Stat. 1972, 43, 1041–1067. [Google Scholar] [CrossRef]
- Ram, D.J.; Sreenivas, T.; Subramaniam, K.G. Parallel simulated annealing algorithms. J. Parallel Distrib. Comput. 1996, 37, 207–212. [Google Scholar] [CrossRef]
- Digabel, H.; Lantuejoul, C. Iterative Algorithms. In Proceedings of the 2nd European Symposium Quantitative Analysis of Microstructures in Material Science, Biology and Medicine, Caen, France, 4–7 October 1977; pp. 85–89. [Google Scholar]
- Vincent, L.; Soille, P. Watersheds in digital spaces: An efficient algorithm based on immersion simulations. IEEE Trans. Pattern Anal. Mach. Intell. 1991, 13, 583–598. [Google Scholar] [CrossRef] [Green Version]








| Label | Interframe Duration | Number of Frames |
|---|---|---|
| BENCH1 | 1 min | 500 |
| BENCH2 | 2 min | 300 |
| BENCH3 | 3 min | 300 |
| BENCH6 | 6 min | 100 |
| Sequence | Pcp-Accuracy | Frames |
|---|---|---|
| BENCH1 | 500 out of 500 | |
| BENCH2 | 298 out of 300 | |
| BENCH2 | 2 out of 300 | |
| BENCH3 | 271 out of 300 | |
| BENCH3 | 17 out of 300 | |
| BENCH3 | 12 out of 300 |
| Registration Accuracy | Number of Frames |
|---|---|
| 55 frames out of 100 | |
| 97% | 40 frames out of 100 |
| 94.5% | 5 frames out of 100 |
| Weights | ||||||||
|---|---|---|---|---|---|---|---|---|
| Value | 3 | 7 | 100 | 4 | 600 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarmadi, S.; Winkle, J.J.; Alnahhas, R.N.; Bennett, M.R.; Josić, K.; Mang, A.; Azencott, R. Stochastic Neural Networks for Automatic Cell Tracking in Microscopy Image Sequences of Bacterial Colonies. Math. Comput. Appl. 2022, 27, 22. https://0-doi-org.brum.beds.ac.uk/10.3390/mca27020022
Sarmadi S, Winkle JJ, Alnahhas RN, Bennett MR, Josić K, Mang A, Azencott R. Stochastic Neural Networks for Automatic Cell Tracking in Microscopy Image Sequences of Bacterial Colonies. Mathematical and Computational Applications. 2022; 27(2):22. https://0-doi-org.brum.beds.ac.uk/10.3390/mca27020022
Chicago/Turabian StyleSarmadi, Sorena, James J. Winkle, Razan N. Alnahhas, Matthew R. Bennett, Krešimir Josić, Andreas Mang, and Robert Azencott. 2022. "Stochastic Neural Networks for Automatic Cell Tracking in Microscopy Image Sequences of Bacterial Colonies" Mathematical and Computational Applications 27, no. 2: 22. https://0-doi-org.brum.beds.ac.uk/10.3390/mca27020022