
Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters


Rare Disease Research Unit, Worldwide Research, Development and Medical, Pfizer Inc., Cambridge, MA 02139, USA
*
Author to whom correspondence should be addressed.
Inorganics 2022, 10(3), 31; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10030031
Submission received: 24 January 2022
/
Revised: 19 February 2022
/
Accepted: 24 February 2022
/
Published: 26 February 2022
(This article belongs to the Special Issue Assembly and Reactivity of Iron–Sulfur Clusters)
Abstract
:Iron–sulfur clusters (Fe–S or ISC) are essential cofactors that function in a wide range of biological pathways. In mammalian cells, Fe–S biosynthesis primarily relies on mitochondria and involves a concerted group of evolutionary-conserved proteins forming the ISC pathway. In the early stage of the ISC pathway, the Fe–S core complex is required for de novo assembly of Fe–S. In humans, the Fe–S core complex comprises the cysteine desulfurase NFS1, the scaffold protein ISCU2, frataxin (FXN), the ferredoxin FDX2, and regulatory/accessory proteins ISD11 and Acyl Carrier Protein (ACP). In recent years, the field has made significant advances in unraveling the structure of the Fe–S core complex and the mechanism underlying its function. Herein, we review the key recent findings related to the Fe–S core complex and its components. We highlight some of the unanswered questions and provide a model of the Fe–S assembly within the complex. In addition, we briefly touch on the genetic diseases associated with mutations in the Fe–S core complex components.
1. Introduction
The mitochondria of mammalian cells are not only the main providers of energy, but also the site of many metabolic processes. The products from these processes include essential cofactors used throughout the cell, such as iron–sulfur clusters (Fe–S or ISC). Fe–S are evolutionary conserved inorganic metallocofactors. They are made from non-heme iron and L-cysteine-derived sulfide and exist in multiple types, primarily as [2Fe–2S], [3Fe-4S], or [4Fe–4S] clusters, although more complex cofactor structures exist [1].
The importance of Fe–S biosynthesis is highlighted by the number and variety of Fe–S cluster-dependent processes in the cell (Table 1 and Table 2). For example, Fe–S clusters are required for lipoic acid synthetase (LIAS), a key enzyme for the synthesis of lipoic acid [2]. Lipoic acid is an essential cofactor that binds and is required for the activity of several mitochondrial complexes such as the pyruvate dehydrogenase (PDH) and the alpha-ketoglutarate dehydrogenase (KGDH) complexes [3]. PDH acts as the gatekeeper of the Krebs cycle, converting pyruvate to acetyl-CoA. KGDH is also a key Krebs cycle enzyme, acting as a sensor of oxidative stress and metabolic flux [4]. Other Fe–S-containing Krebs cycle proteins include aconitase 2 (ACO2), an enzyme that requires [4Fe–4S] to become catalytically active [5], and succinate dehydrogenase subunit B (SDHB), a subunit of mitochondrial respiratory complex II that contains multiple Fe–S clusters [6]. Fe–S are key components of oxidative phosphorylation through their contribution to electron transfers in multiple complexes of the mitochondrial respiratory chain. In addition to SDHB/complex II, Fe–S are present in complex I and in the Rieske Fe–S protein UQCRFS1 of complex III [7]. Therefore, both energy generation and TCA metabolic processes, two important functions of mitochondria, require Fe–S clusters to properly operate. Fe–S are also required outside the mitochondria and are found in nearly every other cellular compartment. A few representative examples include multiple enzymes involved in DNA repair and metabolism that have been recently identified as Fe–S proteins [8,9] (Table 2). In addition, many of the thio-modifications found on tRNA molecules require Fe–S clusters to generate free sulfide to add to tRNA [10].
In mammals, the biosynthesis of Fe–S requires concerted activities in the mitochondrial matrix (the ISC pathway), the mitochondrial membrane (the ISC export) and the cytosol (the cytosolic iron–sulfur cluster assembly (CIA) machinery) to synthesize, traffic, and insert Fe–S onto apo-proteins in the different compartments of the cell (Figure 1) [11,12]. The full process for generating Fe–S clusters can be divided into five distinct steps. First, the de novo synthesis of a [2Fe–2S] cluster in the core complex of the ISC pathway, consisting of the cysteine desulfurase NFS1, the scaffold protein ISCU2, and the accessory/regulatory proteins ISD11 and ACP. This process also requires the activity the ferredoxin-ferredoxin reductase couple (FDX2-FDXR) and frataxin (FXN). Second, the transfer of the newly synthesized cluster on ISCU2 to acceptor proteins, such as glutaredoxin 5 (GLRX5), ISCAs, or directly to [2Fe–2S] client proteins in the mitochondria. The transfer is assisted by the chaperone HSPA9 and the co-chaperone HSCB, which specifically bind to ISCU2. Third, the maturation of [2Fe–2S] clusters into [4Fe–4S] clusters and their transfer to mitochondrial client proteins. This process requires the ISCA and IBA57 proteins. Fourth, the export of a still-unclear molecular entity produced during the early steps of the ISC pathway from the mitochondria to the cytosol via the ABCB7 transporter. Finally, the maturation of Fe–S in the cytosol through the CIA pathway and their transfer to cytosolic and nuclear Fe–S client proteins.
Whereas the ISC export and CIA machinery are specific to eukaryotic cells due to compartmentalization, major components of the mitochondrial ISC pathway are conserved from bacteria to yeast and to human [13,14]. Therefore, mechanisms of the ISC pathway have been studied in prokaryotes as well as in eukaryotic models from Saccharomyces cerevisiae to mouse and human cells over the past 25 years [13,15].
In the more recent years, significant insights into the function and structure of the mitochondrial Fe–S core complex have been obtained, shedding light on key mechanisms of the de novo Fe–S assembly. Herein, we review the data on the mitochondrial Fe–S core complex and its components. In addition, we provide a brief overview of the diseases that are associated with mutations in components of the Fe–S core complex, such as Friedreich’s ataxia, a devastating rare genetic disorder caused by FXN loss of function.
2. The Core Complex of the Mitochondrial ISC Pathway
The mitochondrial Fe–S core complex corresponds to the set of dynamic protein complexes that form and are implicated in the assembly of Fe–S from inorganic iron and sulfur (Figure 1). Six components are part of the human mitochondrial Fe–S core complex: the cysteine desulfurase NFS1, the scaffold ISCU2, frataxin (FXN), ferredoxin 2 (FDX2), and accessory/regulatory proteins ISD11 (also known as LYRM4) and ACP (Table 3). All these proteins are encoded by nuclear genes, which are translated, imported into mitochondria, and eventually processed into their mature form by cleaving the mitochondrial targeting sequence [16,17]. In E. coli, the open reading frames for some of these proteins were found clustered under the isc operon [18].
The Fe–S core protein complex is centered around NFS1, which forms a homodimer considered as the central hub [19,20]. On this dimer, diverse symmetrical complexes assemble, as seen in recent structural studies [21,22,23]. ISCU2, FXN, and potentially FDX2 interact with NFS1 in a pairwise configuration at the outer edge of NFS1, while two ISD11 moieties interact around the NFS1 homodimer interface (Figure 2). Two ACP moieties interact with the core complex by individually binding to one ISD11 apiece. Published structural and biochemical studies of different forms of the complex use various abbreviations to denote which proteins are present in any given experiment. The first nomenclature uses the abbreviation NIA to denote the complex consisting of NFS1, ISD11, and ACP. The second nomenclature instead uses SDA for NFS1, ISD11, and ACP. Both denominations use U and F to represent ISCU2 and Frataxin, respectively. This review utilizes the latter nomenclature system. In addition, the numbering of amino acids in various human proteins refers to the position of the amino acid in the full-length protein before any cleavage of the mitochondrial targeting sequence.
2.1. Cysteine Desulfurase (NFS1)
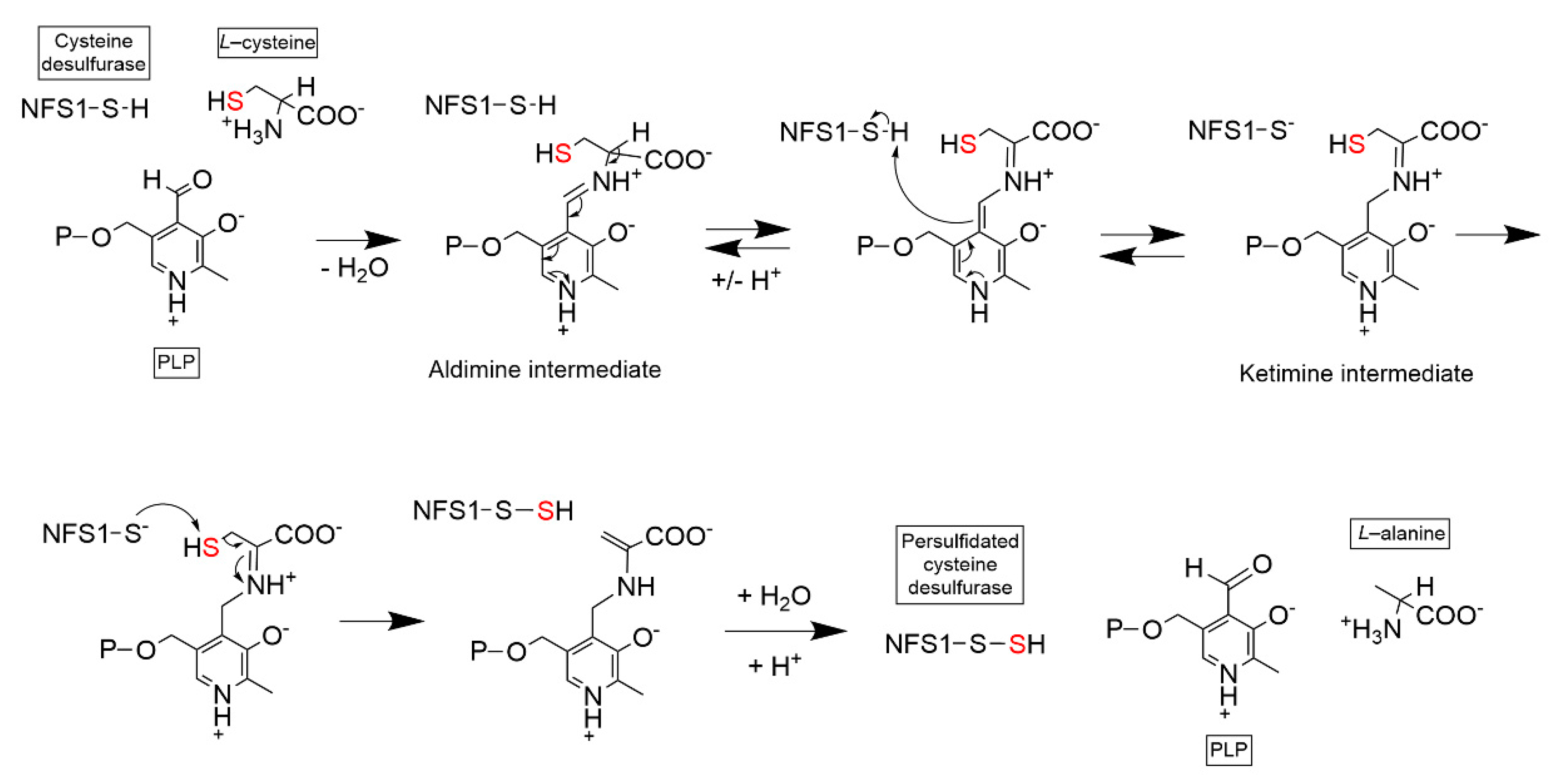
NFS1 is a member of the class 1 pyridoxal phosphate (PLP)-dependent cysteine desulfurase family. NFS1 forms a homodimer and requires pyridoxal phosphate (PLP) to perform its catalytic activity: the transfer of the sulfur moiety on a free L-cysteine to a catalytically conserved cysteine residue in the NFS1 protein (Cys381 in the human protein) [24]. This reaction results in a free alanine amino acid and the formation of a persulfide (-SSH) group on the NFS1 cysteine residue (Figure 3). This catalytic cysteine is located on a flexible loop that transfers the sulfur moiety from the NFS1 catalytic site to the acceptor scaffold protein ISCU2.
NFS1 is the largest protein of the Fe–S core assembly complex and is a ‘platform’ around which the other key components of the complex bind. Crystal and Cryo-EM structures of human (SDA)2, (SDAU)2 and (SDAUF)2 complexes showed that NFS1 binds ISD11 through a hydrophobic interface on the surface of the NFS1 homodimer, which is not present in bacterial homolog IscS [21,22] (Figure 2). Interestingly, a quite different crystal structure of the (SDA)2 complex was obtained by Cory et al. [25]. This structure showed a different organization with dimerization of NFS1 mediated by ISD11 and no significant surface interactions between the two NFS1 monomers. This structure was designated as the ‘open’ conformation of the complex, in contrast to the ‘closed’ conformation obtained by others [21,22]. Because the ‘open’ structure differs from other PLP-dependent transaminases or desulfurases and affects the formation of the PLP-containing catalytic site, it is unclear whether this conformation has any catalytic activity or is even physiologically relevant.
2.2. LYRM Accessory Protein (ISD11)
ISD11 is a member of the LYRM family of accessory proteins and the gene encoding human ISD11 is also known as LYRM4. LYRMs are small (~10–22 kDa) basic mitochondrial proteins characterized by the presence of a conserved LYR/K motif near the N-terminus. Like the other LYRM proteins, the presence of ISD11 is specific to the eukaryotic ISC pathway. Eukaryotic LYRMs were shown to have a role in the assembly and stabilization of multiple mitochondrial complexes including respiratory complexes I, II, III and V, the mitoribosome and the Fe–S core complex with ISD11 [26,27]. Yeast Isd11 was reported to play a major role in NFS1 dimer formation, as the absence of Isd11 promotes Nfs1 aggregation [28]. As a result, yeast mitochondria deleted for Isd11 are not able to generate mature Fe–S-containing proteins or display Fe–S-dependent activities [29]. In human cells, knockdown of ISD11 expression also triggers Fe–S deficiency, leading to activation of cytosolic Iron Regulatory Protein 1 (IRP1) and deregulation of the cellular iron metabolism [30].
Of the recent crystal and Cryo-EM structures of (SDA)2, (SDAU)2 and (SDAUF)2 [21,22], ISD11 was shown to interact with NFS1 on a hydrophobic interface located ~60 Å from the catalytic active site of NFS1 and ISCU2. Like other LYRM protein family members in the mitochondria, ISD11 also directly interacts with the mitochondrial Acyl Carrier Protein (ACP), the most recently identified component of the Fe–S core complex [21,22,25,27,31,32].
2.3. Acyl Carrier Protein (ACP)
Mitochondrial ACP (also known as ACP1 or ACPM in humans) is a bacterial-type acyl carrier protein essential for mitochondrial function. ACP plays a role in mitochondrial fatty acid synthesis (mFAS). It binds acyl chains by utilizing a 4′-phosphopantetheine prosthetic group (4′-PP) covalently bound to a conserved serine (S112 in the human protein) [33]. ACP functions as a scaffold for the de novo synthesis and elongation of fatty acids within the mitochondrial matrix. Particularly, ACP is essential for the synthesis of octanoic acid, the precursor of the essential lipoic acid cofactor. ACP has been shown to interact with the LYRM family of proteins [27,32] and to play a role in the integrity of multiple mitochondrial complexes, including the Fe–S core complex.
Bacterial Acp1 consistently co-purifies with recombinant NFS1-ISD11 complexes and was therefore present in all published (SDA)2, (SDAU)2 and (SDAUF)2 structures [21,22,25]. Attempts to substitute the bacterial Acp1 by co-expression of the human homolog were unsuccessful [21], hence suggesting that both bacterial and human ACPs are interchangeable and may have similar affinities. Although bacterial Acp1 might have initially been thought to be a contaminant, functional studies in yeast and human cells demonstrated it is an important component of the Fe–S core complex [31,32]. In yeast, knockdown of Acp1 results in reduced Fe–S biosynthesis [31]. In human cells, immunoprecipitation and knockdown experiments revealed that endogenous human ACP binds multiple complexes and is essential for their integrity [32]. Furthermore, binding of ACP requires the 4′-PP cofactors, as mutation of the key S112 residue prevents most of these interactions [32].
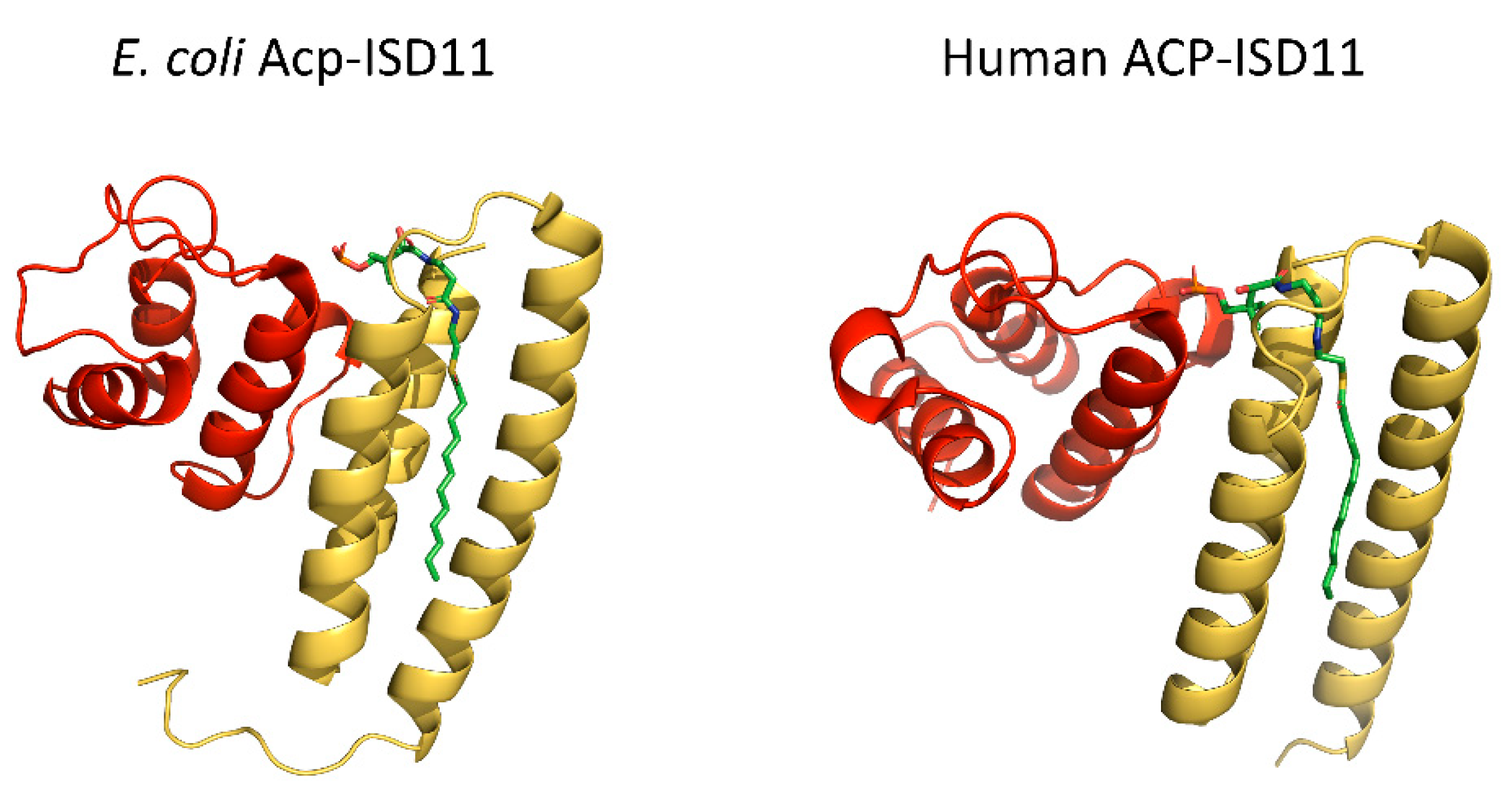
The modality of the interaction between ACP and ISD11 is of particular interest. The binding of ACP involves the docking of ACP-bound long acyl chains into the ISD11 hydrophobic core [32,34] (Figure 4). Studies of different recombinant complexes including bacterial or human ACP identified acyl chains ranging from C14 to C18 [21,22,32,34]. Top-down high-resolution mass spectrometry analyzing human ACP-ISD11 complex, or the bacterial ACP-containing (SDAUF)2 further indicated that the specific interaction is likely mediated by long (C14, C16 and C18) 3-keto acyl chains [21,32]. Because these studies were performed on recombinant complexes produced in bacteria, further investigations are required to validate the nature and relevance of these acyl chains in human cells. However, it is tempting to speculate that this type of binding is likely to occur in other ACP-LYRM interactions, hence pointing to a new key function of ACP and the synthesis of long acyl chains in the physiology of mitochondria. The reason long acyl chains might be required to control the interaction of ACP is intriguing, but ACP might play a role as a metabolic sensor that coordinates multiple key functions within mitochondria.
2.4. Scaffold Protein (ISCU2)
Human ISCU2 is the scaffold protein on which Fe–S is built from free iron and sulfur. The final stage of the assembly is the formation of a [2Fe–2S] cluster on this protein scaffold (Figure 1). Bacterial, yeast, and mammalian ISCUs contain three conserved cysteines (Cys69, Cys95, Cys138 in the human protein) that are required for Fe–S coordination in monomeric or dimeric forms of ISCU [35,36]. Cys138 was shown to be the specific cysteine on which the persulfide produced on NFS1 is transferred during the de novo assembly of Fe–S [37,38]. Characterization of recombinant ISCUs by NMR revealed an equilibrium between a disordered and an ordered structure of the protein, which might play a role in the interaction of ISCU2 with upstream and downstream protein partners [39,40,41]. Interestingly, recombinant ISCU proteins produced in bacteria are purified with Zn2+ bound to conserved residues also involved in Fe–S binding [40,41,42,43]. Binding of zinc stabilizes the ordered conformation of ISCU [39,41,44], but was also reported to inhibit the formation of Fe–S during in vitro reconstitution experiments using recombinant (SDAU)2 [43]. Zn2+-bound ISCU2 was also detected in the structures of (SDAU)2 and (SDAUF)2 [21,22]. Whether the presence of zinc is physiologically relevant is still unknown. However, recent in vitro work with mouse ISCU have shown that zinc is interchangeable with Fe2+ and that iron-bound ISCU2 displays improved ability to assemble Fe–S within the recombinant core complex, thus suggesting that Fe2+-ISCU2 might be the active form of the protein for Fe–S synthesis in vivo [45].
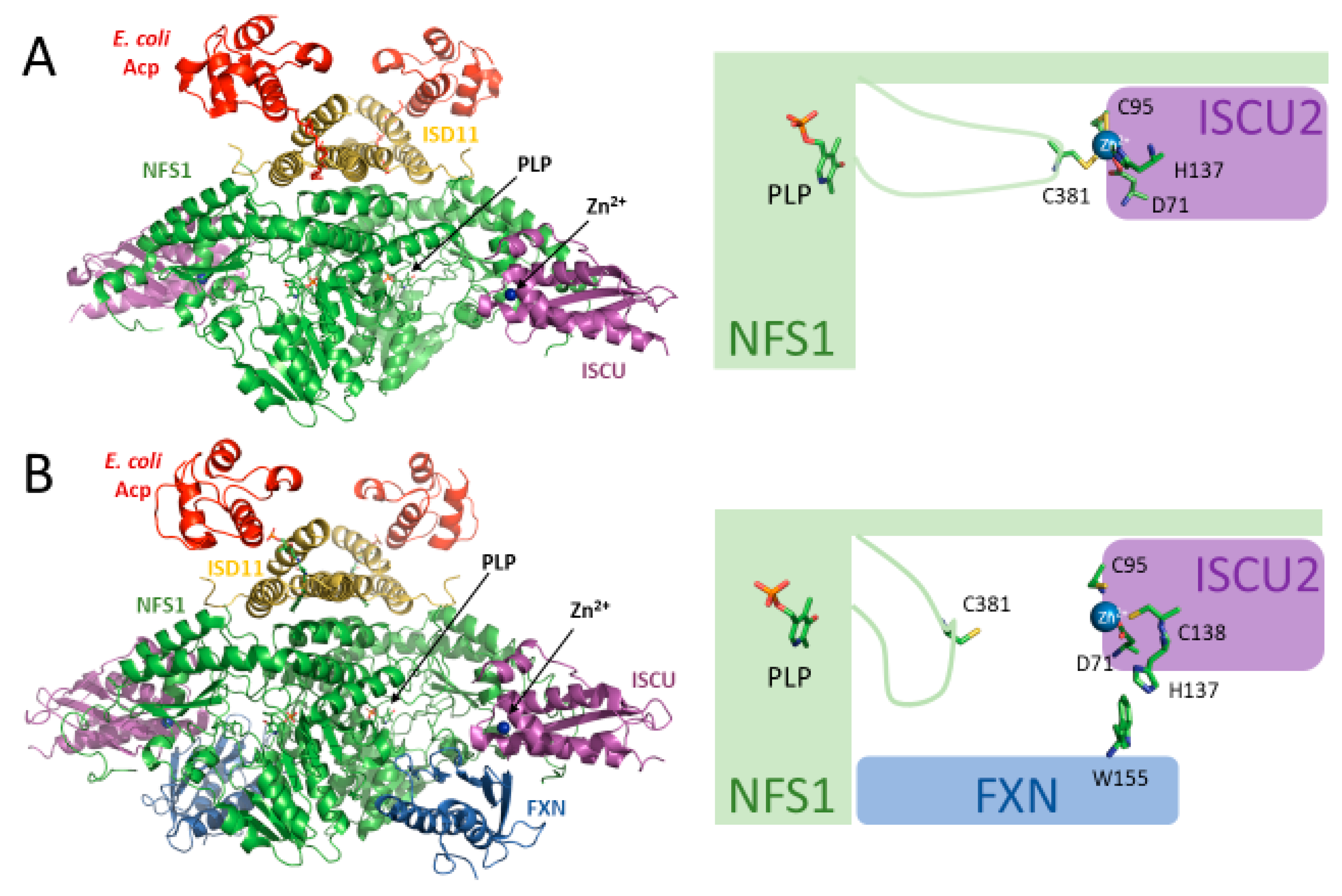
(SDAU)2 and (SDAUF)2 structures show that ISCU2 binds to NFS1 with zinc-coordinating residues facing the NFS1 active site: Asp71, Cys95, His137, and Cys138 [21,22] (Figure 2A). Interestingly, in (SDAU)2, Zn2+ is coordinated by Asp71, Cys95, and His137 of ISCU2 and by Cys381 of the NFS1 flexible loop, which is the catalytic cysteine involved in the transfer of sulfur from NFS1 to ISCU2 [22]. Upon binding of FXN, as shown in the (SDAUF)2 cryo-EM structure, the coordination environment of zinc on ISCU2 is modified: Cys381 is freed and His137 is replaced by Cys138 residue, primarily due to the interaction of ISCU2 His137 with the Trp155 residue of FXN [21] (Figure 2B).
Upstream of the Cys138 residue, ISCU2 contains a conserved LPPVK (131–135) sequence recognized by HSPA9/HSCB chaperones for the transfer of Fe–S from ISCU2 to downstream acceptors [46,47,48]. Very recently, the N-terminal tyrosine Tyr35 was demonstrated to be instrumental in the latest stage of [2Fe–2S] cluster formation, by playing a role in the dimerization of ISCU2 [23]. This residue has been previously shown to be required for proper IscU function in E. coli [49]. By using directed mutagenesis, Freibert et al. could demonstrate that Tyr35 is essential in human cells and that ISCU2 mutants Y35A, Y35K, or Y35D prevent formation of [2Fe–2S] cluster on ISCU2 in in vitro reconstitution experiments without affecting the persulfide transfer from NFS1 to ISCU2 [23]. Quite elegantly, the authors showed that a mixture of negatively charged ISCU2-Y35K and positively charged ISCU2-Y35D mutants was able to restore formation of [2Fe–2S] clusters on ISCU2, hence indicating that ISCU2 dimerization constitutes a critical step in Fe–S formation.
2.5. Frataxin (FXN)
Due to its ability to bind iron in vitro [50,51,52,53,54,55], frataxin was initially suggested to act as the iron donor during Fe–S assembly [56], or as an iron-loaded storage protein in the mitochondrial matrix [57,58]. However, later studies contradicted these claims [59,60]. While the origin of Fe2+ for Fe–S synthesis is still a matter for investigation, studies over the past years have accumulated data demonstrating that mammalian FXN is a modulator of the Fe–S core complex activity [37,45,61,62,63,64]. Specifically, FXN increases the rate at which the persulfide is transferred from NFS1 to ISCU2 [45,63], which was shown to be the rate-limiting step in the persulfidation of ISCU2 in in vitro reconstitution experiments using FDX2-FDXR as reducing system [45].
The interaction of FXN with the Fe–S core complex is highly specific. Schmucker et al. identified FXN functional interactions by combining co-immunoprecipitation and functional experiments in human cells with co-expression studies in bacteria using mouse NFS1, ISD11, and ISCU [62]. The authors showed that FXN main function is driven by its interaction with the preformed (SDAU)2 complex, thus indicating binding of FXN occurs with multiple components of the complex rather than with just one component [62]. In parallel, in vitro studies led to the isolation of mouse and human Fe–S core complexes in the presence or absence of FXN, allowing further mechanistic and structural investigations [61,62,64].
The recently obtained (SDAUF)2 cryo-EM structure have provided new insight into the molecular interaction of FXN with the Fe–S core complex [21]. FXN simultaneously binds to NFS1 and ISCU2, fitting in a pocket close to ISCU2 (Figure 2B). Details of some FXN residues involved in the interaction are provided in Figure 5. A highly conserved acidic ridge on the N-terminal portion of FXN binds to an arginine-rich patch on one NFS1 subunit. FXN associates with the other NFS1 protomer via residues on the β-sheet that contact the NFS1 loop containing the catalytic Cys381 (Figure 5, left). In addition, FXN binds to two ISCU2 regions (Figure 5, right). The first includes a loop containing the conserved Cys69 residue as well as the Asp71 residue involved in zinc coordination. The second interaction is preponderant and contains the LPPVK (131–135) sequence, recognized by HSPA9/HSCB chaperones, and residues His137 and Cys138, a zinc-binding residue in (SDAU)2 and the ISCU2 sulfur acceptor, respectively. As mentioned above, binding of FXN induces a change in the ISCU2 zinc environment, partially driven by the interaction between Trp155 on FXN and His137 on ISCU2 (Figure 2). Consequently, Cys138 residue of ISCU2 becomes accessible. Together with the interaction with the NFS1 Cys381 loop, these modifications might constitute key events explaining how FXN facilitates the transfer of the persulfide from NFS1 to ISCU2.
The implication of the LPPVK sequence in the binding of FXN supports a sequential and dynamic mechanism in which FXN must be released from the complex to allow the downstream transfer of Fe–S, involving the interaction between ISCU2 and HSPA9/HSCB chaperones.
In agreement with the requirement of a dynamic association and dissociation of FXN to support efficient Fe–S assembly and transfer, a recent study demonstrated that AAV-mediated supraphysiological expression of FXN in mice results in a loss of Fe–S client proteins, the same way FXN deficiency does [65]. The hypothesis is that an overabundance of readily available FXN may constrain the core complex in its (SDAUF)2 form, hence preventing ISCU2 dissociation and the downstream delivery of Fe–S.
Interestingly, interaction of FXN within the (SDAUF)2 complex also includes Met140 on ISCU2 [21]. In yeast, mutation of the equivalent Isu1 residue Met141 to Ile, Leu, or Val has been shown to partially bypass the requirement for yeast frataxin (Yfh1) [66,67,68]. How this mutation somehow mimics the function of FXN remains unclear. However, recent in vitro work suggested that the ISCU M140I mutation might have an impact on the transfer of Fe–S from ISCU2 to downstream acceptor proteins rather than on the activity of the core complex [69].
2.6. Ferredoxin-Ferredoxin Reductase (FDX2-FDXR)
To properly assemble Fe–S on ISCU2, the transferred persulfide must be reduced from S−1 to S−2. In in vitro reconstitution experiments using different components of the machinery, the chemical reductant dithiothreitol (DTT) has extensively been utilized to perform that step. In physiological conditions, evidence points to the role of a ferredoxin-ferredoxin reductase couple for the reduction of the persulfide during Fe–S synthesis. Ferredoxins are a group of proteins that contain Fe–S clusters and can act as capacitators to accept or discharge electrons [70]. In E. coli, ferredoxin (Fdx) is part of the isc operon, also expressing the genes involved in the de novo synthesis of Fe–S [18]. In the yeast S. cerevisiae, mitochondrial Yah1 and the ferredoxin reductase Arh1 are essential for Fe–S biogenesis [19,71,72]. In human cells, two mitochondrial ferredoxins, FDX1 and FDX2, with distinct roles exist. By performing knockdown experiments, Sheftel et al. showed that FDX2 is the ferredoxin involved in the biogenesis of Fe–S [73]. More recently, in vitro reconstitution of the de novo Fe–S assembly was efficiently carried out using the ferredoxin-ferredoxin reductase-NADPH system [35,45,74]. The work in more physiological conditions has brought additional information on how FDX2 may proceed during the assembly of Fe–S. Specifically, experiments with the mouse Fe–S core complex has shown that FDX2 selectively and efficiently reduces the persulfide that has been transferred to the iron-loaded ISCU2, allowing the reaction to generate a [2Fe–2S] on the scaffold protein [45].
How ferredoxin interacts with the Fe–S core complex is still unclear. In bacteria, reports indicate that Fdx directly binds the NFS1 homolog IscS [75,76]. Furthermore, the association of Fdx with IscS was seen to compete with the binding of the frataxin homolog CyaY [75,76] and eventually IscU [76]. In yeast, a strong interaction between Yah1 and Isu1 was initially reported [35]. However, a very recent study using the yeast (SDAU)2 complex showed that ferredoxin homolog Yah1 binds to Nfs1 in the same region involved in frataxin (Yfh1) interaction [77], supporting the idea that ferredoxin and frataxin compete to transiently associate with the core Fe–S complex. In human cells, a co-immunoprecipitation of NFS1, ISCU, and ISD11 with FDX2 was reported [78]. A model obtained by small-angle X-ray scattering (SAXS) with the C. thermophilum core complex suggest an alternative binding of ferredoxin [22]. In this model, ferredoxin binds to ISCU and NFS1 in a region distinct from the one recognized by FXN [22], thus implying that both FXN and FDX2 could be present on the complex during Fe–S assembly. To reconcile these data on a potential exclusive or simultaneous binding of FXN and FDX2, additional investigations on the location and nature of FDX2 binding are needed.
Interestingly, a recent in vitro study demonstrated that FDX2-FDXR could also have a role in providing electrons for the maturation of [4Fe–4S] from [2Fe–2S] during the late stage of the ISC pathway, which involves ISCA1, ISCA2, and IBA57 [79].
2.7. Model of Fe–S Assembly within the Core Complex
Combining the past years’ worth of science, the mechanism of Fe–S assembly within the mitochondrial core complex is more understood. Based on the data described above, a model of Fe–S synthesis can be outlined (Figure 6). The heart of the complex consists of a homodimer of NFS1, around which all other proteins interact. Association of the ISD11-ACP complex stabilizes NFS1 dimer, allowing its proper function. In the first step of the model, the iron-loaded scaffold protein ISCU2 binds to NFS1. Upon binding of FXN to the (SDAU)2 complex, the transfer of a persulfide generated by the NFS1 cysteine desulfurase activity to Cys138 of ISCU2 is promoted. FDX2 then associates with the complex and provides electron(s) for the formation of an intermediate Fe–S, potentially [1Fe–1S]. The last step requires ISCU2 dissociation from NFS1 and dimerization, involving the conserved Tyr35 residue, for the formation of a [2Fe–2S] cluster. The newly formed (ISCU2)2-Fe–S or ISCU2-Fe–S is then recognized by the HSPA9/HSCB chaperones for the transfer to downstream Fe–S client proteins.
3. A Complex That Matters: Human Genetic Diseases Affecting the Mitochondrial Fe–S Core Complex
Because Fe–S are essential cofactors, mutations leading to defects in the activity of the Fe–S core complex are associated with cellular dysfunctions and diseases. Friedreich’s ataxia is the most representative, common, and studied human genetic disorder associated with impairment of the early Fe–S assembly in mitochondria. However, with the development of genetic sequencing and diagnosis in the past 20 years, (ultra)rare indications related to mutations in other key components of this complex were unraveled. These diseases are characterized by partial loss of function and mitochondrial dysfunction with diverse presentations and severities. This section briefly summarizes the mutations and their resulting diseases.
A pathological variant in NFS1, c.215G>A, results in mutant protein R72Q and was first identified in 2014 and again in a separate pedigree in 2021 [80,81]. This point mutation reduced NFS1 transcript and protein levels in patient fibroblasts [81], and it is also likely to disrupt NFS1-ISD11 protein–protein interactions based on in silico modeling. Analyses of muscle and liver biopsies revealed deficiencies in mitochondrial respiratory complexes I, II, and III [80]. The resulting disorder, Infantile Mitochondrial Complex II/III Deficiency, is characterized by lactic acidosis, hypotonia, and multisystem organ failure in the first year of life. In both affected pedigrees, one offspring that is homozygous for the mutation suffered an initial crisis ~6 months after birth but survived and now appear asymptomatic.
A pathological variant of ISD11, a homozygous mutation of c.203G>T resulting in a mutated R68L protein, has been reported [82]. While this mutant does not affect the NFS1-ISD11 protein–protein interaction, it did result in more protein aggregation and near-complete loss of NFS1 desulfurase activity from purified proteins. The two affected individuals, who are consanguineous cousins, displayed deficiencies in respiratory complexes I, II, and III in liver and muscle samples. However, the patient with greater severity, including additional complex IV deficiency, died at 2 months of age. The other patient, who displayed severe neonatal lactic acidosis, was reported relatively asymptomatic at time of publication. Like the pedigrees harboring the NFS1 pathological mutation (see above), the first year of life appears to be a critical time for this disease.
A pathological variant of FDX2 has also been reported in a single individual from a consanguineous pedigree [83]. Homozygous mutation of c.12279A>T in the FDX2 gene disrupts the ATG start site for the initiation of translation and results in severe reduction in FDX2 protein levels in patient fibroblasts and muscle samples. Like the variants in NFS1 and ISD11, the patient had deficiencies in respiratory complexes I, II, and III as well as aconitase activity in muscle mitochondria. The patient experiences myoglobinuria and progressive muscular weakness after onset during adolescence.
ISCU myopathy is a disease that has been identified in populations from northern Sweden [84]. Symptoms develop in early childhood and include a severe exercise intolerance characterized by the disruption of muscle iron homeostasis [85]. The genetic cause of the disease is a point mutation g.7044G>C within intron 4 of the ISCU gene [86]. This mutation generates an alternative acceptor splice site between exons 4 and 5 [87]. As a result, the cryptic exon 4A is included in most patient mRNA transcripts isolated from muscle samples, which introduces an early stop codon ahead of exon 5 [87]. Total ISCU mRNA in these samples was significantly reduced compared to controls, suggesting this cryptic exon inclusion leads to mRNA decay. As a result, protein expression in patients’ skeletal muscle samples was also greatly reduced, along with reduced aconitase and SDH activity. Skin fibroblast samples from patients, however, contained normal levels of ISCU mRNA [87], suggesting a tissue-specific pathology.
A more severe form of this disease has also been reported, with increased muscle wasting [88]. The two affected patients were heterozygous for the intronic mutation above as well as a c.149G>A mutation, resulting in a mutant G50E protein. While homozygous patients have relatively normal muscle strength at rest, the more severely affected patients were much weaker and display cardiac hypertrophy, unlike other patients [88]. In homozygous patients, heart and smooth muscles are not affected due to lack of alternative splicing in these tissues [89].
With a prevalence of 1/50,000 in the Caucasian population, Friedreich’s ataxia (FRDA) is the most common disease associated with Fe–S production deficiency [90]. FRDA is caused by a GAA trinucleotide repeat expansion in the first intron of the FXN gene. The mutation leads to heterochromatin formation in the FXN locus [91,92] and results in significant, but not complete, loss of FXN protein expression in cells [93]. All patients carry at least one expanded allele; most are homozygous for an expanded allele, whereas a smaller percentage (~4%) carry one expanded allele and one allele with a mutation that reduces FXN function or expression [62,93,94,95,96]. In most cases, FRDA has an onset prior to 25 years of age. The symptoms are characterized by a mixed sensory and cerebellar ataxia that is progressing with time and resulting from the neurodegeneration of neurons in the cerebellum, spinal cord, and dorsal root ganglia (DRGs) [97,98]. Neurons of the motor cortex are also generally primarily affected [97]. Outside of the nervous system, primary manifestations include hypertrophic cardiomyopathy, which has been estimated as the leading cause of premature death (59), and a higher incidence of diabetes [99,100]. Although patient-derived cells may not spontaneously develop Fe–S deficit in culture, the primary involvement of Fe–S biogenesis in the pathophysiology has been shown in cardiac and neuronal mouse models of the disease [101,102,103,104].
4. Further Reading on Fe–S
This review only encompasses the first step of the full process for generating Fe–S clusters in the mitochondria. Other mechanisms mediate the generation of [4Fe–4S] clusters from two [2Fe–2S] clusters in the mitochondria, as well as the synthesis of Fe–S in the cytoplasm using the proteins of the CIA pathway. Recent reviews cover these additional steps of Fe–S trafficking, export, and maturation [15,105]. Other reviews cover the alternative pathways to generate Fe–S that exist in other species [13,106,107].
5. Conclusions
Recently obtained structural and biochemical data have painted a clearer picture of the mitochondrial Fe–S core complex, its protein interactions, and the steps involved in generating Fe–S. They open a venue for new and exciting research to address unanswered questions. For instance, what is the source of iron in the process? How, where, and when does FDX2 bind to the core complex during Fe–S assembly? What is the nature of the Fe–S intermediate on ISCU2 before its dimerization? How does dimerization of ISCU2 occur? Additionally, the intriguing relationship between ACP, the mitochondrial synthesis of long acyl chain and the Fe–S core complex function deserves particular attention. Importantly, the elucidation of these mechanisms and functions will require complementary experiments to ensure the physiological relevance of in vitro observations.
Author Contributions
Writing—original draft preparation, T.L.P. and A.M.; writing—review and editing, T.L.P. and A.M.; visualization, T.L.P. and A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to thank Christine Bulawa for her assistance with editing the manuscript.
Conflicts of Interest
T.L.P. and A.M. are employees of Pfizer, Inc.
References
- Peters, J.W.; Broderick, J.B. Emerging paradigms for complex iron–sulfur cofactor assembly and insertion. Annu. Rev. Biochem. 2012, 81, 429–450. [Google Scholar] [CrossRef] [PubMed]
- Schonauer, M.S.; Kastaniotis, A.J.; Kursu, V.A.; Hiltunen, J.K.; Dieckmann, C.L. Lipoic acid synthesis and attachment in yeast mitochondria. J. Biol. Chem. 2009, 284, 23234–23242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, E.A.; Snowden, C.K.; Cristea, I.M. Protein lipoylation: An evolutionarily conserved metabolic regulator of health and disease. Curr. Opin. Chem. Biol. 2018, 42, 76–85. [Google Scholar] [CrossRef] [PubMed]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. α-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, L.; Tórtora, V.; Mansilla, S.; Radi, R. Aconitases: Non-redox Iron–Sulfur Proteins Sensitive to Reactive Species. Acc. Chem. Res. 2019, 52, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Maio, N. Biogenesis and functions of mammalian Iron–sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial complex III Rieske Fe–S protein processing and assembly. Cell Cycle 2018, 17, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, J.; Makrantoni, V.; Ingledew, W.J.; Stark, M.J.; White, M.F. The DNA repair helicases XPD and FancJ have essential iron–sulfur domains. Mol. Cell 2006, 23, 801–808. [Google Scholar] [CrossRef]
- Netz, D.J.; Stith, C.M.; Stumpfig, M.; Kopf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2011, 8, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Čavužić, M.; Liu, Y. Biosynthesis of Sulfur-Containing tRNA Modifications: A Comparison of Bacterial, Archaeal, and Eukaryotic Pathways. Biomolecules 2017, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Lill, R.; Dutkiewicz, R.; Freibert, S.A.; Heidenreich, T.; Mascarenhas, J.; Netz, D.J.; Paul, V.D.; Pierik, A.J.; Richter, N.; Stumpfig, M.; et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell Biol. 2015, 94, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Ciofi-Baffoni, S.; Nasta, V.; Banci, L. Protein networks in the maturation of human iron-sulfur proteins. Metallomics 2018, 10, 49–72. [Google Scholar] [CrossRef] [PubMed]
- Braymer, J.J.; Freibert, S.A.; Rakwalska-Bange, M.; Lill, R. Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118863. [Google Scholar] [CrossRef]
- Andreini, C.; Rosato, A.; Banci, L. The Relationship between Environmental Dioxygen and Iron-Sulfur Proteins Explored at the Genome Level. PLoS ONE 2017, 12, e0171279. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmucker, S.; Argentini, M.; Carelle-Calmels, N.; Martelli, A.; Puccio, H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008, 17, 3521–3531. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Nakamura, M. Functional Assignment of the ORF2-iscS-iscU-iscA-hscB-hscA-fdx-0RF3 Gene Cluster Involved in the Assembly of Fe–S Clusters in Escherichia coli. J. Biochem. 1999, 126, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Muhlenhoff, U.; Gerber, J.; Richhardt, N.; Lill, R. Components involved in assembly and dislocation of iron-sulfur clusters on the scaffold protein Isu1p. EMBO J. 2003, 22, 4815–4825. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; White, R.H.; Cash, V.L.; Jack, R.F.; Dean, D.R. Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc. Natl. Acad. Sci. USA 1993, 90, 2754–2758. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boniecki, M.T.; Freibert, S.A.; Muhlenhoff, U.; Lill, R.; Cygler, M. Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun. 2017, 8, 1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freibert, S.A.; Boniecki, M.T.; Stumpfig, C.; Schulz, V.; Krapoth, N.; Winge, D.R.; Muhlenhoff, U.; Stehling, O.; Cygler, M.; Lill, R. N-terminal tyrosine of ISCU2 triggers [2Fe–2S] cluster synthesis by ISCU2 dimerization. Nat. Commun. 2021, 12, 6902. [Google Scholar] [CrossRef] [PubMed]
- Biederbick, A.; Stehling, O.; Rosser, R.; Niggemeyer, B.; Nakai, Y.; Elsasser, H.P.; Lill, R. Role of human mitochondrial Nfs1 in cytosolic iron-sulfur protein biogenesis and iron regulation. Mol. Cell Biol. 2006, 26, 5675–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cory, S.A.; Van Vranken, J.G.; Brignole, E.J.; Patra, S.; Winge, D.R.; Drennan, C.L.; Rutter, J.; Barondeau, D.P. Structure of human Fe–S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP–ISD11 interactions. Proc. Natl. Acad. Sci. USA 2017, 114, E5325–E5334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.X.; Thompson, K.; Taylor, R.W.; Olahova, M. Mitochondrial OXPHOS Biogenesis: Co-Regulation of Protein Synthesis, Import, and Assembly Pathways. Int. J. Mol. Sci. 2020, 21, 3820. [Google Scholar] [CrossRef] [PubMed]
- Dibley, M.G.; Formosa, L.E.; Lyu, B.; Reljic, B.; McGann, D.; Muellner-Wong, L.; Kraus, F.; Sharpe, A.J.; Stroud, D.A.; Ryan, M.T. The Mitochondrial Acyl-carrier Protein Interaction Network Highlights Important Roles for LYRM Family Members in Complex I and Mitoribosome Assembly. Mol. Cell Proteom. 2020, 19, 65–77. [Google Scholar] [CrossRef]
- Adam, A.C.; Bornhovd, C.; Prokisch, H.; Neupert, W.; Hell, K. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 2006, 25, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, N.; Urzica, E.; Guiard, B.; Müller, H.; Lohaus, C.; Meyer, H.E.; Ryan, M.T.; Meisinger, C.; Mühlenhoff, U.; Lill, R.; et al. Essential role of Isd11 in mitochondrial iron–sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 2006, 25, 184–195. [Google Scholar] [CrossRef]
- Shi, Y.; Ghosh, M.C.; Tong, W.H.; Rouault, T.A. Human ISD11 is essential for both iron-sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum. Mol. Genet. 2009, 18, 3014–3025. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Jeong, M.Y.; Wei, P.; Chen, Y.C.; Gygi, S.P.; Winge, D.R.; Rutter, J. The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Majmudar, J.D.; Feng, X.; Fox, N.G.; Nabhan, J.F.; Towle, T.; Ma, T.; Gooch, R.; Bulawa, C.; Yue, W.W.; Martelli, A. 4’-Phosphopantetheine and long acyl chain-dependent interactions are integral to human mitochondrial acyl carrier protein function. Medchemcomm 2019, 10, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Masud, A.J.; Kastaniotis, A.J.; Rahman, M.T.; Autio, K.J.; Hiltunen, J.K. Mitochondrial acyl carrier protein (ACP) at the interface of metabolic state sensing and mitochondrial function. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118540. [Google Scholar] [CrossRef]
- Herrera, M.G.; Noguera, M.E.; Sewell, K.E.; Agudelo Suarez, W.A.; Capece, L.; Klinke, S.; Santos, J. Structure of the Human ACP-ISD11 Heterodimer. Biochemistry 2019, 58, 4596–4609. [Google Scholar] [CrossRef] [PubMed]
- Webert, H.; Freibert, S.A.; Gallo, A.; Heidenreich, T.; Linne, U.; Amlacher, S.; Hurt, E.; Muhlenhoff, U.; Banci, L.; Lill, R. Functional reconstitution of mitochondrial Fe/S cluster synthesis on Isu1 reveals the involvement of ferredoxin. Nat. Commun. 2014, 5, 5013. [Google Scholar] [CrossRef] [Green Version]
- Agar, J.N.; Zheng, L.; Cash, V.L.; Dean, D.R.; Johnson, M.K. Role of the IscU Protein in Iron−Sulfur Cluster Biosynthesis: IscS-mediated Assembly of a [Fe2S2] Cluster in IscU. J. Am. Chem. Soc. 2000, 122, 2136–2137. [Google Scholar] [CrossRef]
- Bridwell-Rabb, J.; Fox, N.G.; Tsai, C.L.; Winn, A.M.; Barondeau, D.P. Human frataxin activates Fe–S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 2014, 53, 4904–4913. [Google Scholar] [CrossRef] [Green Version]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autreaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Fuzery, A.K.; Tonelli, M.; Ta, D.T.; Westler, W.M.; Vickery, L.E.; Markley, J.L. Structure and dynamics of the iron-sulfur cluster assembly scaffold protein IscU and its interaction with the cochaperone HscB. Biochemistry 2009, 48, 6062–6071. [Google Scholar] [CrossRef] [Green Version]
- Cai, K.; Frederick, R.O.; Kim, J.H.; Reinen, N.M.; Tonelli, M.; Markley, J.L. Human mitochondrial chaperone (mtHSP70) and cysteine desulfurase (NFS1) bind preferentially to the disordered conformation, whereas co-chaperone (HSC20) binds to the structured conformation of the iron-sulfur cluster scaffold protein (ISCU). J. Biol. Chem. 2013, 288, 28755–28770. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Kelly, G.; Pastore, A. The Scaffold Protein IscU Retains a Structured Conformation in the FeS Cluster Assembly Complex. ChemBioChem 2014, 15, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Tonelli, M.; Kim, T.; Markley, J.L. Three-dimensional structure and determinants of stability of the iron-sulfur cluster scaffold protein IscU from Escherichia coli. Biochemistry 2012, 51, 5557–5563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, N.G.; Martelli, A.; Nabhan, J.F.; Janz, J.; Borkowska, O.; Bulawa, C.; Yue, W.W. Zinc(II) binding on human wild-type ISCU and Met140 variants modulates NFS1 desulfurase activity. Biochimie 2018, 152, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, C.; Adrover, M.; Puglisi, R.; Yan, R.; Temussi, P.A.; Pastore, A. The role of zinc in the stability of the marginally stable IscU scaffold protein. Protein Sci. 2014, 23, 1208–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Muller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.I.; Dizin, E.; Yoon, T.; Cowan, J.A. Kinetic and structural characterization of human mortalin. Protein Expr. Purif. 2010, 72, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.G.; Silberg, J.J.; Vickery, L.E. Interaction of the iron-sulfur cluster assembly protein IscU with the Hsc66/Hsc20 molecular chaperone system of Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 7790–7795. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.G.; Cupp-Vickery, J.R.; Vickery, L.E. Contributions of the LPPVK motif of the iron-sulfur template protein IscU to interactions with the Hsc66-Hsc20 chaperone system. J. Biol. Chem. 2003, 278, 37582–37589. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Yuda, E.; Fujishiro, T.; Hirabayashi, K.; Wada, K.; Takahashi, Y. Identification of IscU residues critical for de novo iron–sulfur cluster assembly. Mol. Microbiol. 2019, 112, 1769–1783. [Google Scholar] [CrossRef]
- Cook, J.D.; Bencze, K.Z.; Jankovic, A.D.; Crater, A.K.; Busch, C.N.; Bradley, P.B.; Stemmler, A.J.; Spaller, M.R.; Stemmler, T.L. Monomeric Yeast Frataxin Is an Iron-Binding Protein†. Biochemistry 2006, 45, 7767–7777. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Gakh, O.; Mooney, S.M.; Isaya, G. The Ferroxidase Activity of Yeast Frataxin. J. Biol. Chem. 2002, 277, 38589–38595. [Google Scholar] [CrossRef] [Green Version]
- Yoon, T.; Cowan, J.A. Iron−Sulfur Cluster Biosynthesis. Characterization of Frataxin as an Iron Donor for Assembly of [2Fe–2S] Clusters in ISU-Type Proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Adinolfi, S.; Pastore, A.; Laue, T.M.; Dennis Chasteen, N. Iron binding and oxidation kinetics in frataxin CyaY of Escherichia coli. J. Mol. Biol. 2004, 341, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Dizin, E.; Cowan, J.A. N-terminal iron-mediated self-cleavage of human frataxin: Regulation of iron binding and complex formation with target proteins. J. Biol. Inorg. Chem. 2007, 12, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dizin, E.; Cowan, J.A. Mapping iron binding sites on human frataxin: Implications for cluster assembly on the ISU Fe–S cluster scaffold protein. J. Biol. Inorg. Chem. 2008, 13, 825–836. [Google Scholar] [CrossRef]
- Gerber, J.; Muhlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Cavadini, P.; O’Neill, H.A.; Benada, O.; Isaya, G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Gakh, O.; O’Neill, H.A.; Mangravita, A.; Nichol, H.; Ferreira, G.C.; Isaya, G. Yeast frataxin sequentially chaperones and stores iron by coupling protein assembly with iron oxidation. J. Biol. Chem. 2003, 278, 31340–31351. [Google Scholar] [CrossRef] [Green Version]
- Aloria, K.; Schilke, B.; Andrew, A.; Craig, E.A. Iron-induced oligomerization of yeast frataxin homologue Yfh1 is dispensable in vivo. EMBO Rep. 2004, 5, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Seguin, A.; Sutak, R.; Bulteau, A.L.; Garcia-Serres, R.; Oddou, J.L.; Lefevre, S.; Santos, R.; Dancis, A.; Camadro, J.M.; Latour, J.M.; et al. Evidence that yeast frataxin is not an iron storage protein in vivo. Biochim. Biophys. Acta 2010, 1802, 531–538. [Google Scholar] [CrossRef]
- Tsai, C.L.; Barondeau, D.P. Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhofer-Donze, M.; Reutenauer, L.; Puccio, H. Mammalian frataxin: An essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS ONE 2011, 6, e16199. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.; Barondeau, D.P. Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc. Natl. Acad. Sci. USA 2019, 116, 19421–19430. [Google Scholar] [CrossRef] [Green Version]
- Colin, F.; Martelli, A.; Clemancey, M.; Latour, J.M.; Gambarelli, S.; Zeppieri, L.; Birck, C.; Page, A.; Puccio, H.; Ollagnier de Choudens, S. Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J. Am. Chem. Soc. 2013, 135, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Huichalaf, C.; Perfitt, T.L.; Kuperman, A.; Gooch, R.; Kovi, R.C.; Brenneman, K.A.; Chen, X.; Hirenallur-Shanthappa, D.; Ma, T.; Assaf, B.T.; et al. In vivo overexpression of frataxin causes toxicity mediated by iron-sulfur cluster deficiency. Mol. Ther.-Methods Clin. Dev. 2022. [Google Scholar] [CrossRef]
- Yoon, H.; Knight, S.A.; Pandey, A.; Pain, J.; Zhang, Y.; Pain, D.; Dancis, A. Frataxin-bypassing Isu1: Characterization of the bypass activity in cells and mitochondria. Biochem. J. 2014, 459, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.; Knight, S.A.; Pandey, A.; Pain, J.; Turkarslan, S.; Pain, D.; Dancis, A. Turning Saccharomyces cerevisiae into a Frataxin-Independent Organism. PLoS Genet. 2015, 11, e1005135. [Google Scholar] [CrossRef]
- Yoon, H.; Golla, R.; Lesuisse, E.; Pain, J.; Donald, J.E.; Lyver, E.R.; Pain, D.; Dancis, A. Mutation in the Fe–S scaffold protein Isu bypasses frataxin deletion. Biochem. J. 2012, 441, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Das, D.; Patra, S.; Bridwell-Rabb, J.; Barondeau, D.P. Mechanism of frataxin “bypass” in human iron-sulfur cluster biosynthesis with implications for Friedreich’s ataxia. J. Biol. Chem. 2019, 294, 9276–9284. [Google Scholar] [CrossRef]
- Bruschi, M.; Guerlesquin, F. Structure, function and evolution of bacterial ferredoxins. FEMS Microbiol. Rev. 1988, 4, 155–175. [Google Scholar] [CrossRef]
- Li, J.; Saxena, S.; Pain, D.; Dancis, A. Adrenodoxin reductase homolog (Arh1p) of yeast mitochondria required for iron homeostasis. J. Biol. Chem. 2001, 276, 1503–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, H.; Kaut, A.; Kispal, G.; Lill, R. A mitochondrial ferredoxin is essential for biogenesis of cellular iron-sulfur proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1050–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Elsasser, H.P.; Muhlenhoff, U.; Webert, H.; Hobler, A.; Hannemann, F.; Bernhardt, R.; Lill, R. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 11775–11780. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Adinolfi, S.; Pastore, A. Ferredoxin, in conjunction with NADPH and ferredoxin-NADP reductase, transfers electrons to the IscS/IscU complex to promote iron–sulfur cluster assembly. Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 1113–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Konarev, P.V.; Iannuzzi, C.; Adinolfi, S.; Roche, B.; Kelly, G.; Simon, L.; Martin, S.R.; Py, B.; Barras, F.; et al. Ferredoxin competes with bacterial frataxin in binding to the desulfurase IscS. J. Biol. Chem. 2013, 288, 24777–24787. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Frederick, R.O.; Reinen, N.M.; Troupis, A.T.; Markley, J.L. [2Fe–2S]-Ferredoxin Binds Directly to Cysteine Desulfurase and Supplies an Electron for Iron–Sulfur Cluster Assembly but Is Displaced by the Scaffold Protein or Bacterial Frataxin. J. Am. Chem. Soc. 2013, 135, 8117–8120. [Google Scholar] [CrossRef] [PubMed]
- Uzarska, M.A.; Grochowina, I.; Soldek, J.; Jelen, M.; Schilke, B.; Marszalek, J.; Craig, E.A.; Dutkiewicz, R. During FeS-cluster biogenesis ferredoxin and frataxin use overlapping bindings sites on yeast cysteine desulfurase Nfs1. J. Biol. Chem. 2022, 298, 101570. [Google Scholar] [CrossRef]
- Beilschmidt, L.K.; Ollagnier de Choudens, S.; Fournier, M.; Sanakis, I.; Hograindleur, M.A.; Clemancey, M.; Blondin, G.; Schmucker, S.; Eisenmann, A.; Weiss, A.; et al. ISCA1 is essential for mitochondrial Fe4S4 biogenesis in vivo. Nat. Commun. 2017, 8, 15124. [Google Scholar] [CrossRef]
- Weiler, B.D.; Brück, M.-C.; Kothe, I.; Bill, E.; Lill, R.; Mühlenhoff, U. Mitochondrial [4Fe–4S] protein assembly involves reductive [2Fe–2S] cluster fusion on ISCA1–ISCA2 by electron flow from ferredoxin FDX2. Proc. Natl. Acad. Sci. USA 2020, 117, 20555–20565. [Google Scholar] [CrossRef]
- Hershkovitz, T.; Kurolap, A.; Tal, G.; Paperna, T.; Mory, A.; Staples, J.; Brigatti, K.W.; Regeneron Genetics, C.; Gonzaga-Jauregui, C.; Dumin, E.; et al. A recurring NFS1 pathogenic variant causes a mitochondrial disorder with variable intra-familial patient outcomes. Mol. Genet. Metab. Rep. 2021, 26, 100699. [Google Scholar] [CrossRef]
- Farhan, S.M.K.; Wang, J.; Robinson, J.F.; Lahiry, P.; Siu, V.M.; Prasad, C.; Kronick, J.B.; Ramsay, D.A.; Rupar, C.A.; Hegele, R.A. Exome sequencing identifies NFS 1 deficiency in a novel Fe–S cluster disease, infantile mitochondrial complex II/III deficiency. Mol. Genet. Genom. Med. 2014, 2, 73–80. [Google Scholar] [CrossRef]
- Lim, S.C.; Friemel, M.; Marum, J.E.; Tucker, E.J.; Bruno, D.L.; Riley, L.G.; Christodoulou, J.; Kirk, E.P.; Boneh, A.; DeGennaro, C.M.; et al. Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum. Mol. Genet. 2013, 22, 4460–4473. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Saada, A.; Halvardson, J.; Soiferman, D.; Shaag, A.; Edvardson, S.; Horovitz, Y.; Khayat, M.; Shalev, S.A.; Feuk, L.; et al. Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur. J. Hum. Genet. 2014, 22, 902–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugge, U.; Holmberg, M.; Holmgren, G.; Almay, B.G.; Linderholm, H. Hereditary myopathy with lactic acidosis, succinate dehydrogenase and aconitase deficiency in northern Sweden: A genealogical study. J. Med. Genet. 1995, 32, 344–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, R.G.; Henriksson, K.G.; Jorfeldt, L.; Hultman, E.; Wibom, R.; Sahlin, K.; Areskog, N.H.; Gunder, M.; Ayyad, K.; Blomqvist, C.G.; et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Investig. 1991, 88, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, A.; Lind, L.; Thornell, L.E.; Holmberg, M. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum. Mol. Genet. 2008, 17, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Knight, M.A.; Tong, W.H.; Hernandez, D.; Ayyad, K.; Taivassalo, T.; Andersen, P.M.; Singleton, A.; Rouault, T.A.; Fischbeck, K.H.; et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 2008, 82, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollberg, G.; Tulinius, M.; Melberg, A.; Darin, N.; Andersen, O.; Holmgren, D.; Oldfors, A.; Holme, E. Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain 2009, 132, 2170–2179. [Google Scholar] [CrossRef] [Green Version]
- Nordin, A.; Larsson, E.; Thornell, L.E.; Holmberg, M. Tissue-specific splicing of ISCU results in a skeletal muscle phenotype in myopathy with lactic acidosis, while complete loss of ISCU results in early embryonic death in mice. Hum. Genet. 2011, 129, 371–378. [Google Scholar] [CrossRef]
- Beilschmidt, L.K.; Puccio, H.M. Mammalian Fe–S cluster biogenesis and its implication in disease. Biochimie 2014, 100, 48–60. [Google Scholar] [CrossRef]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Saveliev, A.; Everett, C.; Sharpe, T.; Webster, Z.; Festenstein, R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 2003, 422, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Tsai, C.L.; Bridwell-Rabb, J.; Barondeau, D.P. Friedreich’s ataxia variants I154F and W155R diminish frataxin-based activation of the iron-sulfur cluster assembly complex. Biochemistry 2011, 50, 6478–6487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridwell-Rabb, J.; Winn, A.M.; Barondeau, D.P. Structure-function analysis of Friedreich’s ataxia mutants reveals determinants of frataxin binding and activation of the Fe–S assembly complex. Biochemistry 2011, 50, 7265–7274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossee, M.; Durr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschutter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206. [Google Scholar] [CrossRef]
- Harding, I.H.; Lynch, D.R.; Koeppen, A.H.; Pandolfo, M. Central Nervous System Therapeutic Targets in Friedreich Ataxia. Hum. Gene Ther. 2020, 31, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256 (Suppl. 1), 3–8. [Google Scholar] [CrossRef]
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 94–102. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Stork, S.; Liu, D.; Hu, K.; Herrmann, S.; Ertl, G.; Niemann, M. Cardiomyopathy of Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 88–93. [Google Scholar] [CrossRef] [PubMed]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Wattenhofer-Donze, M.; Schmucker, S.; Bouvet, S.; Reutenauer, L.; Puccio, H. Frataxin is essential for extramitochondrial Fe–S cluster proteins in mammalian tissues. Hum. Mol. Genet. 2007, 16, 2651–2658. [Google Scholar] [CrossRef] [PubMed]
- Seznec, H.; Simon, D.; Bouton, C.; Reutenauer, L.; Hertzog, A.; Golik, P.; Procaccio, V.; Patel, M.; Drapier, J.C.; Koenig, M.; et al. Friedreich ataxia: The oxidative stress paradox. Hum. Mol. Genet. 2005, 14, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piguet, F.; de Montigny, C.; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. 2018, 26, 1940–1952. [Google Scholar] [CrossRef] [Green Version]
- Hinton, T.V.; Batelu, S.; Gleason, N.; Stemmler, T.L. Molecular characteristics of proteins within the mitochondrial Fe–S cluster assembly complex. Micron 2021, 153, 103181. [Google Scholar] [CrossRef] [PubMed]
- Baussier, C.; Fakroun, S.; Aubert, C.; Dubrac, S.; Mandin, P.; Py, B.; Barras, F. Making iron-sulfur cluster: Structure, regulation and evolution of the bacterial ISC system. Adv. Microb. Physiol. 2020, 76, 1–39. [Google Scholar] [CrossRef]
- Srour, B.; Gervason, S.; Monfort, B.; D’Autreaux, B. Mechanism of Iron-Sulfur Cluster Assembly: In the Intimacy of Iron and Sulfur Encounter. Inorganics 2020, 8, 55. [Google Scholar] [CrossRef]
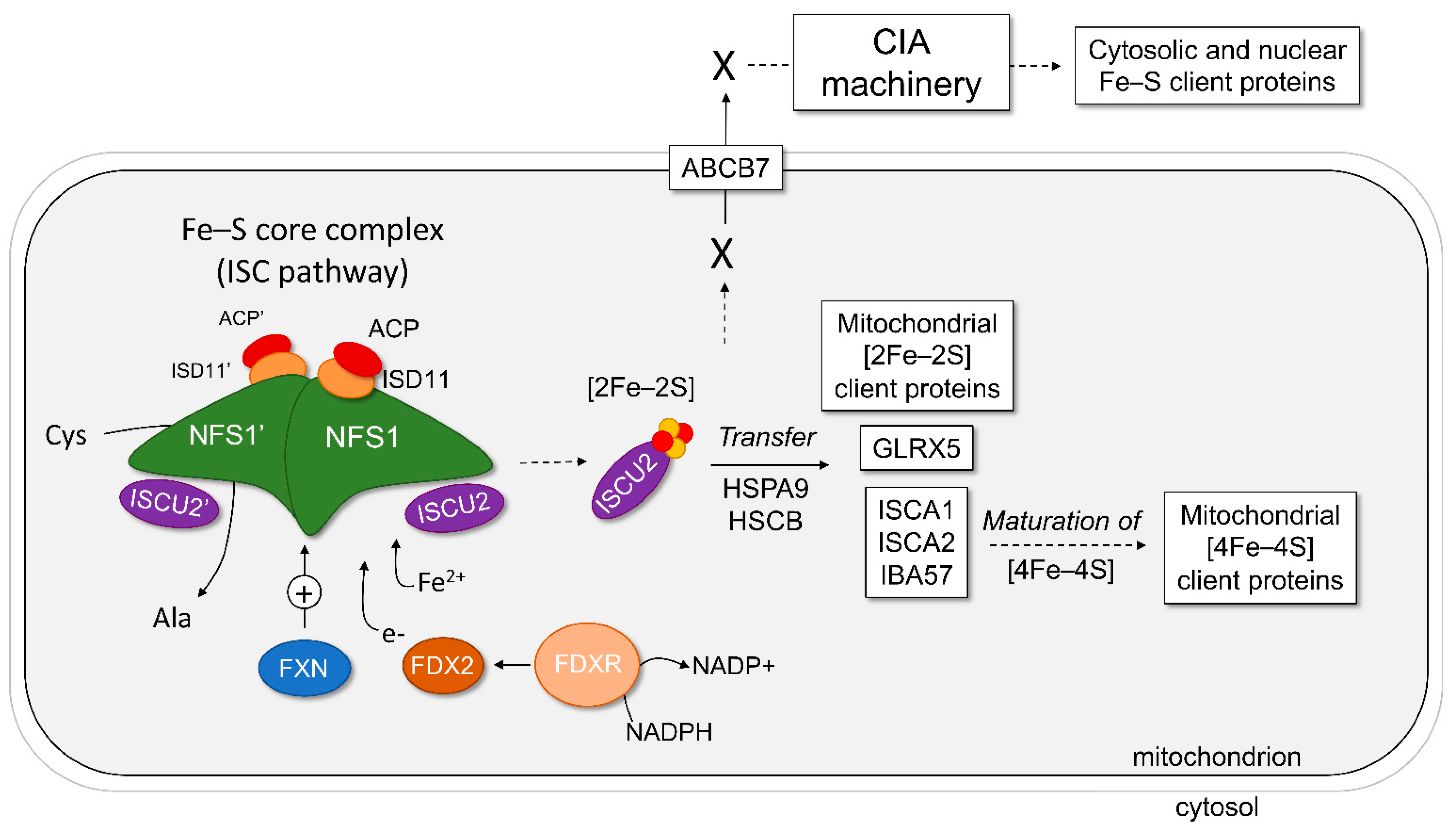
Figure 1.
Representation of the human mitochondrial and cytosolic iron–sulfur cluster biosynthesis. The proteins of the Fe–S core complex are shown as drawn molecules, whereas the proteins of downstream pathways are listed in boxes. The first step of Fe–S biosynthesis involves the Fe–S core complex of the ISC pathway, which is built around the NFS1 dimer with two subunits of ISD11, ACP, and ISCU2. Frataxin (FXN) can bind the complex and stimulate (+) the formation of Fe–S on ISCU2, while electrons are donated via the ferredoxin 2 (FDX2)–ferredoxin reductase (FDXR)–NADPH pathway. This generates a [2Fe–2S] cluster bound to ISCU2. The second step is the transfer of Fe–S from ISCU2, with the help of chaperone proteins HSPA9 and HSCB, to mitochondrial client proteins as listed. The third step is the maturation of [2Fe–2S] to [4Fe–4S] clusters in the mitochondria and subsequent delivery to mitochondrial client proteins. The fourth step is the export of an unknown precursor molecule (X) generated by the ISC pathway to the cytoplasm via ABCB7 and other components of the ISC export. Finally, this precursor molecule is processed by the CIA pathway to mature and deliver Fe–S to client proteins in nearly every other cellular compartment.
Figure 1.
Representation of the human mitochondrial and cytosolic iron–sulfur cluster biosynthesis. The proteins of the Fe–S core complex are shown as drawn molecules, whereas the proteins of downstream pathways are listed in boxes. The first step of Fe–S biosynthesis involves the Fe–S core complex of the ISC pathway, which is built around the NFS1 dimer with two subunits of ISD11, ACP, and ISCU2. Frataxin (FXN) can bind the complex and stimulate (+) the formation of Fe–S on ISCU2, while electrons are donated via the ferredoxin 2 (FDX2)–ferredoxin reductase (FDXR)–NADPH pathway. This generates a [2Fe–2S] cluster bound to ISCU2. The second step is the transfer of Fe–S from ISCU2, with the help of chaperone proteins HSPA9 and HSCB, to mitochondrial client proteins as listed. The third step is the maturation of [2Fe–2S] to [4Fe–4S] clusters in the mitochondria and subsequent delivery to mitochondrial client proteins. The fourth step is the export of an unknown precursor molecule (X) generated by the ISC pathway to the cytoplasm via ABCB7 and other components of the ISC export. Finally, this precursor molecule is processed by the CIA pathway to mature and deliver Fe–S to client proteins in nearly every other cellular compartment.

Figure 2.
The (SDAU)2 and (SDAUF)2 structures in the presence of zinc. (A) Crystal structure of (SDAU)2 complex in the presence of Zn2+ (5WLW, [22]). In this structure, Zn2+ is ligated by residues Asp71, Cys95, and His137 of ISCU2 and Cys381 of the NFS1 flexible loop. Cys381 is the catalytic residue implicated in the transfer of persulfide from NFS1 to ISCU2. (B) Cryo-EM structure of the (SDAUF)2 complex in the presence of Zn2+ (6NZU, [21]). In this structure, FXN residue Trp155 interacts with ISCU2 His137, and instead ISCU2 Cys138 binds to Zn2+. NFS1 Cys381 does not bind to Zn2+ in this structure and is therefore free to perform its catalytic activity.
Figure 2.
The (SDAU)2 and (SDAUF)2 structures in the presence of zinc. (A) Crystal structure of (SDAU)2 complex in the presence of Zn2+ (5WLW, [22]). In this structure, Zn2+ is ligated by residues Asp71, Cys95, and His137 of ISCU2 and Cys381 of the NFS1 flexible loop. Cys381 is the catalytic residue implicated in the transfer of persulfide from NFS1 to ISCU2. (B) Cryo-EM structure of the (SDAUF)2 complex in the presence of Zn2+ (6NZU, [21]). In this structure, FXN residue Trp155 interacts with ISCU2 His137, and instead ISCU2 Cys138 binds to Zn2+. NFS1 Cys381 does not bind to Zn2+ in this structure and is therefore free to perform its catalytic activity.

Figure 3.
Enzymatic mechanism of PLP–dependent NFS1 cysteine desulfuration. PLP cofactor is attached to NFS1 via Schiff base at NFS1 Lys258. PLP reacts with free L-cysteine to generate an intermediate aldimine adduct and a ketimine adduct. The catalytic Cys381 residue becomes persulfidated using PLP–cysteine as a substrate. Hydrolysis releases L–alanine from PLP. NFS1: NFS1 protein highlighting Cys381 residue; P: Phosphate group.
Figure 3.
Enzymatic mechanism of PLP–dependent NFS1 cysteine desulfuration. PLP cofactor is attached to NFS1 via Schiff base at NFS1 Lys258. PLP reacts with free L-cysteine to generate an intermediate aldimine adduct and a ketimine adduct. The catalytic Cys381 residue becomes persulfidated using PLP–cysteine as a substrate. Hydrolysis releases L–alanine from PLP. NFS1: NFS1 protein highlighting Cys381 residue; P: Phosphate group.

Figure 4.
The ACP–ISD11 heterodimer. Comparison of E. coli Acp–ISD11 from SDAUF Cryo-EM structure (6NZU, [21], left) and human ACP–ISD11 heterodimer crystal structure (6ODD, [34], right). Both structures contain a long acyl chain (green) attached to ACP (red) that interacts with the ISD11 (yellow) hydrophobic core.
Figure 4.
The ACP–ISD11 heterodimer. Comparison of E. coli Acp–ISD11 from SDAUF Cryo-EM structure (6NZU, [21], left) and human ACP–ISD11 heterodimer crystal structure (6ODD, [34], right). Both structures contain a long acyl chain (green) attached to ACP (red) that interacts with the ISD11 (yellow) hydrophobic core.

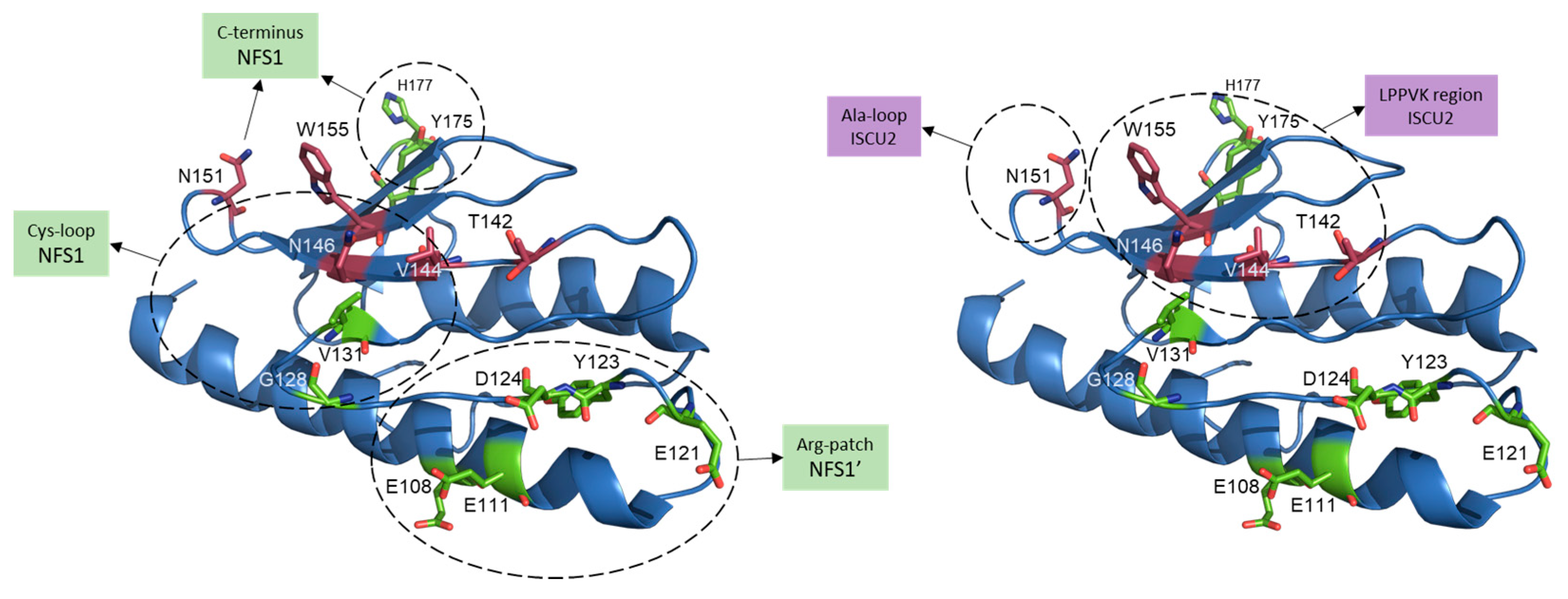
Figure 5.
Residues involved in FXN binding to the mitochondrial Fe–S core complex. Human FXN structure from SDUAF Cryo–EM structure (6NZU, [21]) with some of the key residues involved in the interaction with ISCU2 (in magenta) and with both NFS1 subunits (NFS1 and NFS1′, in green). Dashed circles indicate residues interacting with different regions of NFS1 or NFS1’ (left), or with different regions of ISCU2 (right). N151, N146 and V144 residues are likely involved in the interaction with both ISCU and NFS1.
Figure 5.
Residues involved in FXN binding to the mitochondrial Fe–S core complex. Human FXN structure from SDUAF Cryo–EM structure (6NZU, [21]) with some of the key residues involved in the interaction with ISCU2 (in magenta) and with both NFS1 subunits (NFS1 and NFS1′, in green). Dashed circles indicate residues interacting with different regions of NFS1 or NFS1’ (left), or with different regions of ISCU2 (right). N151, N146 and V144 residues are likely involved in the interaction with both ISCU and NFS1.

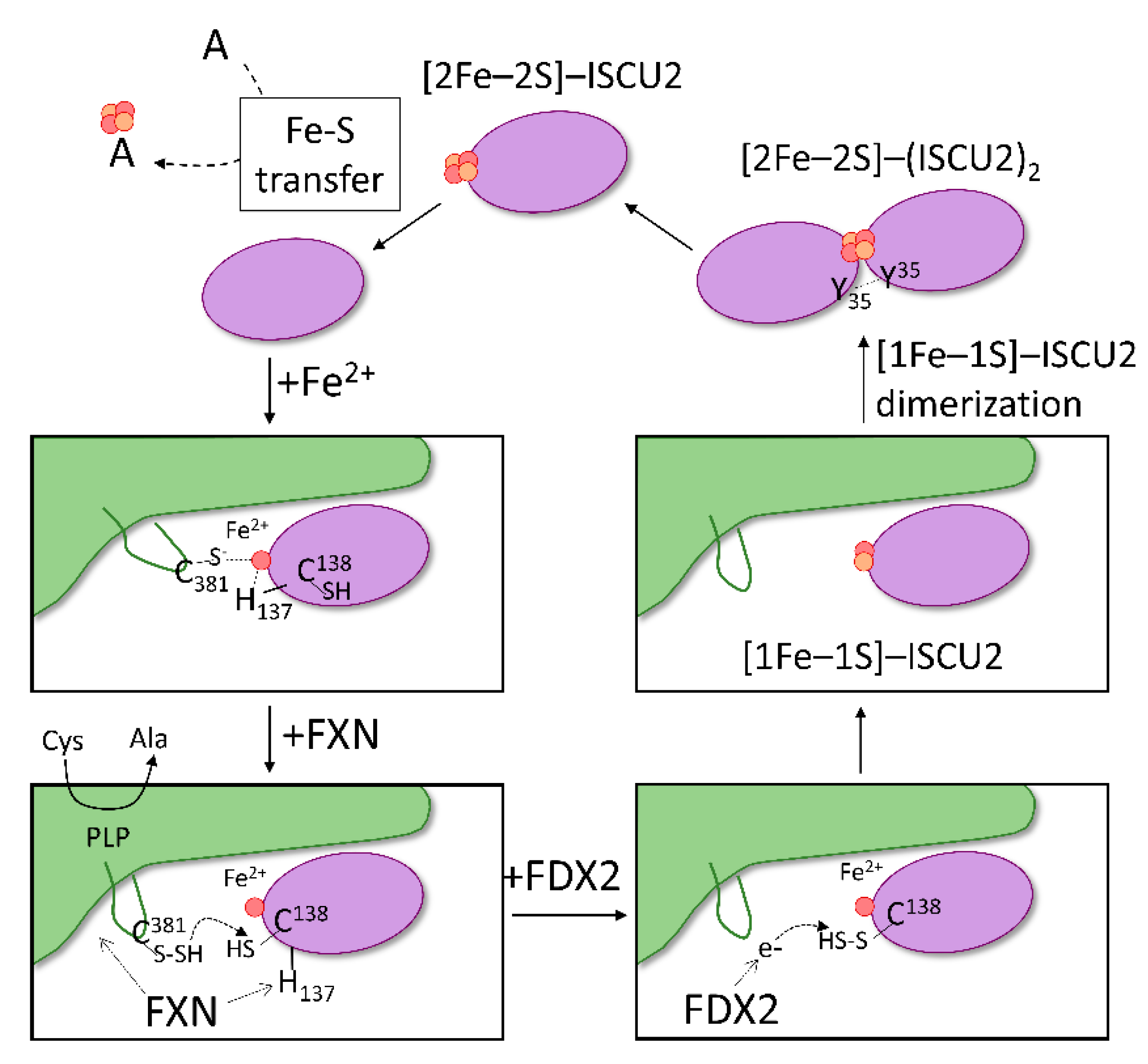
Figure 6.
Proposed model of Fe–S assembly in the Fe–S core complex. The proposed mechanism begins with binding of iron to ISCU2 (purple) and the association of Fe2+–ISCU2 to NFS1 (green). In the formed SDAU complex, coordination of Fe2+ site potentially includes the catalytic Cys381 of NFS1 and His137 of ISCU2. Interaction of FXN to both NFS1 and ISCU2 modifies the Fe2+ environment by shifting ISCU2 His137 away from Fe2+, allowing Cys138 to ligate instead. NFS1 Cys381 is free to perform the catalytic generation of persulfide from free cysteine and to transfer it to ISCU2 Cys138. Reduced FDX2 then binds and donates electron to reduce the ISCU2 persulfide. Once sulfur is reduced to sulfide, a [1Fe–1S] precursor is formed on ISCU2. Dimerization of ISCU2, mediated by Tyr35 residues, brings two precursor molecules together to form a [2Fe–2S] cluster on ISCU2. The generated [2Fe–2S] is then available for downstream transfer to acceptors (A).
Figure 6.
Proposed model of Fe–S assembly in the Fe–S core complex. The proposed mechanism begins with binding of iron to ISCU2 (purple) and the association of Fe2+–ISCU2 to NFS1 (green). In the formed SDAU complex, coordination of Fe2+ site potentially includes the catalytic Cys381 of NFS1 and His137 of ISCU2. Interaction of FXN to both NFS1 and ISCU2 modifies the Fe2+ environment by shifting ISCU2 His137 away from Fe2+, allowing Cys138 to ligate instead. NFS1 Cys381 is free to perform the catalytic generation of persulfide from free cysteine and to transfer it to ISCU2 Cys138. Reduced FDX2 then binds and donates electron to reduce the ISCU2 persulfide. Once sulfur is reduced to sulfide, a [1Fe–1S] precursor is formed on ISCU2. Dimerization of ISCU2, mediated by Tyr35 residues, brings two precursor molecules together to form a [2Fe–2S] cluster on ISCU2. The generated [2Fe–2S] is then available for downstream transfer to acceptors (A).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Fe–S client proteins found in the human mitochondria.
| Mitochondrial Compartment | ||||
|---|---|---|---|---|
| Fe–S Protein | Full Name | Pathway | Cluster | Function |
| SDHB | Succinate dehydrogenase iron–sulfur subunit B | Cellular Respiration, TCA Cycle | [4Fe–4S] [3Fe–4S] [2Fe–2S] | Subunit of SDH that is involved in Complex II function |
| Rieske protein UQCRFS1/RISP | Cytochrome b-c1 complex subunit Rieske | Cellular Respiration | [2Fe–2S] | Component of Complex III |
| NDUFS1 | NADH-ubiquinone oxidoreductase, 75 kDa subunit | Cellular Respiration | [4Fe–4S] [2Fe–2S] | Subunit of Complex I |
| NDUFS8 | NADH dehydrogenase iron–sulfur protein 8 | Cellular Respiration | [4Fe–4S] | Subunit of Complex I |
| NDUFS7 | NADH dehydrogenase iron–sulfur protein 7 | Cellular Respiration | [4Fe–4S] | Subunit of Complex I |
| NDUFV2 | NADH dehydrogenase flavoprotein 2 | Cellular Respiration | [2Fe–2S] | Subunit of Complex I |
| NDUFV1 | NADH dehydrogenase flavoprotein 1 | Cellular Respiration | [4Fe–4S] | Subunit of Complex I |
| ACO2 | Aconitate hydratase, mitochondrial | TCA Cycle | [4Fe–4S] | Enzyme that generates isocitrate in step 2 of TCA cycle |
| ETFDH | Electron transfer flavoprotein-ubiquinone oxidoreductase, mitochondrial | Fatty acid oxidation | [2Fe–2S] | Accepts electrons from ETF, reduces ubiquinone |
| LIAS | Lipoyl synthase | Lipoic acid synthesis | [4Fe–4S] | Converts octanoylated domains into lipoylated derivatives |
| MOCS1 | Molybdenum cofactor biosynthesis protein 1 | MoCo biosynthesis | [4Fe–4S] | Catalyzes the conversion of 5′-GTP to cyclic pyranopterin monophosphate |
| GLRX2 | Glutaredoxin-2 | Redox homeostasis | [2Fe–2S] | Maintains mitochondrial redox homeostasis upon induction of apoptosis |
| IND1 | Mitochondrial P-loop NTPase | Complex I assembly | [4Fe–4S] | Scaffold for transfer of Fe–S to Complex I |
Table 2.
Fe–S client proteins found in the human nucleus and cytosol.
| Nuclear Compartment | ||||
|---|---|---|---|---|
| Fe–S Protein | Full Name | Pathway | Cluster | Function |
| MUTYH | Adenine DNA glycosylase | DNA repair | [4Fe–4S] | Involved in oxidative DNA damage repair |
| EXO5 | Exonuclease V | DNA repair | [4Fe–4S] | Exonuclease involved in DNA repair |
| NTHL1 | Endonuclease III-like protein 1 | DNA repair | [4Fe–4S] | Catalyzes the first step in base excision repair |
| PRIM2 | DNA primase large subunit | DNA replication | [4Fe–4S] | DNA primase |
| POLA1 | DNA polymerase alpha catalytic subunit | DNA replication | [4Fe–4S] | Catalytic subunit of DNA polymerase alpha |
| POLD1 | DNA polymerase delta catalytic subunit | DNA replication | [4Fe–4S] | Catalytic subunit of DNA polymerase delta |
| POLE | DNA polymerase epsilon catalytic subunit A | DNA replication | [4Fe–4S] | Catalytic subunit of DNA polymerase epsilon |
| POLZ | DNA polymerase zeta catalytic subunit | Translesion DNA synthesis | [4Fe–4S] | Catalytic subunit of DNA polymerase zeta |
| DDX11 | ATP-dependent DNA helicase DDX11 | DNA replication and repair | [4Fe–4S] | DNA-dependent ATPase and ATP-dependent DNA helicase |
| Cytosolic Compartment | ||||
| Fe–S Protein | Full Name | Pathway | Cluster | Function |
| IRP1/ACO1 | Iron regulatory protein 1,cytosolic aconitase 1 | Iron metabolism, TCA cycle | [4Fe–4S] | Regulates iron levels through mRNA binding |
| KIF4A | Chromosome-associated kinesin | Mitosis | [4Fe–4S] | Motor protein involved in metaphase to anaphase to transition |
| TYW1 | S-adenosyl-L-methionine-dependent tRNA 4-demethylwyosine synthase | tRNA modification | [4Fe–4S] | Enzyme involved in tRNA(Phe) biosynthesis |
| CDKAL1 | Threonylcarbamoyladenosine tRNA methylthiotransferase | tRNA modification | [4Fe–4S] | Catalyzes the methylthiolation of t6A |
| ELP3 | Elongator complex protein 3 | Transcription | [4Fe–4S] | Catalytic histone acetyltransferase subunit of the RNA polymerase II elongator complex |
| GPAT | Amidophosphoribosyltransferase, Atase | Nucleotide metabolism | [4Fe–4S] | Involved in the synthesis of N(1)-(5-phospho-D-ribosyl)glycinamide during IMP biosynthesis |
| DPYD | Dihydropyrimidine dehydrogenase [NADP(+)] | Nucleotide metabolism | [4Fe–4S] | Catalyzes the reduction of uracil and thymine |
| XDH | Xanthine dehydrogenase | Nucleotide metabolism | [2Fe–2S] | Involved in purine degradation |
| ABCE1/RLI1 | ATP-binding cassette sub-family E member 1 | Protein translation | [4Fe–4S] | Splitting of ribosome subunits during translation termination |
| DPH1/2 | 2-(3-amino-3-carboxypropyl)histidine synthase subunit 1/2 | Protein translation | [4Fe–4S] | Involved in the post-translational modification of histidine to diphthamide |
Table 3.
List of conserved proteins required for the function of the mitochondrial Fe–S core complex. Note that Ferredoxin Reductase does not bind the Fe–S core complex but is required for its function.
Table 3.
List of conserved proteins required for the function of the mitochondrial Fe–S core complex. Note that Ferredoxin Reductase does not bind the Fe–S core complex but is required for its function.
| Protein | Human Protein | Yeast Protein | Bacterial Protein | Other Names | Required Cofactor | Function in Fe–S Core Complex |
|---|---|---|---|---|---|---|
| Cysteine Desulfurase | NFS1 | Nfs1 | IscS | - | PLP | Persulfide production and transfer to scaffold |
| LYRM Protein | ISD11 | Isd11 | - | LYRM4 | - | Stabilizes cysteine desulfurase complex |
| Acyl Carrier Protein | NDUFAB1 | Acp1 | Acp | ACP1, ACPm | 4′PP, acyl chain | Binds to LYRM protein for complex stability |
| Scaffold Protein | ISCU | Isu1/2 | IscU | - | - | Receives persulfide from cysteine desulfurase and assembles Fe–S |
| Frataxin | FXN | Yfh1 | CyaY | - | - | Modulates persulfide transfer rate |
| Ferredoxin | FDX2 | Yah1 | Fdx | FDX1L | [2Fe–2S] | Provides electrons to reduce persulfide |
| Ferredoxin Reductase | FDXR | Arh1 | Fpr | - | NADPH+ | Reduces [2Fe–2S]-ferredoxin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Perfitt, T.L.; Martelli, A. Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters. Inorganics 2022, 10, 31. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10030031
AMA Style
Perfitt TL, Martelli A. Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters. Inorganics. 2022; 10(3):31. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10030031
Chicago/Turabian StylePerfitt, Tyler L., and Alain Martelli. 2022. "Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters" Inorganics 10, no. 3: 31. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10030031
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.