2.3.1. QTAIM Derived Topological Properties
In order to further investigate the covalent character of M–Cl in these systems, the quantum theory of atoms in molecules (QTAIM) was employed. QTAIM is able to provide a robust and quantitative measure of the covalent contribution to bonding in the f-element complexes under consideration here.
Table 4 summarises the key topological properties of the electron density at the M–Cl bond critical point (BCP). A key parameter is the magnitude of the electron density at the M–Cl BCP (
): this metric has been used extensively in the quantification of covalency, and provides a measure of the accumulation of electron density in the chemical bonding region. Broadly speaking, values of
greater than 0.2 are indicative of covalent interactions, while values lower than 0.1 indicate predominant ionic character. The variation of this metric can therefore provide information regarding the variation of covalency in the systems under consideration.
Table 4.
Values of the electron density () and its Laplacian () at the M–Cl critical point of metal hexachlorides, evaluated at theoretically optimised geometries. All values are in a.u.
Table 4.
Values of the electron density () and its Laplacian () at the M–Cl critical point of metal hexachlorides, evaluated at theoretically optimised geometries. All values are in a.u.
| System | | |
|---|
| CASPT2 | RASPT2 | B3LYP | PBE | CASPT2 | RASPT2 | B3LYP | PBE |
|---|
| [UCl6] | 0.105 | 0.102 | 0.099 | 0.096 | +0.148 | +0.126 | +0.156 | +0.159 |
| [UCl6]− | 0.086 | 0.084 | 0.082 | 0.081 | +0.148 | +0.150 | +0.145 | +0.146 |
| [UCl6]2− | 0.064 | 0.059 | 0.063 | 0.065 | +0.154 | +0.144 | +0.130 | +0.140 |
| [UCl6]3− | 0.038 | 0.037 | 0.041 | 0.044 | +0.121 | +0.111 | +0.119 | +0.124 |
| [CeCl6]2− | 0.064 | 0.061 | 0.058 | 0.060 | +0.144 | +0.141 | +0.122 | +0.128 |
| [CeCl6]3− | 0.037 | 0.036 | 0.040 | 0.044 | +0.105 | +0.104 | +0.101 | +0.107 |
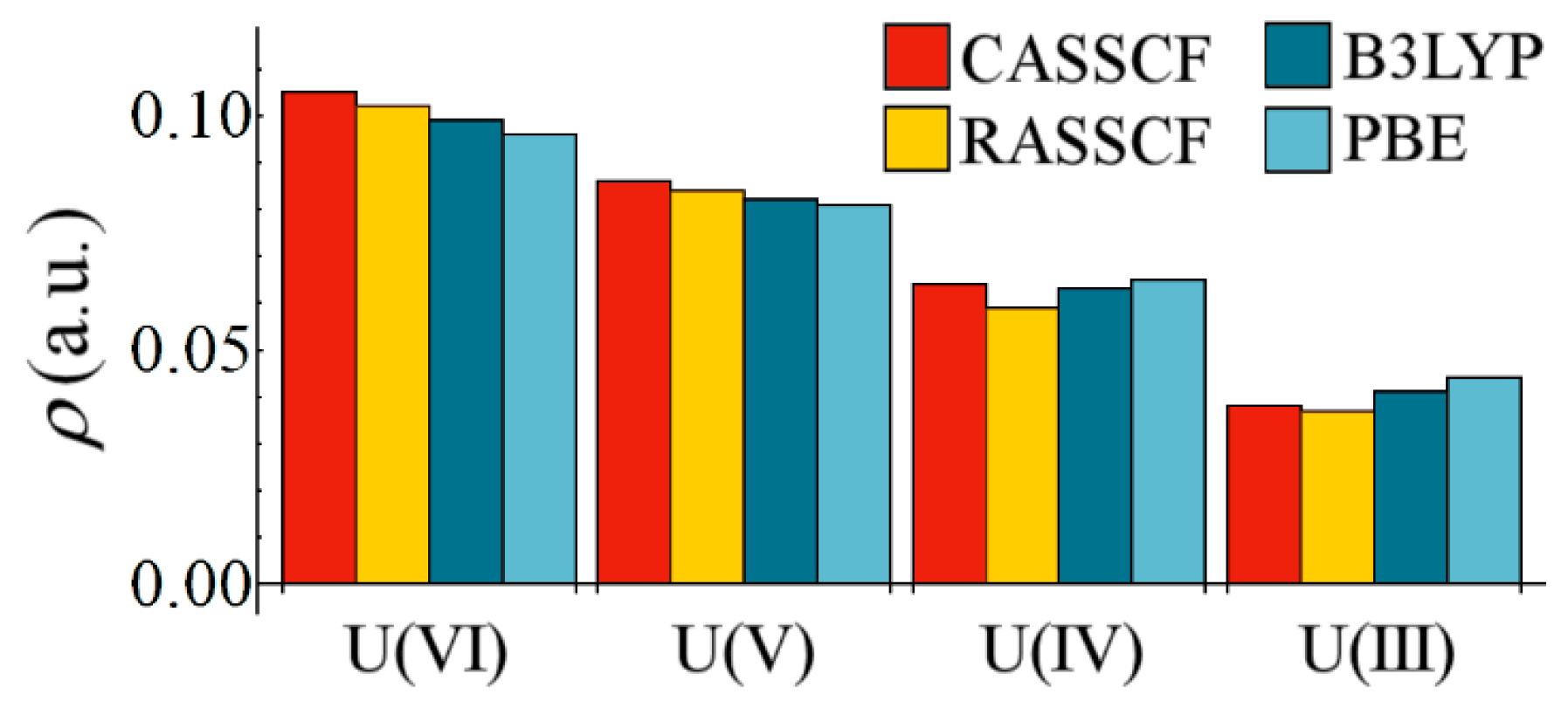
The low values of
reported in
Table 4 demonstrate that, unsurprisingly, M–Cl bonds in all complexes should be considered primarily ionic in character. This assertion is supported by the values of
, which are expected to be positive for predominantly ionic interactions. However,
Figure 2 demonstrates that, when considering the variation in
with respect to uranium oxidation state, all methodologies employed here display a clear and common trend, namely a reduction in magnitude from U(VI) to U(III). All methodologies are in broad agreement, with the correlated wavefunction methods demonstrating the greatest variation, commensurate with the greater variation in U–Cl bond lengths at this level of theory. The reduction in covalent character is partly due to the increasing M–Cl bond length as the oxidation state is lowered, but
Figure 1 implies that this reduction may also be due to increased energetic mismatch between metal and ligand orbitals in lower oxidation state complexes.
Figure 2.
Variation of at the U–Cl bond critical point as a function of oxidation state.
Figure 2.
Variation of at the U–Cl bond critical point as a function of oxidation state.
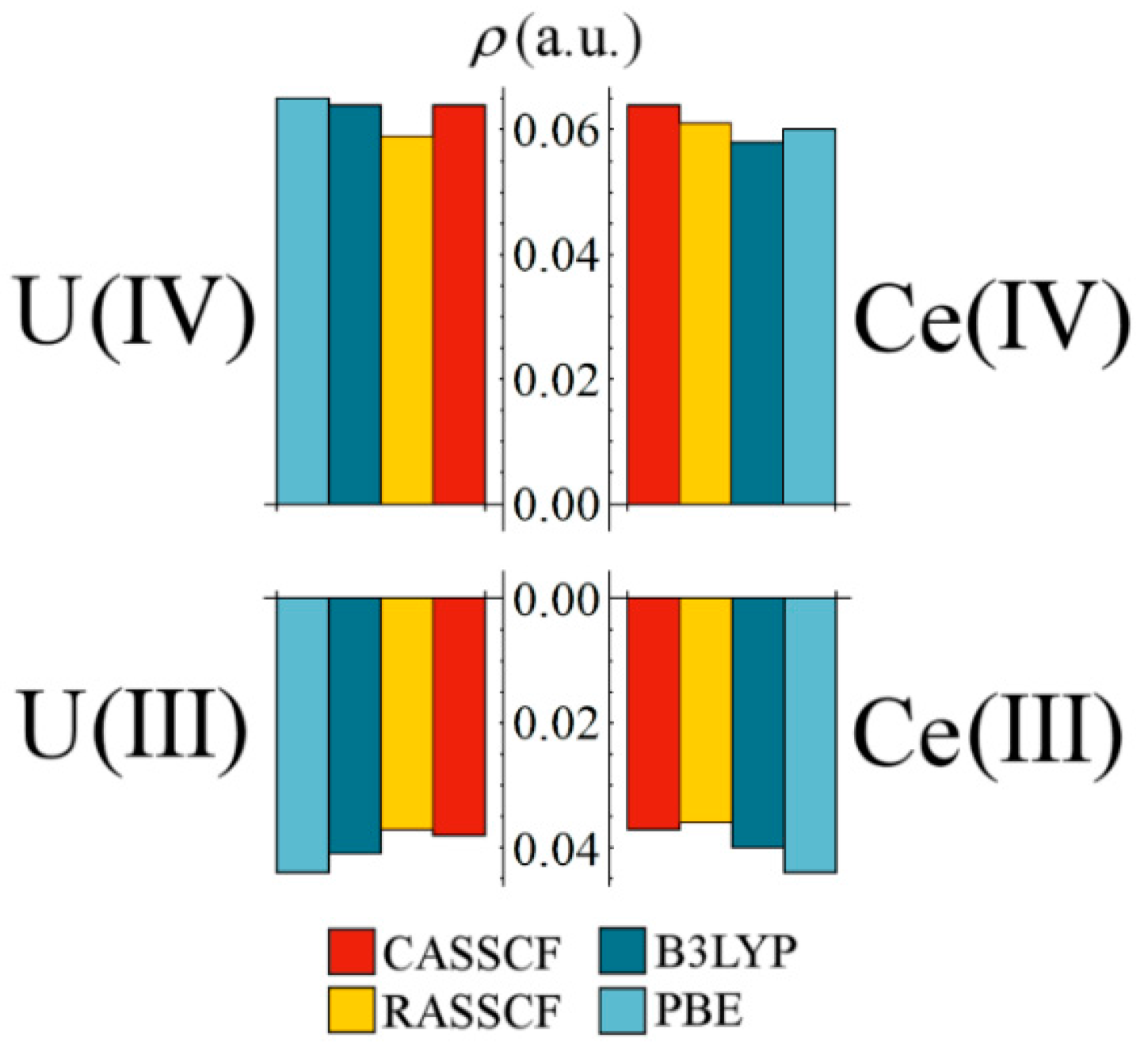
Figure 3 compares the magnitude of
at the M–Cl BCP for uranium and cerium complexes in the same oxidation state. Remarkably, Ce values are almost identical to those of the U complexes, and with the same decrease when moving from the M(IV) to M(III) oxidation state. These data are a clear indicator of uranium-like levels of covalency in an analogous cerium complex. In previous QTAIM studies of Ce and U complexes [
4,
9,
12] cerium covalency has always been noticeably lower, with
values approximately 80% of those obtained for analogous uranium complexes. For the systems under consideration here, Ce–Cl
values are, on average, 96.6% of those obtained for the analogous U complexes.
Figure 3.
Comparison of at the M–Cl bond critical point for tri- and tetravalent cerium and uranium complexes.
Figure 3.
Comparison of at the M–Cl bond critical point for tri- and tetravalent cerium and uranium complexes.
2.3.2. QTAIM Derived Integrated Properties
In addition to the topological properties of the electron density considered in the previous section, integrated properties can also be evaluated. Of interest here is the atomic electron population, a one-electron property obtained by integrating the electron density over a given atomic basin, and from which atomic charges can be derived. In addition to this, a pair of related two-electron properties, the localisation (λ) and delocalisation (δ) indices, can also be analysed in order to give detailed information regarding the nature and magnitude of bonding interactions: our previous research has identified a strong relationship between localisation index and oxidation state [
4,
12], while the delocalisation index, which quantifies the degree of electron sharing between a pair of atoms [
27,
28] can be considered as an alternative measure of covalency to
[
4,
12,
15,
27], providing data regarding electron delocalisation between atomic centres, which may occur independently of charge accumulation in the bonding region.
Table 5 summarises the QTAIM derived atomic charges and reveals that metal charges obtained from correlated wavefunction calculations are noticeably higher than those calculated using DFT. Although it is tempting to interpret this as being indicative of increased ionic character when employing wavefunction based methodologies, this is in contradiction to the topological data of
Table 4, which demonstrates that all methodologies predict similar degrees of covalent character. This issue will be revisited in light of integrated two-electron data. There is a clear trend in all methodologies of increased electronic charge located on the Cl ion as the oxidation state is lowered, commensurate with the increased total electronic charge. A proportion of this charge, however, is found to be located on the uranium centre, whose charge decreases with decreasing oxidation state. Finally, it should be noted that, when considering equivalent oxidation states, cerium and uranium charges are very similar, again indicating similar electronic structures.
Table 5.
QTAIM derived atomic charges of metal hexachlorides, evaluated at theoretically optimised geometries. All values are in a.u.
Table 5.
QTAIM derived atomic charges of metal hexachlorides, evaluated at theoretically optimised geometries. All values are in a.u.
| System | q(M) | q(Cl) |
|---|
| CASPT2 | RASPT2 | B3LYP | PBE | CASPT2 | RASPT2 | B3LYP | PBE |
|---|
| [UCl6] | +3.01 | +3.25 | +2.55 | +2.36 | −0.50 | −0.54 | −0.42 | −0.39 |
| [UCl6]− | +3.02 | +3.23 | +2.52 | +2.33 | −0.67 | −0.71 | −0.59 | −0.55 |
| [UCl6]2− | +2.88 | +2.95 | +2.39 | +2.24 | −0.81 | −0.82 | −0.73 | −0.71 |
| [UCl6]3− | +2.38 | +2.41 | +2.11 | +2.00 | −0.90 | −0.90 | −0.85 | −0.83 |
| [CeCl6]2− | +2.81 | +2.86 | +2.26 | +1.97 | −0.80 | −0.81 | −0.71 | −0.68 |
| [CeCl6]3− | +2.39 | +2.41 | +2.10 | +2.07 | −0.90 | −0.90 | −0.85 | −0.83 |
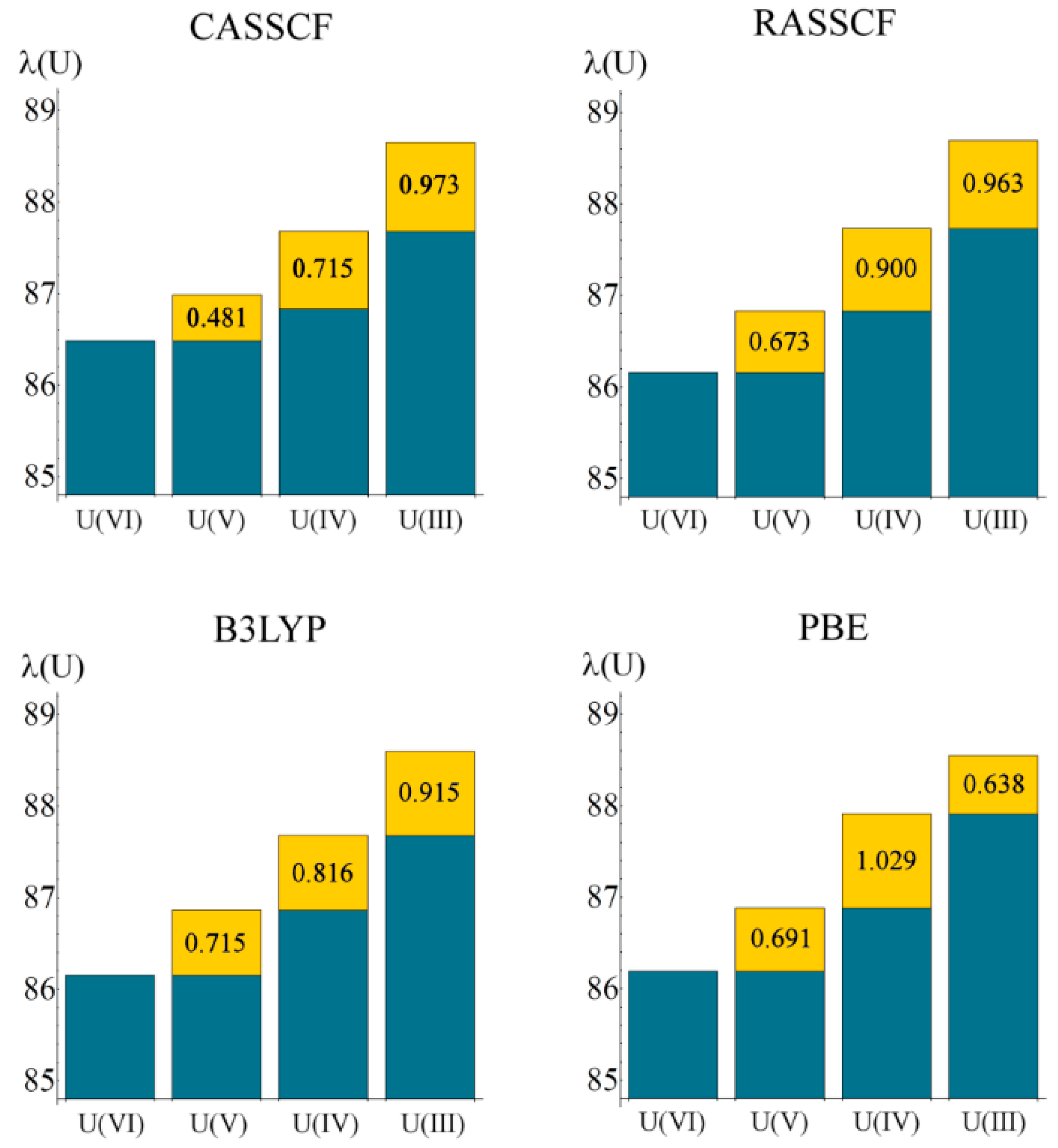
Localisation indices, λ(M), are summarised in
Table 6. Here, a trend reflecting the change in oxidation state is much more pronounced that of the atomic charges, with λ increasing significantly as the uranium oxidation state reduces from VI to III (see
Figure 4). In a purely ionic system, it might be expected that λ(U) would increase by unity for each change in oxidation state: in a previous study [
12] we considered the variation in λ across the actinide series in the largely ionic An(C
8H
8)
2 (An = Th–Cm) and found changes close to unity. In the present work, variation in λ(U) is less marked, implying greater covalency in the interactions.
Table 6.
QTAIM derived localisation (λ) along with Z(M)–λ(M), the number of electron donated to, or shared with, the ligand set by the metal centre evaluated at theoretically optimised geometries. Figures in parentheses are rounded to the nearest integer. All values are in a.u.
Table 6.
QTAIM derived localisation (λ) along with Z(M)–λ(M), the number of electron donated to, or shared with, the ligand set by the metal centre evaluated at theoretically optimised geometries. Figures in parentheses are rounded to the nearest integer. All values are in a.u.
| System | λ(M) | Z(M)–λ(M) |
|---|
| CASPT2 | RASPT2 | B3LYP | PBE | CASPT2 | RASPT2 | B3LYP | PBE |
|---|
| [UCl6] | 86.48 | 86.16 | 86.15 | 86.19 | 5.52 (6) | 5.84 (6) | 5.85 (6) | 5.81 (6) |
| [UCl6]− | 86.96 | 86.83 | 86.86 | 86.88 | 5.04 (5) | 5.17 (5) | 5.14 (5) | 5.12 (5) |
| [UCl6]2− | 87.68 | 87.73 | 87.68 | 87.91 | 4.32 (4) | 4.27 (4) | 4.32 (4) | 4.09 (4) |
| [UCl6]3− | 88.65 | 88.69 | 88.60 | 88.55 | 3.35 (3) | 3.31 (3) | 3.40 (3) | 3.45 (3) |
| [CeCl6]2− | 53.74 | 53.79 | 53.79 | 53.78 | 4.26 (4) | 4.21 (4) | 4.21 (4) | 4.22 (4) |
| [CeCl6]3− | 54.72 | 54.75 | 54.71 | 54.69 | 3.28 (3) | 3.25 (3) | 3.29 (3) | 3.31 (3) |
Figure 4.
Localisation index as a function of oxidation state, calculated at all levels of theory. Values in yellow boxes indicate increase in electron localisation as oxidation state is lowered. All values are in atomic units.
Figure 4.
Localisation index as a function of oxidation state, calculated at all levels of theory. Values in yellow boxes indicate increase in electron localisation as oxidation state is lowered. All values are in atomic units.
In order to make comparison between the cerium and uranium complexes under consideration, a more useful measure is
Z(M)–λ(M), which gives the number of electrons donated to, or shared with, the ligand set by the metal centre. We expect this measure to correlate with oxidation state, as we have previously reported [
4,
12].
Table 6 shows that this correlation is also present in systems considered here: rounding
Z(M)–λ(M) to the nearest integer returns the formal oxidation state for all systems at all levels of theory. The similarity between uranium and cerium complexes is again pronounced, with a maximum difference of just 0.15 a.u. and, on average, differences approximately half of this.
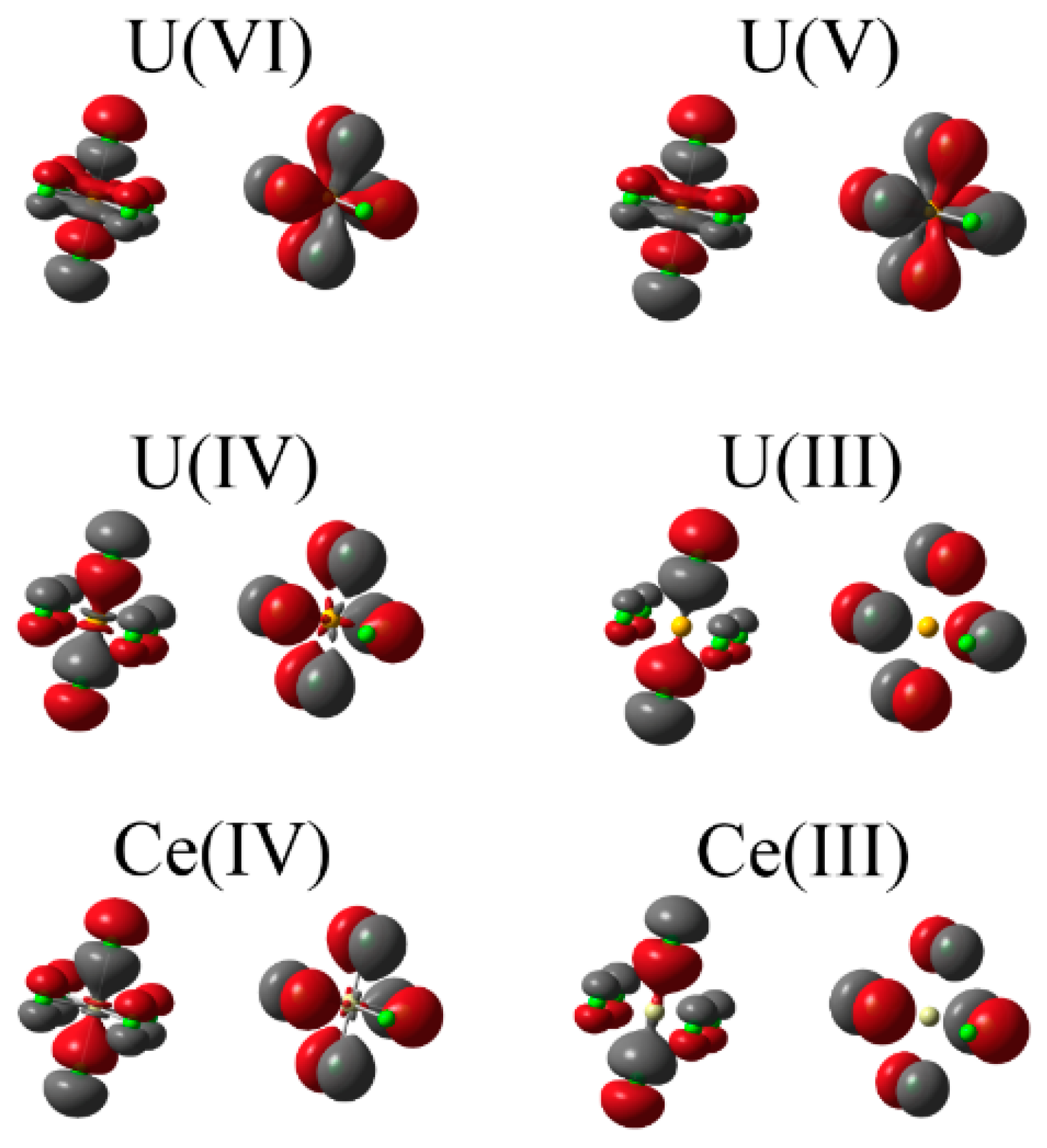
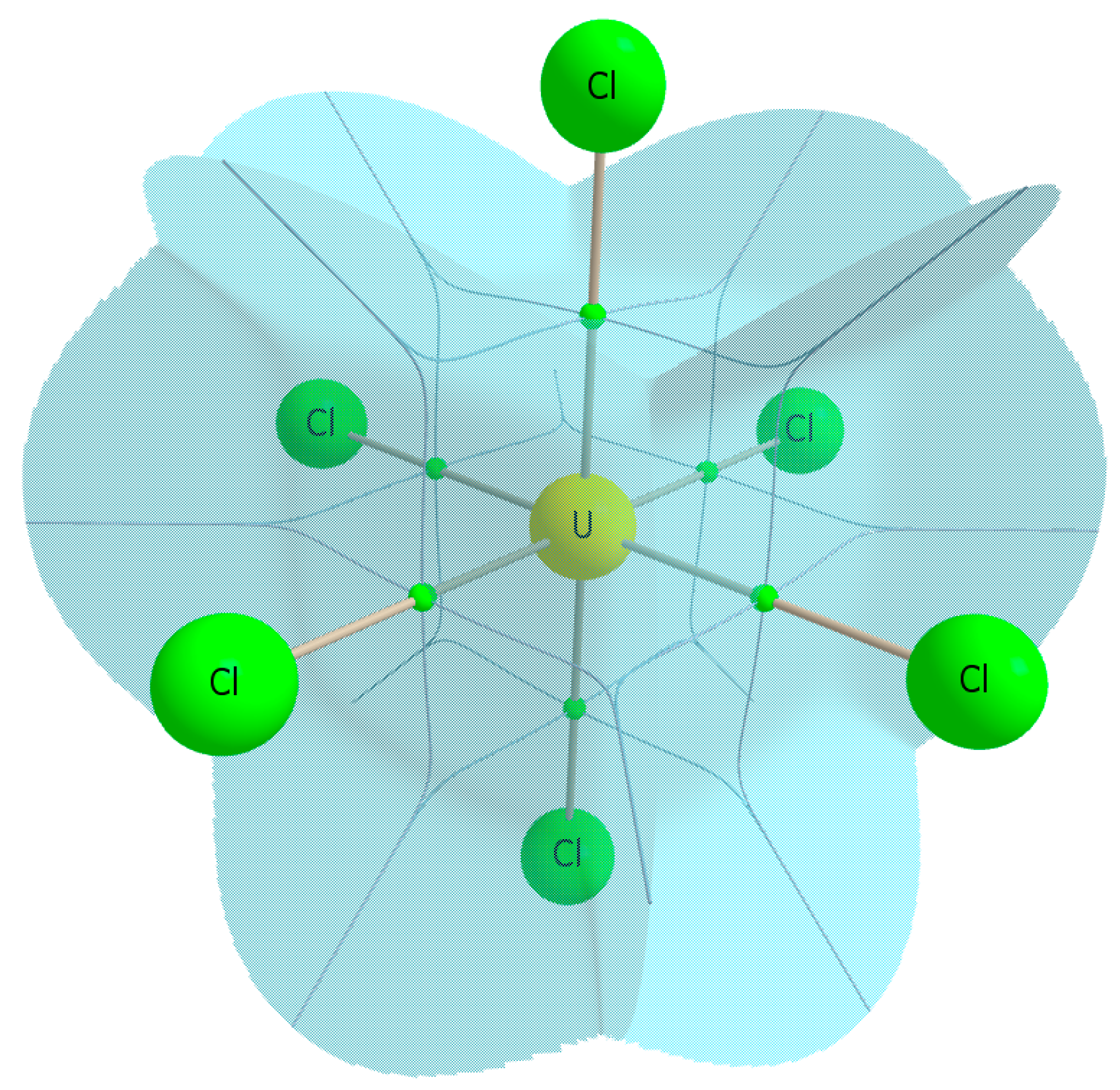
Finally we consider the delocalisation indices as an alternative measure of covalency. The high (O
h) symmetry of the systems (and therefore the atomic basins of the central ions: see
Figure 5) under consideration here, along with the formal definition of the delocalisation index [
27], allows us to decompose the total index into
gerade and
ungerade parity contributions. Since orbitals comprising the d-manifold have
gerade parity and those comprising the f-manifold have
ungerade parity, this decomposition provides a mechanism for assessing the d- and f-contributions to electron sharing. Total and decomposed delocalisation indices are given in
Table 7 and data corresponding to uranium complexes is visualised in
Figure 6.
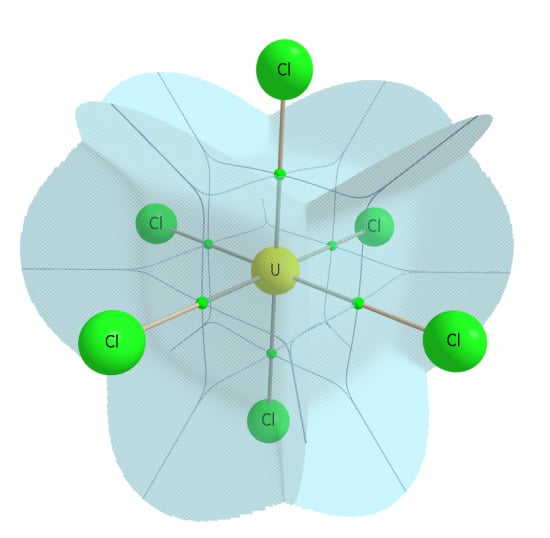
Figure 5.
QTAIM calculated atomic basin of uranium in UCl6, illustrating the Oh symmetry of the basin.
Figure 5.
QTAIM calculated atomic basin of uranium in UCl6, illustrating the Oh symmetry of the basin.
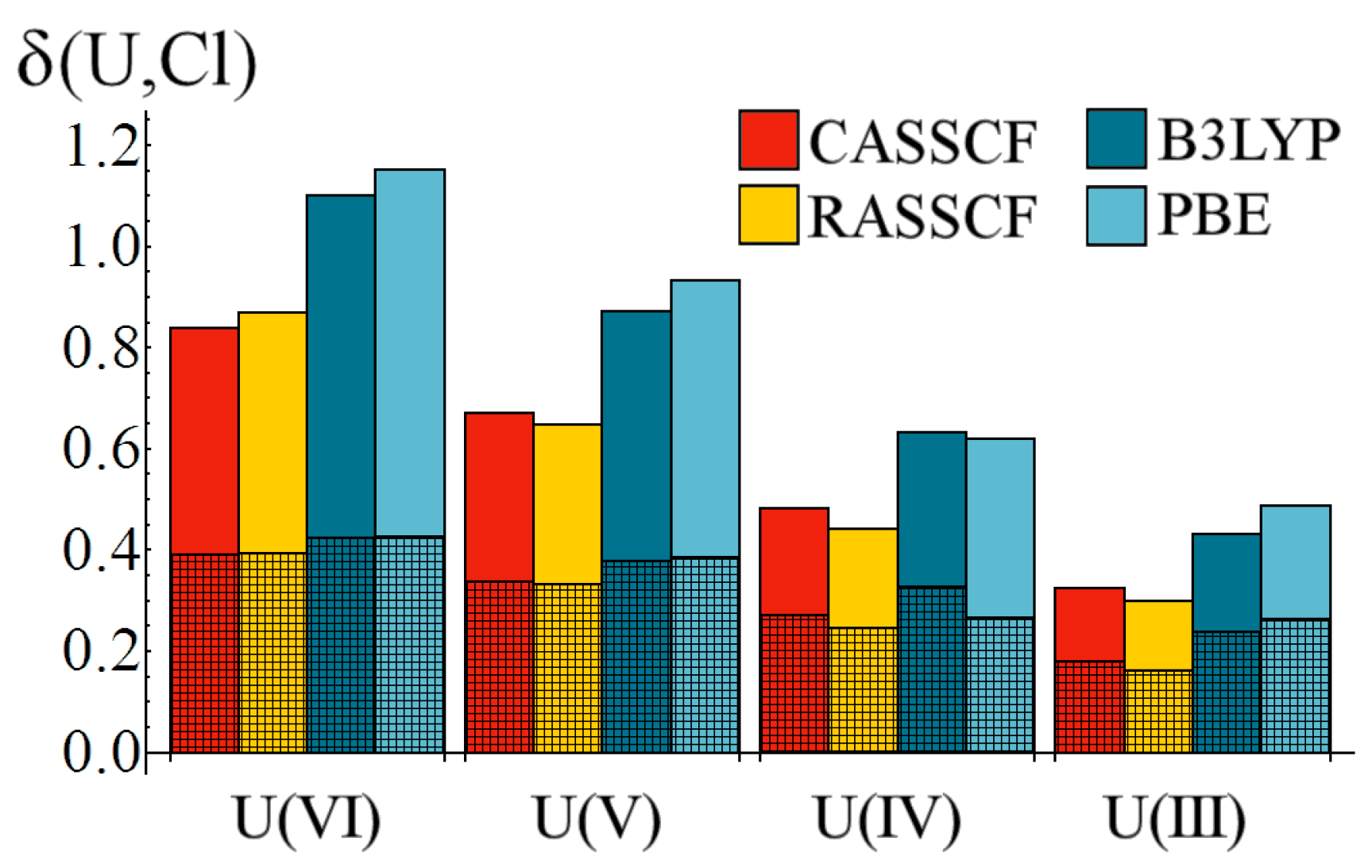
Figure 6.
Variation of the U–Cl delocalisation index, δ, as a function of oxidation state. Cross-hatched regions correspond to the gerade contributions to δ. All values are given in a.u.
Figure 6.
Variation of the U–Cl delocalisation index, δ, as a function of oxidation state. Cross-hatched regions correspond to the gerade contributions to δ. All values are given in a.u.
Table 7.
Delocalisation indices, along with decomposed gerade (g) and ungerade (u) contributions, evaluated at theoretically optimised geometries. All values are in a.u.
Table 7.
Delocalisation indices, along with decomposed gerade (g) and ungerade (u) contributions, evaluated at theoretically optimised geometries. All values are in a.u.
| System | δ(M,Cl) |
|---|
| CASPT2 | RASPT2 | B3LYP | PBE |
|---|
| Total | u | g | Total | u | g | Total | u | g | Total | u | g |
|---|
| [UCl6] | 0.838 | 0.447 | 0.391 | 0.868 | 0.475 | 0.393 | 1.102 | 0.678 | 0.424 | 1.151 | 0.725 | 0.426 |
| [UCl6]− | 0.673 | 0.335 | 0.338 | 0.647 | 0.316 | 0.331 | 0.872 | 0.494 | 0.378 | 0.932 | 0.547 | 0.385 |
| [UCl6]2− | 0.482 | 0.210 | 0.272 | 0.442 | 0.197 | 0.245 | 0.633 | 0.307 | 0.326 | 0.619 | 0.353 | 0.266 |
| [UCl6]3− | 0.324 | 0.145 | 0.179 | 0.300 | 0.138 | 0.162 | 0.432 | 0.193 | 0.239 | 0.487 | 0.224 | 0.263 |
| [CeCl6]2− | 0.485 | 0.202 | 0.283 | 0.450 | 0.184 | 0.266 | 0.651 | 0.435 | 0.216 | 0.716 | 0.482 | 0.234 |
| [CeCl6]3− | 0.297 | 0.124 | 0.173 | 0.279 | 0.122 | 0.157 | 0.395 | 0.163 | 0.232 | 0.446 | 0.195 | 0.251 |
The delocalisation index data bears some resemblance to the
data of
Figure 2, in that there is a clear reduction in the degree of electron sharing between the metal and chloride ions as the oxidation state is lowered. However, whereas quantitative agreement between correlated wavefunction and density functional methods was found with respect to
,
Figure 6 reveals a significant disparity: density functional methods predict significantly larger M–Cl delocalisation indices than those obtained from correlated wavefunctions approaches. Decomposition of these indices shows that, for the U(VI) and U(V) oxidation states, this disparity is due to increased
ungerade, e.g., f-electron, contributions, whereas for the lower U(IV) and U(III) the disparity has a growing
gerade contribution. Kohn-Sham DFT suffers from the self-interaction problem, in which the interaction of a given electron with itself is not identically equal to zero (as is the case in correlated wavefunction approaches). This self-interaction error can lead to unexpected apparent electron delocalisation which vanishes when a correlated wavefunction approach is employed [
29,
30]. We therefore suggest that the origin of the enhanced delocalisation in DFT-calculated densities is due to self-interaction, a view supported by the fact that this effect is less pronounced in B3LYP-derived densities: B3LYP would be expected to be less prone to self-interaction error due to its incorporation of a component of exact exchange. We can deduce from our
data, however, that the enhanced delocalisation does not lead to an increase in electron density at the U–Cl bond-critical point.
Focusing on the correlated wavefunction data in
Table 6, we find that the degree of total electron sharing in the U(III) complex is just 39% (35%) of that found in the U(VI) complex at the CASSCF (RASSCF) level. The
ungerade contribution reduces to 32% (29%) and the
gerade contribution reduces to 46% (41%) at the CASSCF (RASSCF) level. This shows that f-electron contributions to covalency, as quantified by this measure, drop more rapidly than those to d-electron contributions as the oxidation state is lowered.
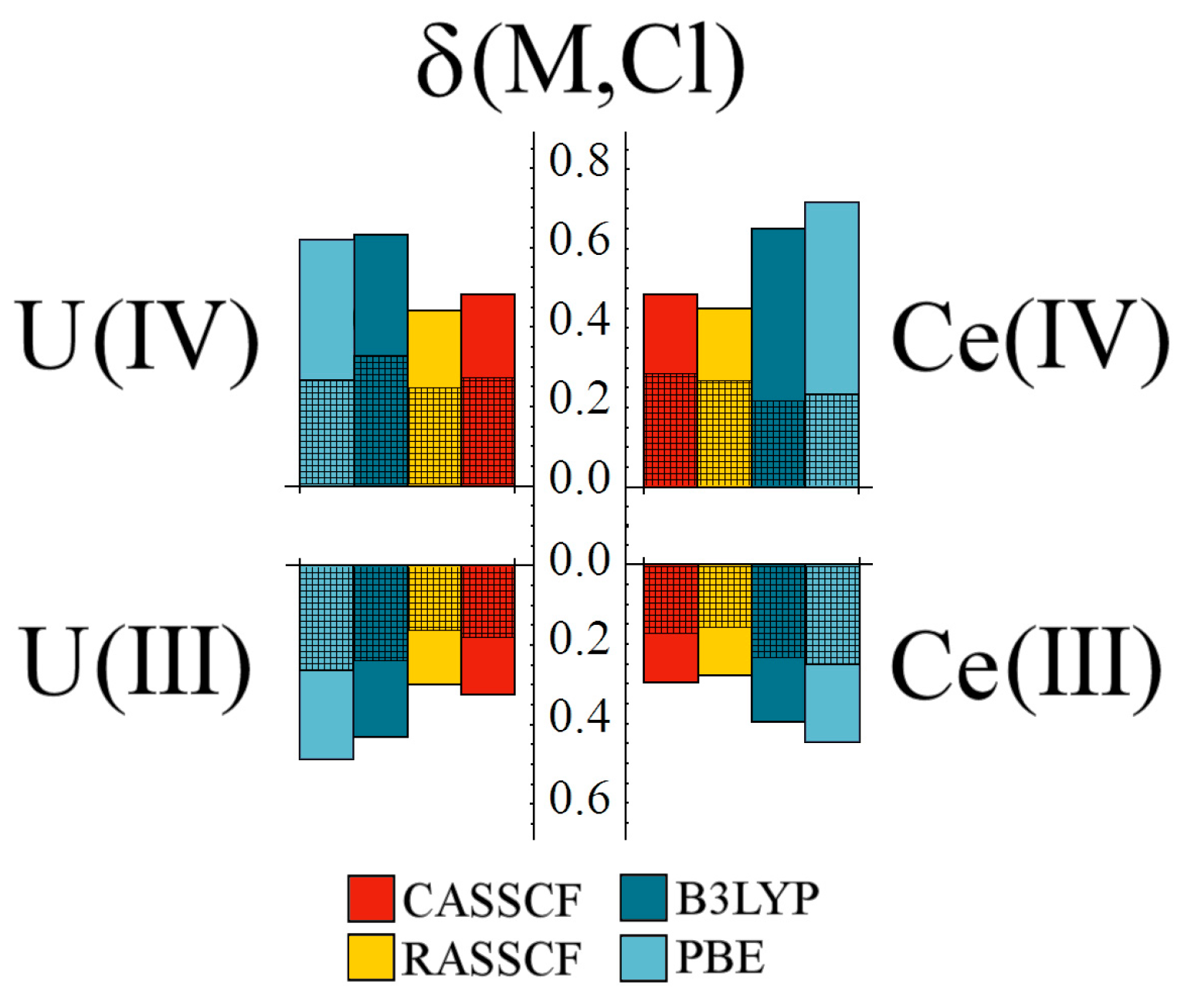
Comparing uranium and cerium complexes, delocalisation indices largely mirror
, with all methodologies giving comparable values for the same oxidation state (see
Figure 7). Correlated wavefunction approaches, in particular, give almost identical results for the uranium and cerium complexes.
When considering
gerade and
ungerade contribution to δ(M,Cl), similar behaviour is again seen, with the exception being the Ce(IV) DFT-generated densities.
Table 1 shows that both exchange-correlation functionals overestimate the Ce–Cl bond length in this system, which presumably accounts for the
gerade contributions.
Figure 7.
Comparison of M–Cl delocalisation index, δ, for tri- and tetravalent uranium and cerium hexahalides. Cross-hatched regions correspond to the gerade contributions to δ. All values are given in a.u.
Figure 7.
Comparison of M–Cl delocalisation index, δ, for tri- and tetravalent uranium and cerium hexahalides. Cross-hatched regions correspond to the gerade contributions to δ. All values are given in a.u.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






