
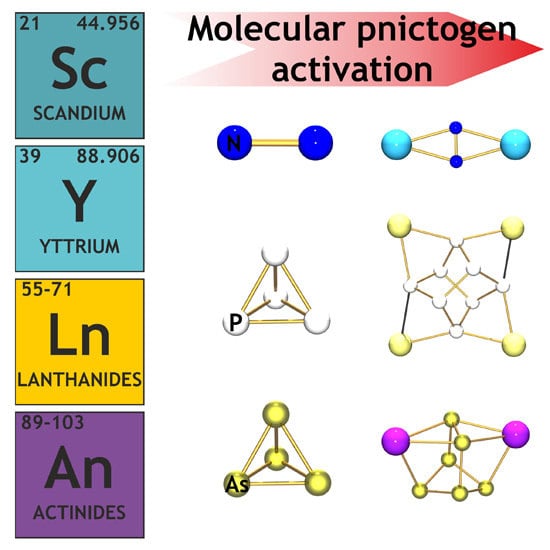
Molecular Pnictogen Activation by Rare Earth and Actinide Complexes

Abstract
:
1. Introduction
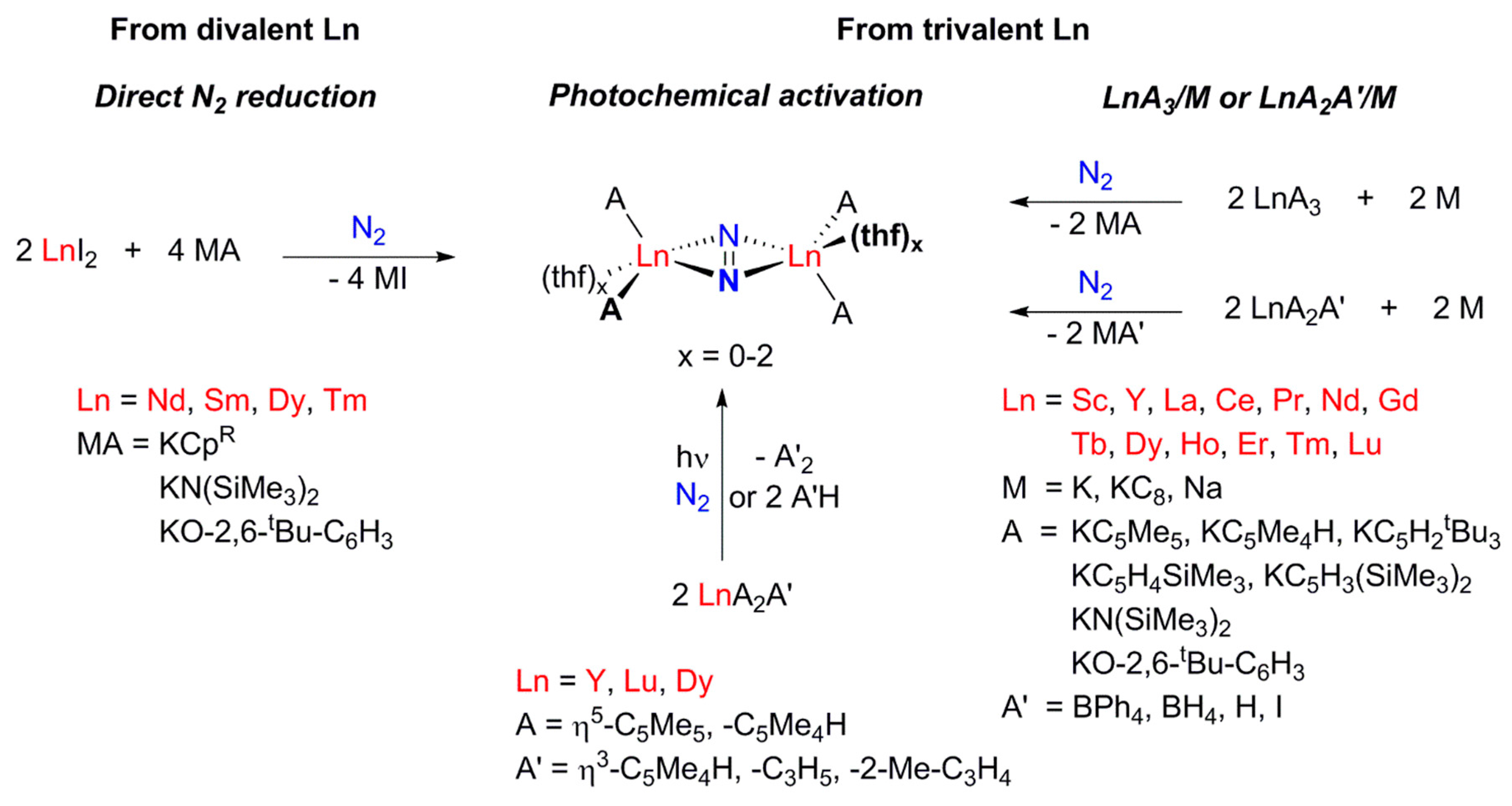
2. Dinitrogen Activation by Rare Earth Complexes
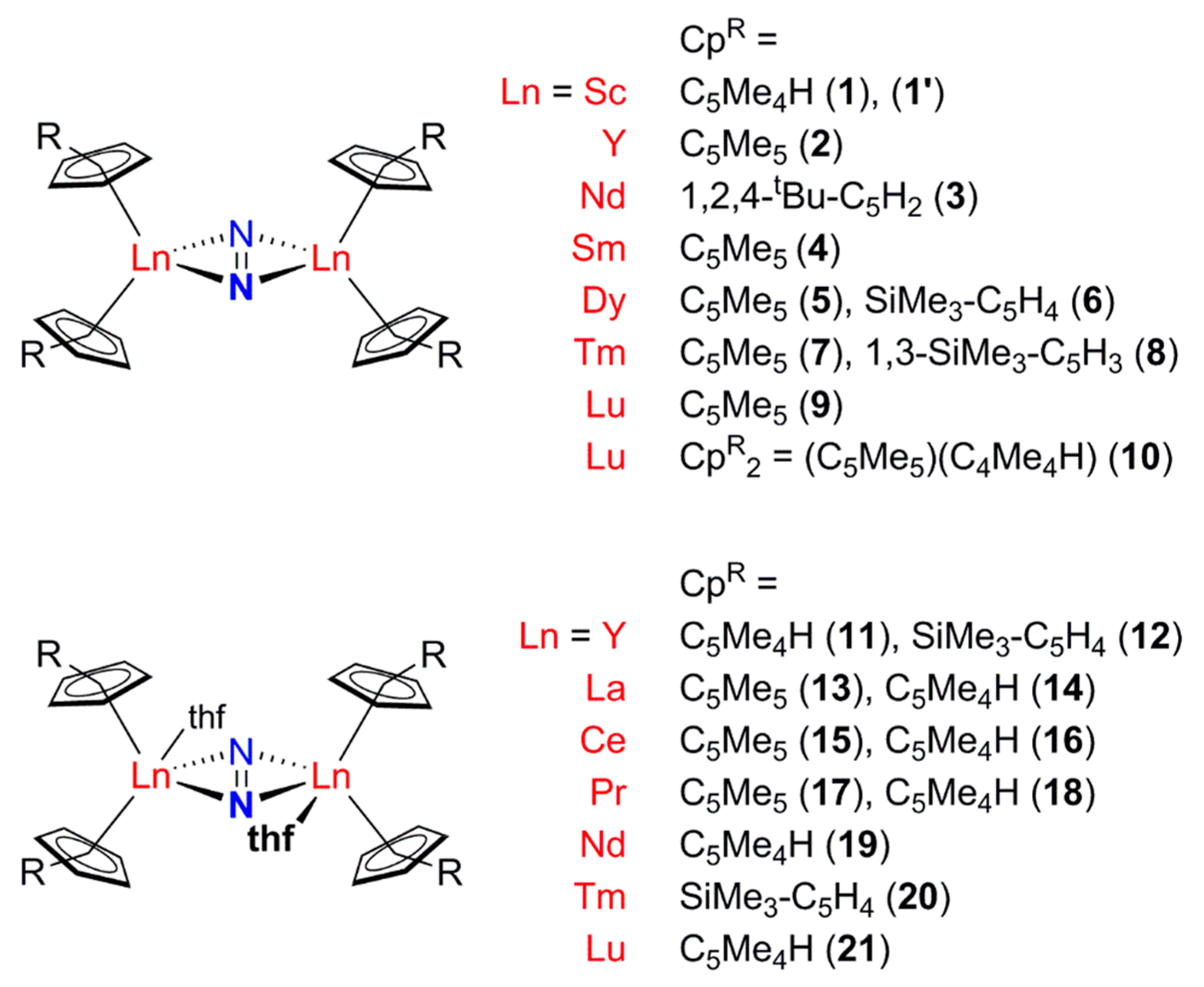
2.1. Complexes Containing a Formal N22− Ligand


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex (#) [Reference] | N–N Bond Length (Å) | Ln–N(N2) Bond Lengths (Å) | N–N Frequency (cm−1) | 14/15N-NMR Spectroscopy (ppm) a |
|---|---|---|---|---|
| N2 | 1.0975 [66] | - | 2331 [67] | −75 [68] |
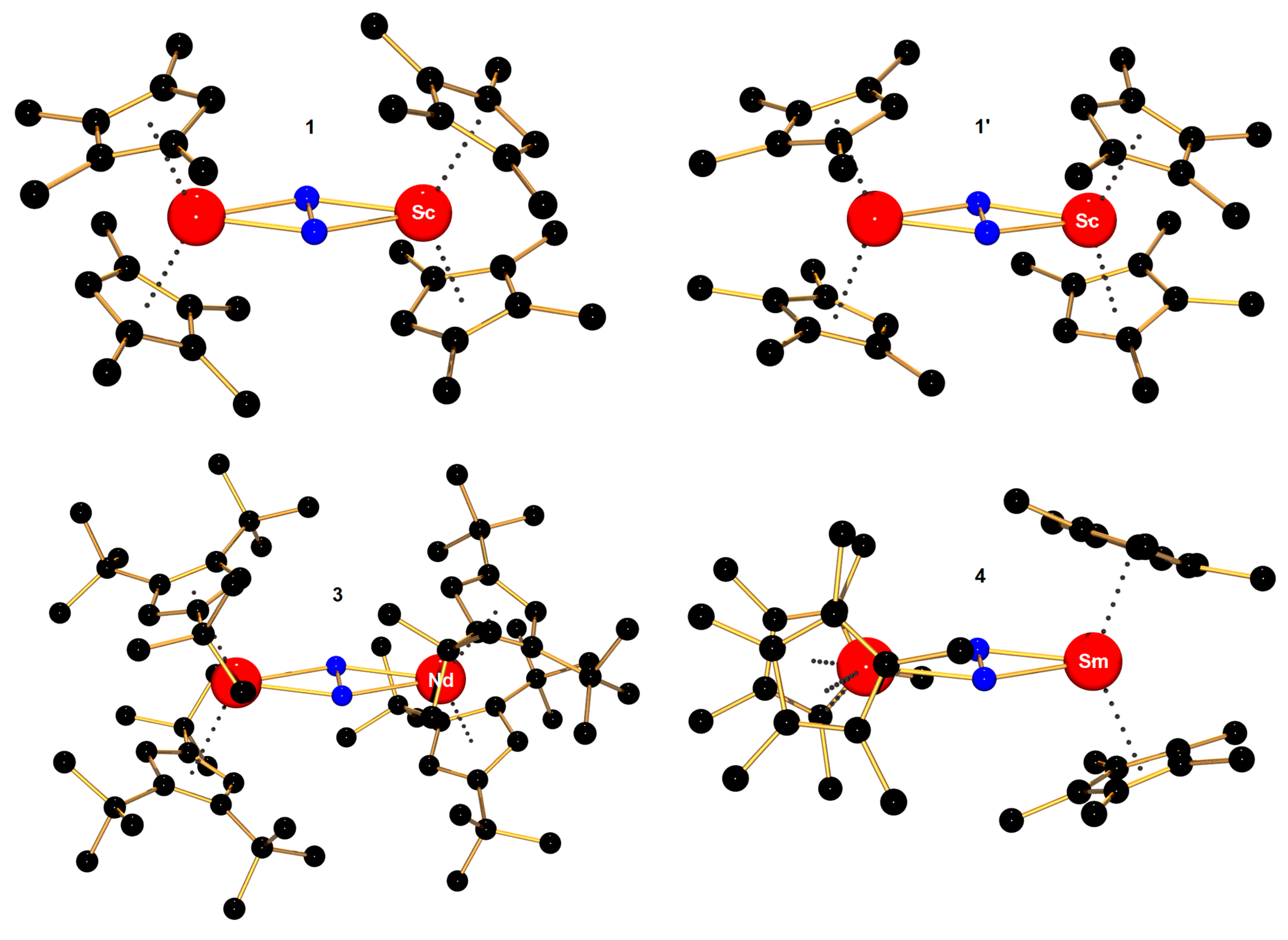
| [(η5-C5Me4H)2Sc]2(μ-η2:η2-N2) (1) [69] | 1.239(3) | 2.216(1) | - | - |
| 2.220(1) | ||||
| [(η5-C5Me4H)2Sc]2(μ-η2:η2-N2) (1′) [70] | 1.229(3) | 2.197(2) | - | 385 |
| 2.179(2) | ||||
| [(η5-C5Me5)2Y]2(μ-η2:η2-N2) (2) [71] | 1.172(6) | 2.279(3) | - | 496 |
| 2.292(3) | ||||
| [(η5-1,2,4-tBu–C5H2)2Nd]2(μ-η2:η2-N2) (3) [67] | 1.226(12) | 2.495(2) | 1622 (14N2) | - |
| 2.497(2) | 1569 (15N2) | |||
| [(η5-C5Me5)2Sm]2(μ-η2:η2-N2) b (4) [72] | 1.088(12) | 2.348(6) | - | −117 (263 K) |
| 2.367(6) | −161 (203 K) | |||
| [(η5-C5Me5)2Dy]2(μ-η2:η2-N2) (5) [73] | - | - | - | - |
| [(η5-SiMe3–C5H4)2Dy]2(μ-η2:η2-N2) (6) [74] | Connectivity only | - | ||
| [(η5-C5Me5)2Tm]2(μ-η2:η2-N2) (7) [75] | Connectivity only | - | ||
| [(η5-1,3-SiMe3–C5H3)2Tm]2(μ-η2:η2-N2) (8) [75] | 1.259(4) | 2.273(2) | - | - |
| 2.272(2) | ||||
| [(η5-C5Me5)2Lu]2(μ-η2:η2-N2) (9) [71] | Connectivity only | 527 | ||
| [(η5-C5Me5)(η5-C5Me4H)Lu]2(μ-η2:η2-N2) (10) [76] | 1.275(3) | 2.291(3) | 1736 (14N2) | - |
| 2.295(3) | 1678 (15N2) | |||
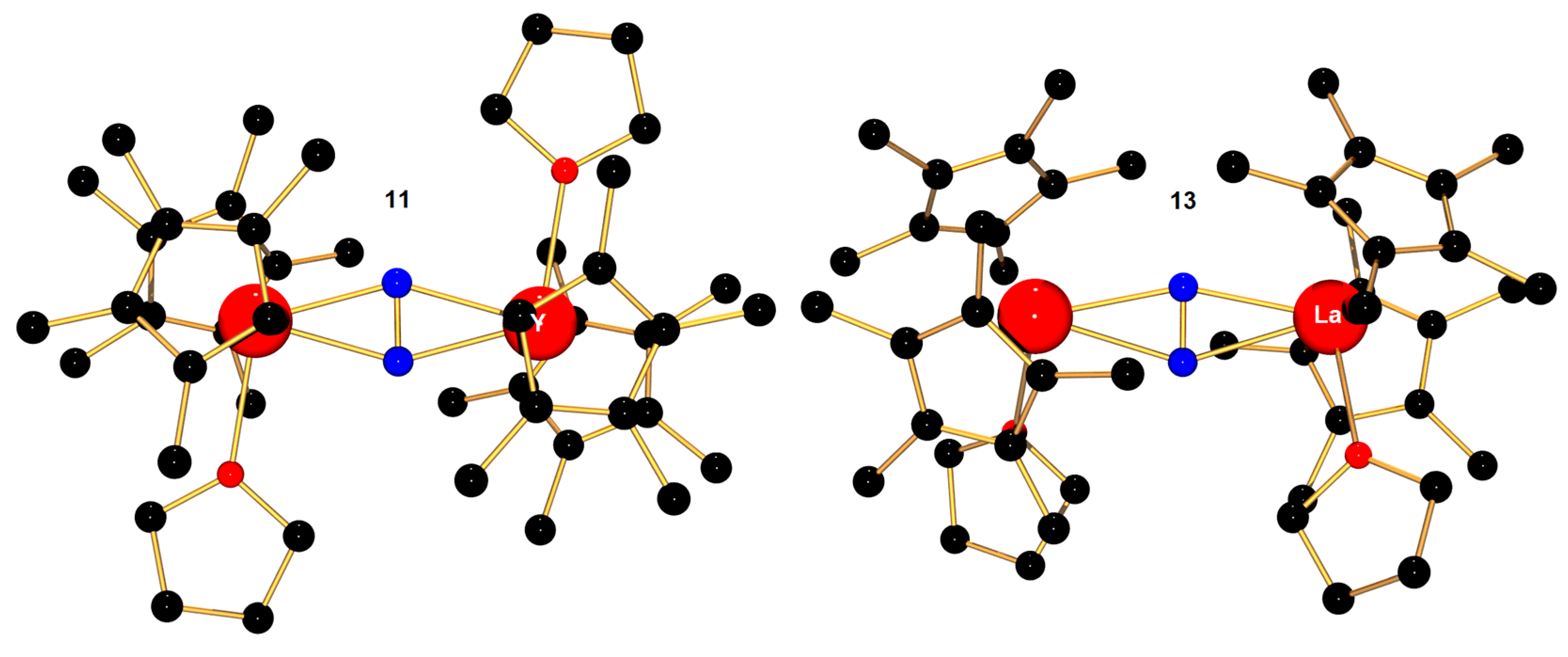
| [(η5-C5Me4H)2Y(thf)]2(μ-η2:η2-N2) c (11) [77] | 1.252(5) | 2.338(3) | - | 468 |
| 2.370(3) | ||||
| [(η5-SiMe3–C5H4)2Y(thf)]2(μ-η2:η2-N2) c (12) [78] | 1.244(2) | 2.3214(14) | - | - |
| 2.3070(14) | ||||
| [(η5-C5Me5)2La(thf)]2(μ-η2:η2-N2) d (13) [79] | 1.233(5) | 2.537(4) | - | 569 |
| 2.478(4) | ||||
| [(η5-C5Me4H)2La(thf)]2(μ-η2:η2-N2) c (14) [79] | 1.243(4) | 2.457(2) | - | 495 |
| 2.503(2) | ||||
| [(η5-C5Me5)2Ce(thf)]2(μ-η2:η2-N2) c (15) [68] | 1.258(9) | 2.4548(15) | - | 871 |
| 2.542(2) | ||||
| [(η5-C5Me4H)2Ce(thf)]2(μ-η2:η2-N2) d (16) [68] | 1.235(6) | 2.428(3) | - | 1001 |
| 2.475(3) | ||||
| [(η5-C5Me5)2Pr(thf)]2(μ-η2:η2-N2) d (17) [68] | 1.242(9) | 2.4459(14) | - | 2231 |
| 2.512(2) | ||||
| [(η5-C5Me4H)2Pr(thf)]2(μ-η2:η2-N2) c (18) [68] | 1.235(7) e | 2.418(4) e | - | 2383 |
| 2.455(3) e | ||||
| [(η5-C5Me4H)2Nd(thf)]2(μ-η2:η2-N2) c (19) [79] | 1.241(5) e | 2.404(3) e | - | - |
| 2.451(2) e | ||||
| [(η5-SiMe3–C5H4)2Tm(thf)]2(μ-η2:η2-N2) c (20) [75] | 1.236(8) | 2.274(4) | - | - |
| 2.302(4) | ||||
| [(η5-C5Me4H)2Lu(thf)]2(μ-η2:η2-N2) c (21) [80] | 1.243(12) | 2.290(6) | - | 521 |
| 2.311(6) | ||||
| [{(Me3Si)2N}2Y(thf)]2(μ-η2:η2-N2) c (22) [81] | 1.274(3) | 2.297(2) | 1425 (14N2) | +513 (t) |
| 2.308(2) | 1377 (15N2) | |||
| [{(Me3Si)2N}2La(thf)]2(μ-η2:η2-N2) (23) [81] | - | - | - | 516 |
| [{(Me3Si)2N}2Nd(thf)]2(μ-η2:η2-N2) c (24) [81] | 1.258(3) | 2.3758(16) | - | - |
| 2.3938(16) | ||||
| [{(Me3Si)2N}2Gd(thf)]2(μ-η2:η2-N2) c (25) [81] | 1.278(4) | 2.326(2) | - | - |
| 2.353(2) | ||||
| [{(Me3Si)2N}2Tb(thf)]2(μ-η2:η2-N2) c (26) [81] | 1.271(4) | 2.301(2) | - | - |
| 2.328(2) | ||||
| [{(Me3Si)2N}2Dy(thf)]2(μ-η2:η2-N2) c (27) [82] | 1.305(6) | 2.287(3) | - | - |
| 2.312(3) | ||||
| [{(Me3Si)2N}2Ho(thf)]2(μ-η2:η2-N2) c (28) [83] | 1.264(4) | 2.296(2) | - | - |
| 2.315(2) | ||||
| [{(Me3Si)2N}2Er(thf)]2(μ-η2:η2-N2) c (29) [81] | 1.276(5) | 2.271(3) | - | - |
| 2.302(3) | ||||
| [{(Me3Si)2N}2Tm(thf)]2(μ-η2:η2-N2) c (30) [82] | 1.261(4) | 2.271(2) | - | - |
| 2.296(2) | ||||
| [{(Me3Si)2N}2Lu(thf)]2(μ-η2:η2-N2) c (31) [83] | 1.285(4) | 2.241(2) | 1451 (14N2) | 557 |
| 2.272(2) | ||||
| [{(Me3Si)2N}2Y(PhCN)]2(μ-η2:η2-N2) c (32) [84] | 1.258(2) | 2.2848(13) | - | - |
| 2.3092(13) | ||||
| [{(Me3Si)2N}2Y(C5H5N)]2(μ-η2:η2-N2) c (33) [84] | 1.255(3) | 2.2917(16) | - | - |
| 2.3107(17) | ||||
| [{(Me3Si)2N}2Y(4-NMe2–C5H4N)]2(μ-η2:η2-N2) c (34) [84] | 1.259(2) | 2.2979(12) | - | - |
| 2.3132(12) | ||||
| [{(Me3Si)2N}2Y(Ph3PO)]2(μ-η2:η2-N2) c (35) [84] | 1.262(2) | 2.3000(14) | - | - |
| 2.3022(14) | ||||
| [{(Me3Si)2N}2Y(Me3NO)]2(μ-η2:η2-N2) c (36) [84] | 1.198(3) | 2.2925(17) | - | - |
| 2.2941(18) | ||||
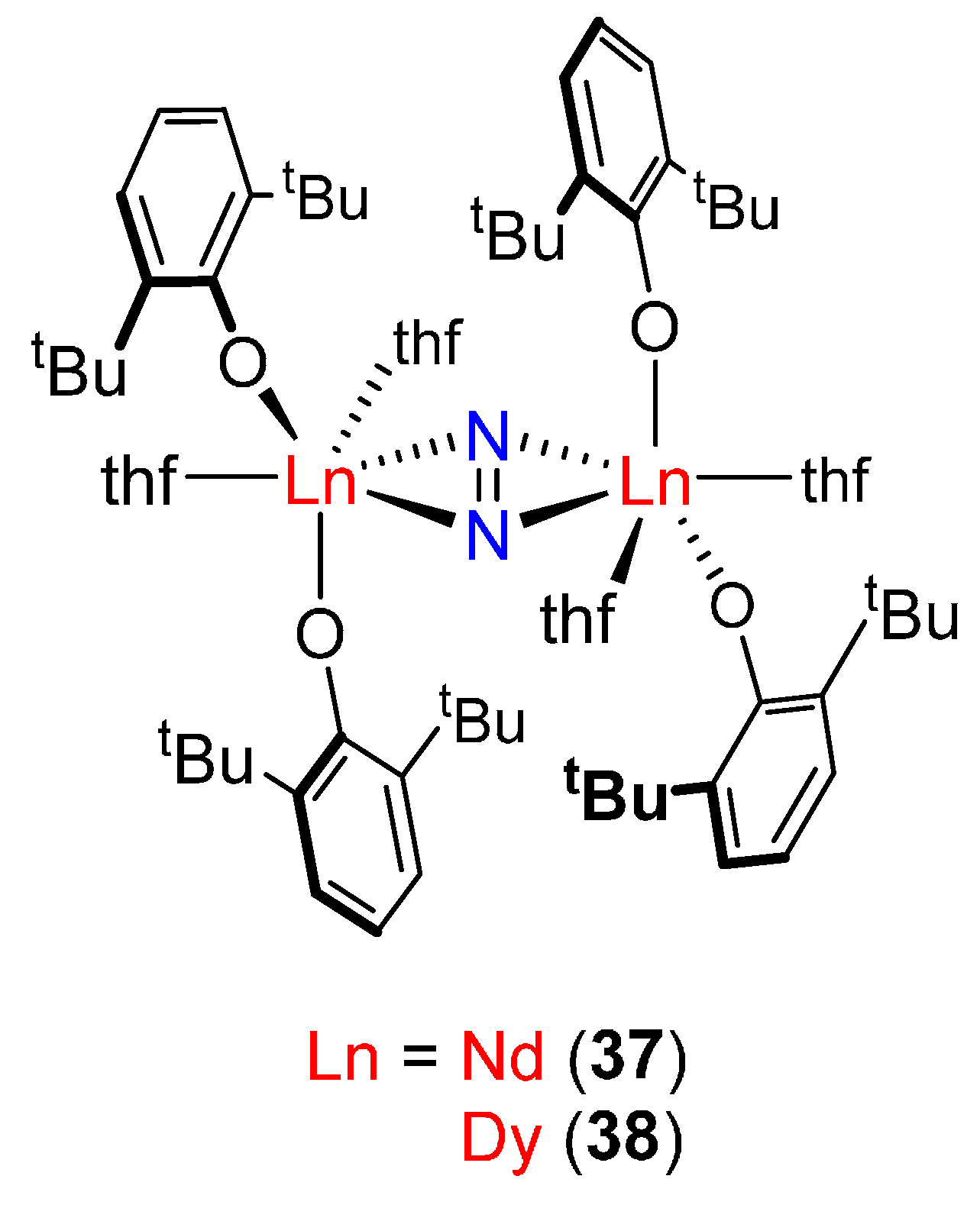
| [(2,6-tBu–C6H3O)2Nd(thf)2]2(μ-η2:η2-N2) (37) [82] | 1.242(7) | 2.397(4) | - | - |
| 2.401(3) | ||||
| [(2,6-tBu–C6H3O)2Dy(thf)2]2(μ-η2:η2-N2) (38) [82] | 1.257(7) f | 2.328(4) f | 1526 (14N2) | - |
| 2.340(4) f | ||||
| 1.256(9) g | 2.336(5) g | |||
| 2.336(5) g | ||||
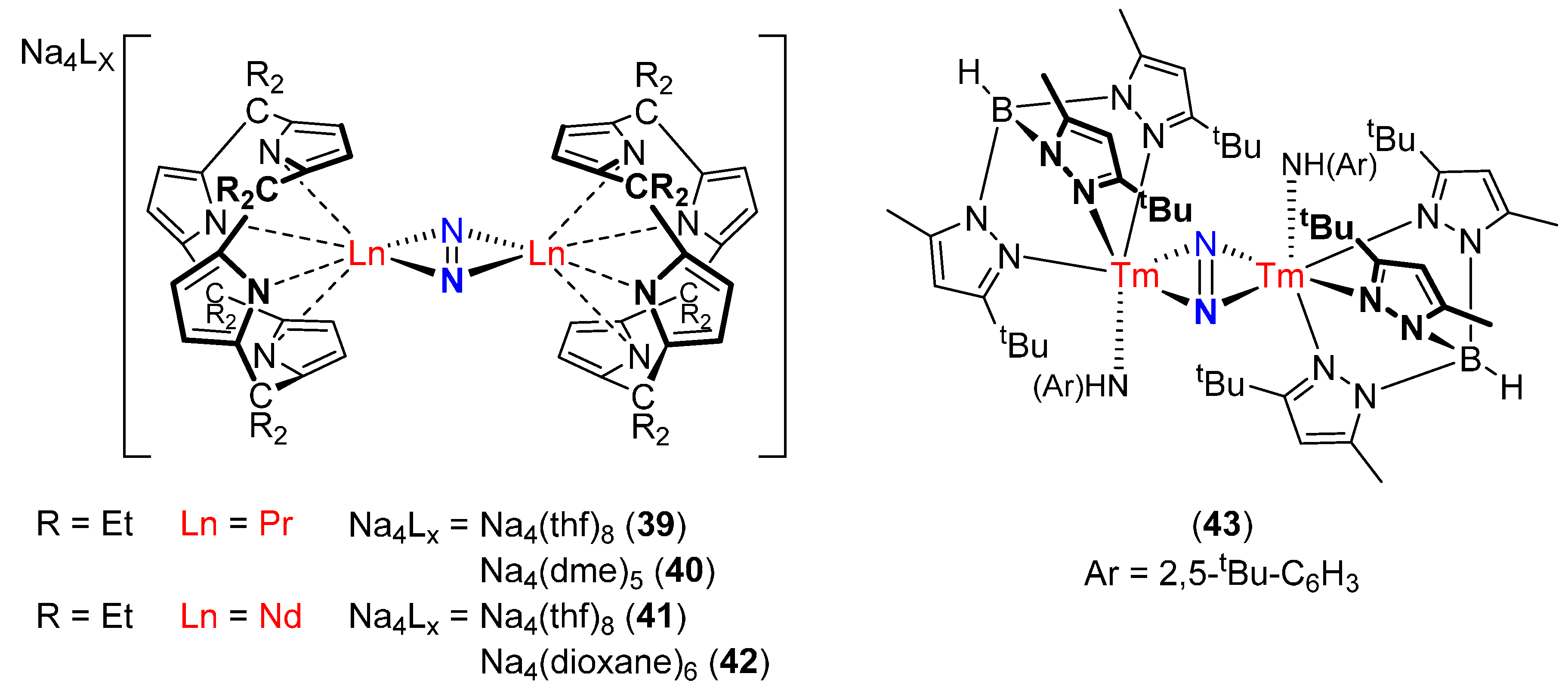
| [Na4(thf)8][(η5:η1:η5:η1-Et2calix[4]pyrrole)Pr]2(μ-η2:η2-N2) (39) [85] | - | - | - | - |
| [Na4(dme)5][(η5:η1:η5:η1-Et2calix[4]pyrrole)Pr]2(μ-η2:η2-N2) (40) [85] | 1.254(7) | 2.414(5) | - | - |
| 2.457(5) | ||||
| [Na4(thf)8][(η5:η1:η5:η1-Et2calix[4]pyrrole)Nd]2(μ-η2:η2-N2) (41) [85] | - | - | - | - |
| [Na4(dioxane)6][(η5:η1:η5:η1-Et2calix[4]pyrrole)Nd]2(μ-η2:η2-N2) (42) [85] | 1.234(8) | 2.511(4) | - | - |
| 2.508(4) | ||||
| [{HB(3-tBu-5-Me–pz)}Tm{NH(2,5-tBu–C6H3)}]2(μ-η2:η2-N2) (43) [86] | 1.215(10) | 2.274(8) | - | - |
| 2.286(9) | ||||
2.1.1. Cyclopentadienyl Ancillary Ligands



2.1.2. Amide Ancillary Ligands

2.1.3. Aryloxide Ancillary Ligands

2.1.4. Multidentate Ancillary Ligands

2.2. Complexes Containing a Formal N23− Ligand
2.2.1. Amide Ancillary Ligands
| Complex (#) [Reference] | N–N Bond Length (Å) | Ln–N (N2) Bond Lengths (Å) | Ln–N–N–Ln Torsion Angle (°) | N–N Frequency (cm−1) |
|---|---|---|---|---|
| N2 | 1.0975 [66] | - | - | 2331 [67] |
| [K(thf)6][{(Me3Si)2N}2Y(thf)]2(μ-η2:η2-N2) b (44) [93] | 1.401(6) | 2.194(3) | 0 | 1002 (14N2) (calculated) |
| 2.218(3) | ||||
| 1.401(6) a | 2.190(3) a | |||
| 2.213(3) a | ||||
| [K(thf)6][{(Me3Si)2N}2La(thf)]2(μ-η2:η2-N2) (45) [65] | - | - | 0 | - |
| [K(thf)6][{(Me3Si)2N}2Lu(thf)]2(μ-η2:η2-N2) b (46) [65] | 1.414(8) | 2.163(4) | 0 | 979 (14N2) |
| 2.180(4) | ||||
| [K][{(Me3Si)2N}2Y(thf)]2(μ3-η2:η2:η2-N2) b (47) [93] | 1.405(3) | 2.225(2) | 14.22 | 989 (14N2) |
| 2.242(2) | 956 (15N2) | |||
| [K][{(Me3Si)2N}2Gd(thf)]2(μ3-η2:η2:η2-N2) b (48) [97] | 1.395(3) | 2.248(2) | 13.64 | - |
| 2.274(2) | ||||
| [K][{(Me3Si)2N}2Tb(thf)]2(μ3-η2:η2:η2-N2) b (49) [97] | 1.401(3) | 2.235(2) | 16.12 | - |
| 2.260(2) | ||||
| [K][{(Me3Si)2N}2Dy(thf)]2(μ3-η2:η2:η2-N2) b (50) [97] | 1.404(5) | 2.229(4) | 15.27 | - |
| 2.242(4) | ||||
| [K(18c6)(thf)2][{(Me3Si)2N}2Y(thf)]2(μ-η2:η2-N2) b (51) [97] | 1.396(3) | 2.1909(17) | 0 | - |
| 2.2136(16) | ||||
| [K(18c6)(thf)2][{(Me3Si)2N}2Gd(thf)]2(μ-η2:η2-N2) b (52) [94] | 1.401(4) | 2.224(2) | 0 | - |
| 2.249(2) | ||||
| [K(18c6)(thf)2][{(Me3Si)2N}2Tb(thf)]2(μ-η2:η2-N2) b (53) [94] | 1.394(3) | 2.2056(15) | 0 | - |
| 2.2345(15) | ||||
| [K(18c6)(thf)2][{(Me3Si)2N}2Dy(thf)]2(μ-η2:η2-N2) b (54) [94] | 1.393(7) | 2.199(4) | 0 | - |
| 2.213(4) | ||||
| [K(18c6)(thf)2][{(Me3Si)2N}2Ho(thf)]2(μ-η2:η2-N2) b (55) [95] | 1.404(4) | 2.188(2) | 0 | - |
| 2.210(2) | ||||
| [K(18c6)(thf)2][{(Me3Si)2N}2Er(thf)]2(μ-η2:η2-N2) b (56) [65] | 1.409(4) | 2.178(2) | 0 | - |
| 2.204(2) | ||||
| [Na(thf)6][{(Me3Si)2N}2Y(thf)]2(μ-η2:η2-N2) b (57) [65] | 1.393(7) | 2.199(4) | 0 | - |
| 2.213(4) | ||||
| [Na(thf)6][{(Me3Si)2N}2Er(thf)]2(μ-η2:η2-N2) b (58) [65] | 1.403(4) | 2.1817(19) 2.2019(19) | 0 | - |
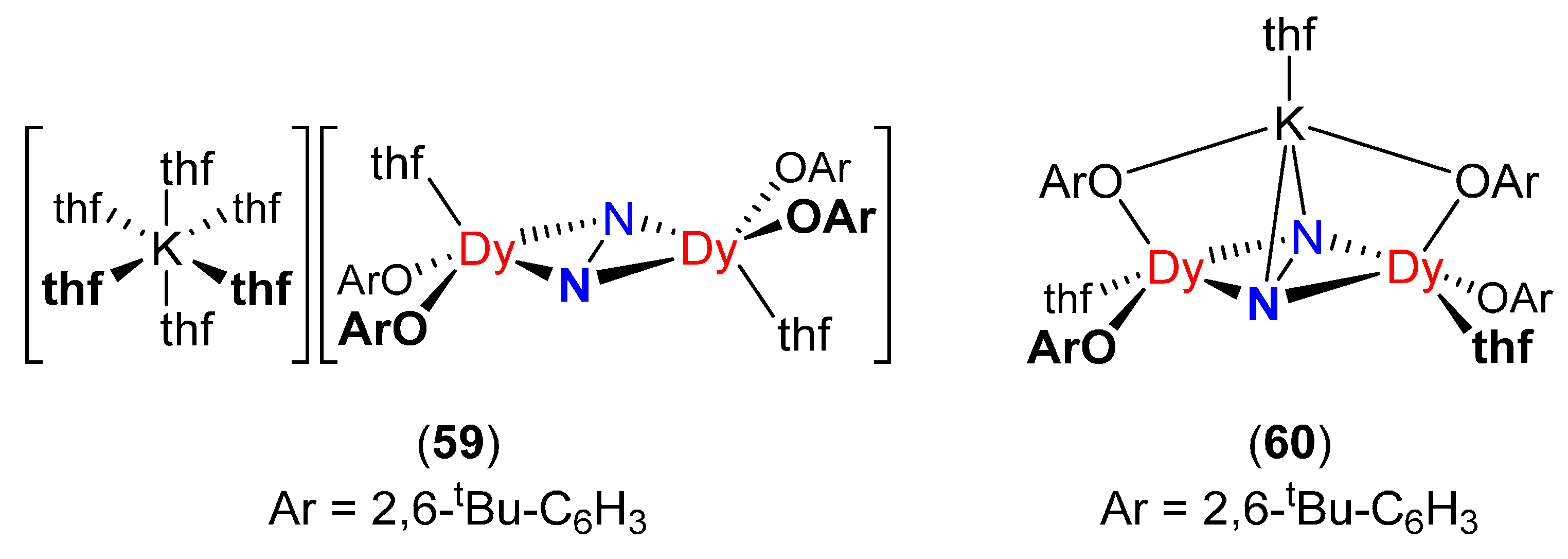
| [K(thf)6][(2,6-tBu–C6H3O)2Dy(thf)]2(μ-η2:η2-N2) (59) [93] | 1.396(7) | 2.197(3) | 0 | 962 (14N2) |
| 2.203(4) | ||||
| [K(thf)][(2,6-tBu–C6H3O)2Dy(thf)]2(μ3-η2:η2:η2-N2) (60) [93] | 1.402(7) | 2.235(5) | 6.59 | - |
| 2.209(5) |

2.2.2. Aryloxide Ancillary Ligands

2.3. Complexes Containing a Formally N24− Ligand
| Complex (#) [Reference] | N–N Bond Length (Å) | Ln–N (N2) Bond Lengths (Side-on) (Å) | Ln–N (N2) Bond Lengths (End-on) (Å) |
|---|---|---|---|
| N2 | 1.0975 [66] | - | - |
| [Li(thf)2]2[(Et2calix[4]pyrrole)Sm]2(N2Li4) (61) [101] | 1.525(4) | 2.357(2) | - |
| 2.342(2) | |||
| [{Ph2C(C4H3N)2}Sm(thf)]4(μ4-η1:η1:η2:η2-N2) (62) [102] | 1.412(17) | 2.327(3) | 2.177(8) |
| 2.327(3) | 2.177(8) | ||
| [{CyC(C4H3N)2}Sm(thf)]4(μ4-η1:η1:η2:η2-N2) (63) [103] | 1.392(16) | 2.339(3) | 2.160(8) |
| 2.339(3) | 2.160(8) | ||
| [{Et2C(C4H3N)2}Sm(thf)]4(μ4-η1:η1:η2:η2-N2) (64) [104] | 1.415(4) | 2.328(3) | 2.145(3) |
| 2.342(3) | |||
| [{Ph(Me)C(C4H3N)2}Sm(dme)]4(μ4-η1:η1:η2:η2-N2) (65) [104] | 1.42(2) | 2.316(13) | 2.149(11) |
| 2.316(12) | |||
| [Na(thf)]2[{CyC(C4H3N)2}Sm(thf)]4(μ6-η1:η1:η1:η1:η2:η2-N2) (66) [103] | 1.371(16) | 2.332(11) | 2.178(10) |
| 2.324(11) | |||
| [{Li(thf)}3(μ3-Cl)][(Cycalix[4]pyrrole)Sm]2(μ-η2:η2-N2) (67) [105] | 1.08(3) | 2.880(18) | - |
| 2.974(18) | |||
| [(Li(thf)2][(Cycalix[4]pyrrole)2Sm3Li2](μ5-η1:η1:η2:η2:η2-N2) (68) [105] | 1.502(5) | 2.249(4) (Sm(1)–N(1)) | - |
| 2.253(4) (Sm(1)–N(1)) | |||
| 2.355(4) (Sm(2)–N(1)) | |||
| 2.370(4) (Sm(2)–N(1)) | |||
| 2.398(4) (Sm(3)–N(1)) | |||
| 2.376(4) (Sm(3)–N(1)) |

3. Dinitrogen Activation by Actinide Complexes
3.1. Complexes Containing an Activated N2 Ligand

| Complex (#) [Reference] | Stability | N–N Bond Length (Å) | N–N Frequency (cm−1) | 14/15N-NMR Spectroscopy (ppm) a |
|---|---|---|---|---|
| N2 | - | 1.0975 [66] | 2331 [67] | −75 [68] |
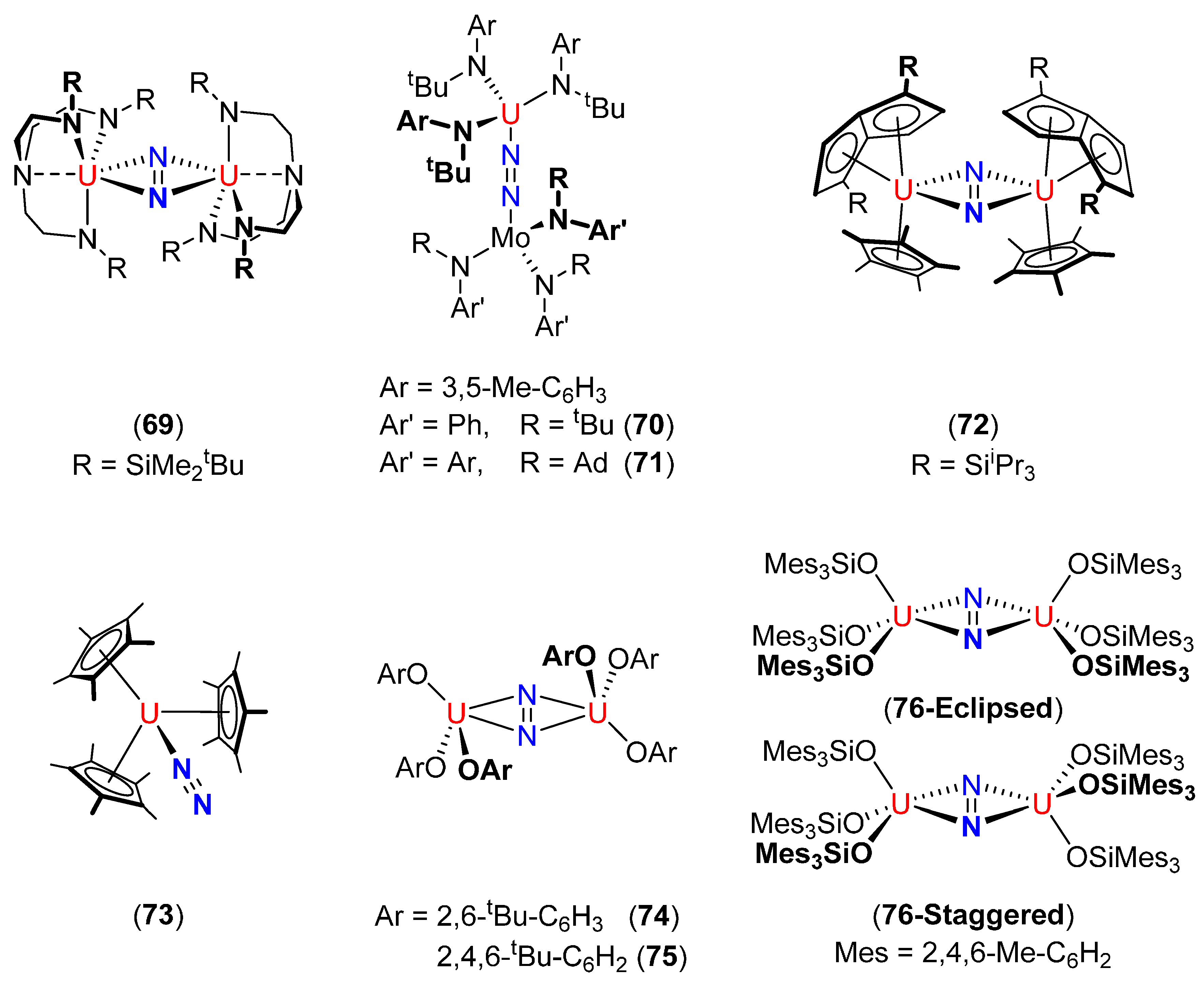
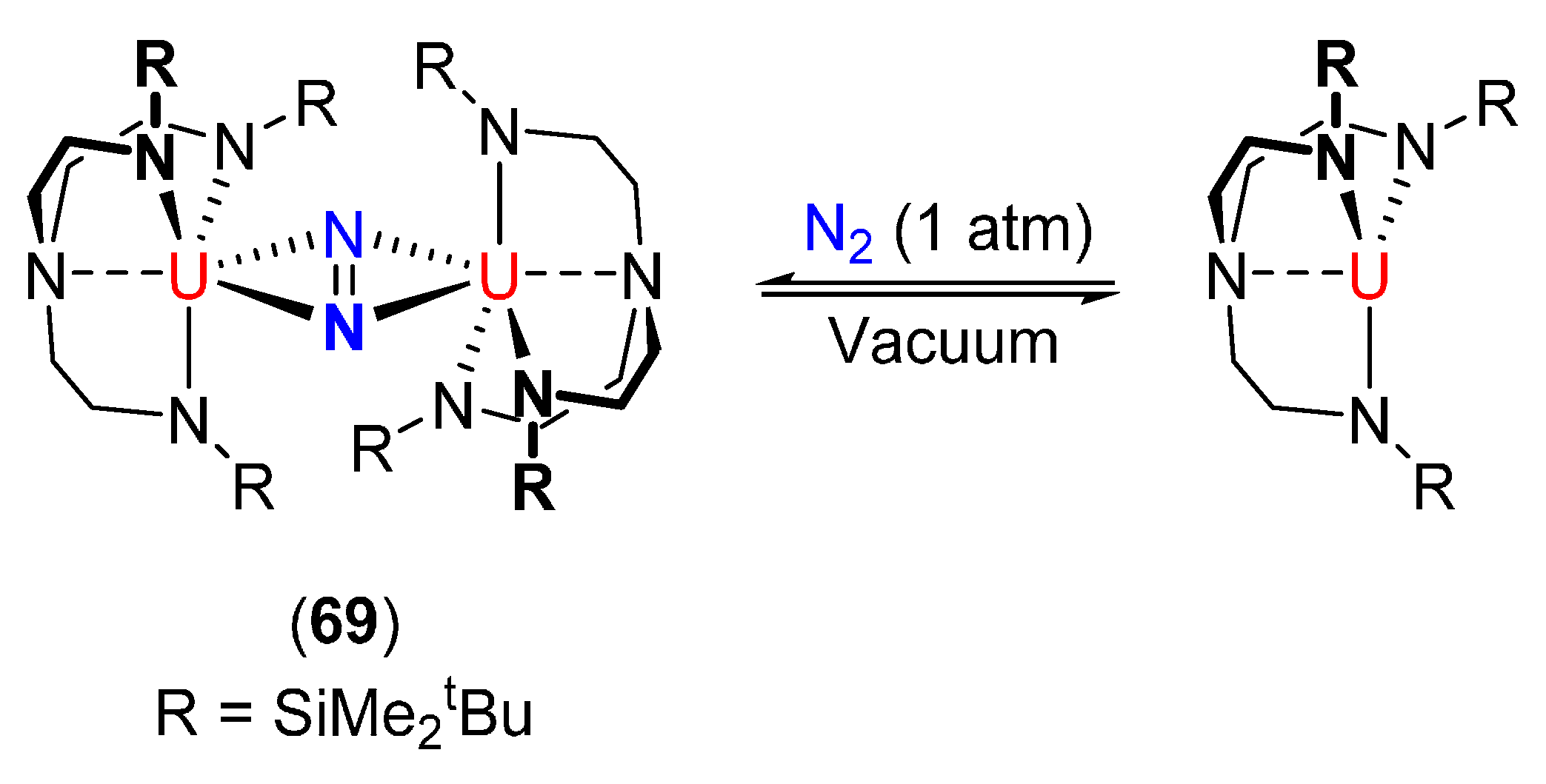
| [{N(CH2CH2NSiMe2tBu)3}U]2(μ-η2:η2-N2) (69) [109] | Stable under N2 (1 atm) | 1.109(7) | - | - |
| N2 dissociation in vacuo | ||||
| {Ph(tBu)N}3Mo(μ2-η1:η1-N2)U{N(tBu)(3,5-Me–C6H3)}3 (70) [110] | Stable in vacuo at 25 °C | 1.232(11) | - (14N2) | - |
| “Thermally stable” | 1547 (15N2) | |||
| {(3,5-Me-C6H3)(Ad)N}3Mo(μ2-η1:η1-N2)U{N(tBu)(3,5-Me–C6H3)}3 (71) [110] | Stable in vacuo at 25 °C | 1.23(2) | 1568 (14N2) | - |
| “Thermally stable” | 1527 (15N2) | |||
| [(η5-C5Me5)(η8-1,4-SiiPr3–C8H4)U]2(μ-η2:η2-N2) (72) [111] | 75% conversion to 72 at 50 psi N2 | 1.232(10) | - | - |
| N2 dissociation in vacuo, in solution and solid state | ||||
| (η5-C5Me5)3U(η1-N2) (73) [112] | Crystallisation at 80 psi N2 | 1.120(14) | 2207 (14N2) | - |
| N2 dissociation in vacuo or in solution under N2 (1 atm) | 2134 (15N2) | |||
| [(2,6-tBu–C6H3O)3U]2(μ-η2:η2-N2) (74) [88] | N2 dissociation in vacuo and in solution at 25 °C | 1.163(19) | - | - |
| 1.204 (17) | ||||
| 1.201(19) | ||||
| [(2,4,6-tBu–C6H2O)3U]2(μ-η2:η2-N2) (75) [88] | Stable in vacuo at 25 °C | 1.236(5) | 1451 (14N2) | - |
| N2 dissociation at 80 °C in solution | 1404 (15N2) | |||
| [{(Mes)3SiO}3U]2(μ-η2:η2-N2) (76) [87] | Stable in vacuo at 25 °C | 1.124(12) (eclipsed) | 1437 (14N2) | 4213.5 |
| Slowly forms U{OSi(Mes)3}4 at 100 °C in solution | 1.080(11) (staggered) | 1372 (15N2) |

3.2. Complexes Resulting from N2 Cleavage


4. White Phosphorus Activation by Rare Earth Complexes


| Complex (#) [Reference] | Average P–P Bond Lengths (Å) | Average M–P Bond Length (Å) | 31P-NMR Spectroscopy (298 K) (ppm) a |
|---|---|---|---|
| P4 | 2.21 [121] | - | −488 to −527 [122] |
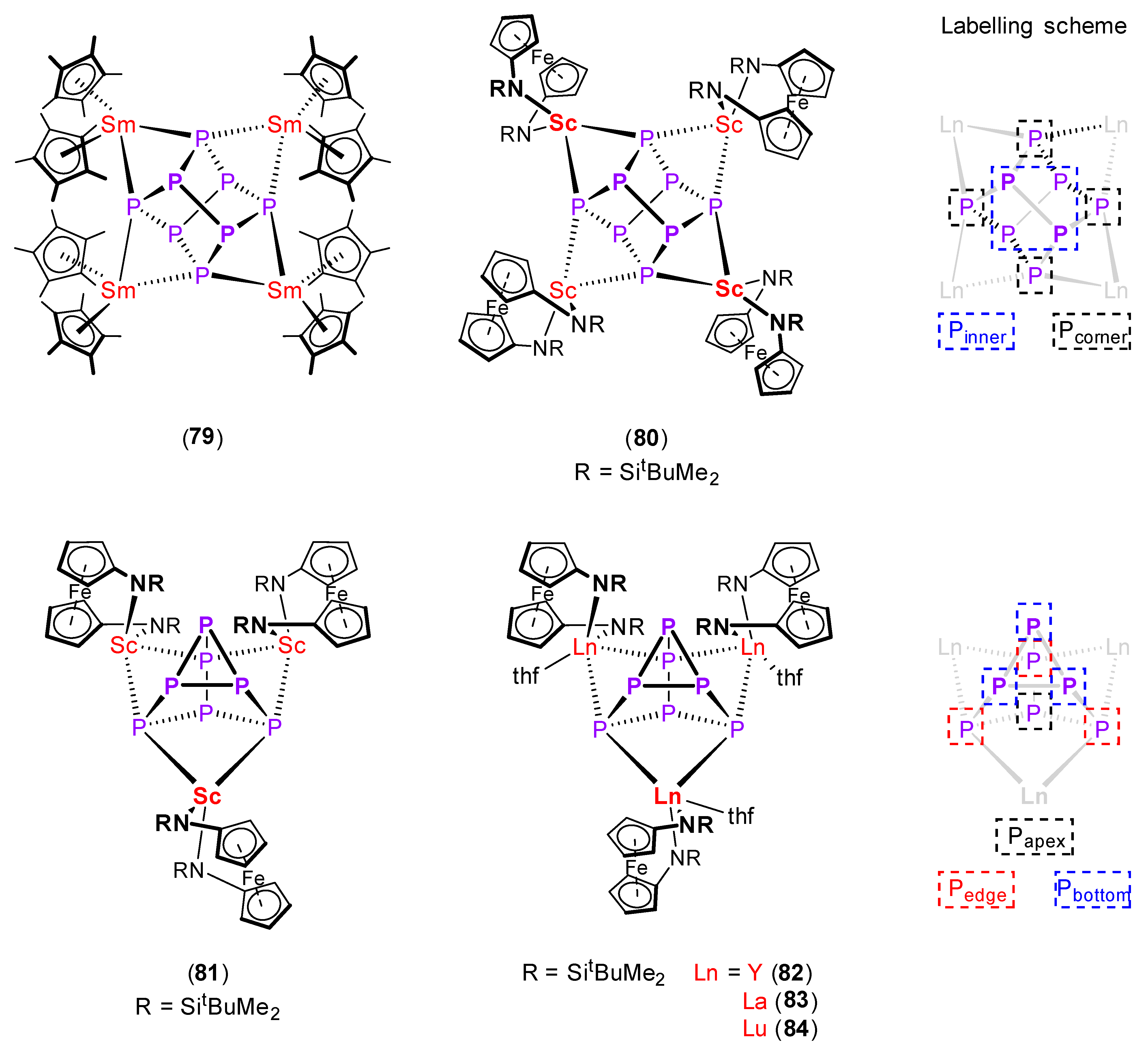
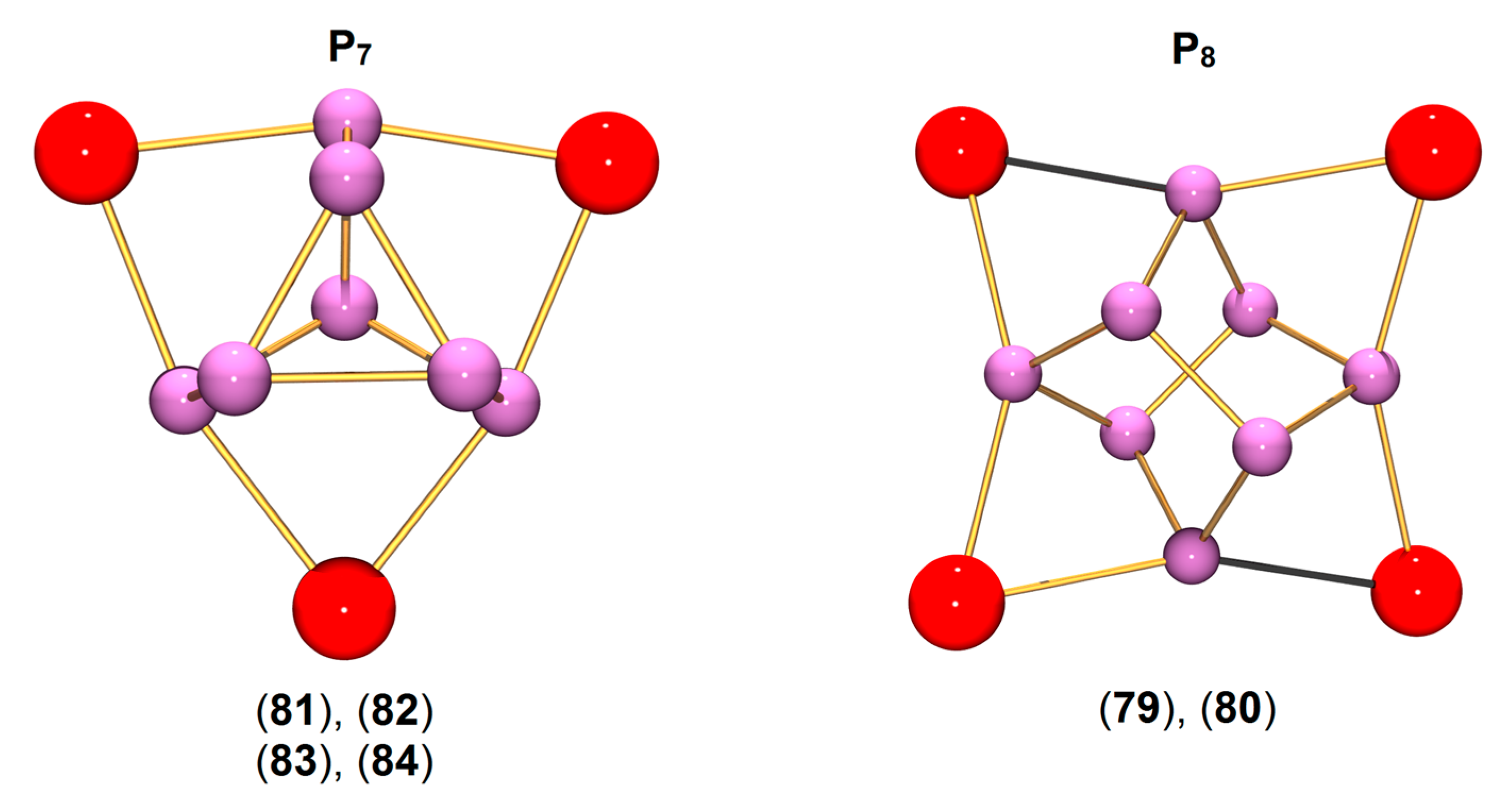
| [(η5-C5Me5)2Sm]4(μ4-η2:η2:η2:η2-P8) (79) [123] | 2.195 (Pcorner–Pinner) | 3.047 | - |
| 2.291 (Pinner–Pinner) | |||
| [{Fe(1-NSitBuMe2–C5H4)2}Sc]4(μ4-η2:η2:η2:η2-P8) (80) [124] | 2.204 (Pcorner–Pinner) | 2.768 | +45.7 |
| 2.308 (Pinner–Pinner) | +96.2 | ||
| [{Fe(1-NSitBuMe2–C5H4)2}Sc]3(μ3-η2:η2:η2-P7) (81) [124] | 2.229 (Pbottom–Pbottom) | 2.750 | +23.1 |
| 2.197 (Pedge–Pbottom) | −118.9 | ||
| 2.201 (Papex–Pedge) | −131.4 | ||
| [{Fe(1-NSitBuMe2–C5H4)2}Y(thf)]3(μ3-η2:η2:η2-P7) (82) [124] | 2.238 (Pbottom–Pbottom) | 2.950 | −21.1 |
| 2.176 (Pedge–Pbottom) | −82.4 | ||
| 2.188 (Papex–Pedge) | −130.3 | ||
| [{Fe(1-NSitBuMe2–C5H4)2}La(thf)]3(μ3-η2:η2:η2-P7) (83) [125] | 2.258 (Pbottom–Pbottom) | 3.120 | −75 |
| 2.161 (Pedge–Pbottom) | |||
| 2.191 (Papex–Pedge) | |||
| [{Fe(1-NSitBuMe2–C5H4)2}Lu(thf)]3(μ3-η2:η2:η2-P7) (84) [125] | 2.233 (Pbottom–Pbottom) | 2.893 | +0.8 |
| 2.181 (Pedge–Pbottom) | −96.8 | ||
| 2.183 (Papex–Pedge) | −133.3 |

5. White Phosphorus Activation by Actinide Complexes
| Complex (#) [Reference] | Average P–P Bond Lengths (Å) | Average An–P Bond Length (Å) | 31P-NMR Spectroscopy (ppm) a |
|---|---|---|---|
| P4 | 2.21 [121] | - | −488 to −527 [122] |
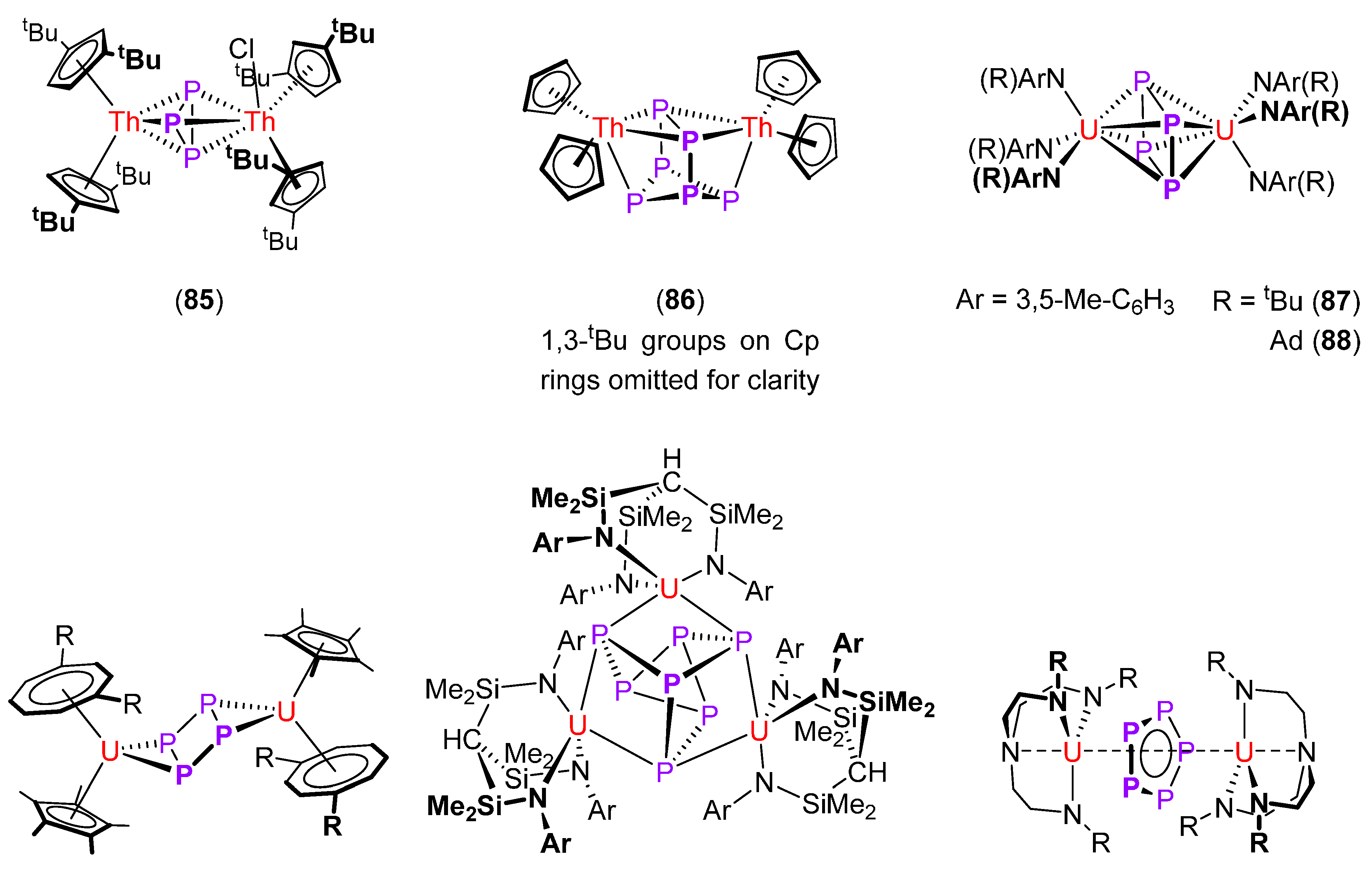
| [(η5-1,3-tBu–C5H3)2Th](μ-η3:η3-cyclo-P3)[(η5-1,3-tBu–C5H3)2ThCl] (85) [128] | 2.185 | 2.913 | −75.7 (293 K) |
| [(η5-1,3-tBu–C5H3)2Th]2(μ-η3:η3-P6) (86) [128] | 2.234 | 2.904 (Th-η2-P) | +125.4 (293 K) |
| 2.844 (Th-η1-P) | −41.9 (293 K) | ||
| [{(3,5-Me–C6H3)(tBu)N}3U](μ-η4:η4-cyclo-P4) (87) [129] | 2.160 | 3.127 | +794 |
| [{(3,5-Me–C6H3)(Ad)N}3U](μ-η4:η4-cyclo-P4) (88) [129] | 2.159 | 3.124 | +803 |
| [(η5-C5Me5)(η8-1,4-SiiPr3–C8H6)U]2(μ-η2:η2-cyclo-P4) (89) [130] | 2.150 | 2.977 | +718 |
| [HC(SiMe2N-4-Me–C6H4)3U]3(μ3-η2:η2:η2-P7) (90) [130] | 2.249 (Pbottom–Pbottom) | 2.990 | - |
| 2.187 (Pedge–Pbottom) | |||
| 2.209 (Papex–Pedge) | |||
| [{N(CH2CH2NSiiPr3)3}U]2(μ-η5:η5-cyclo-P5) (91) [131] | 2.006 | 3.280 | - |


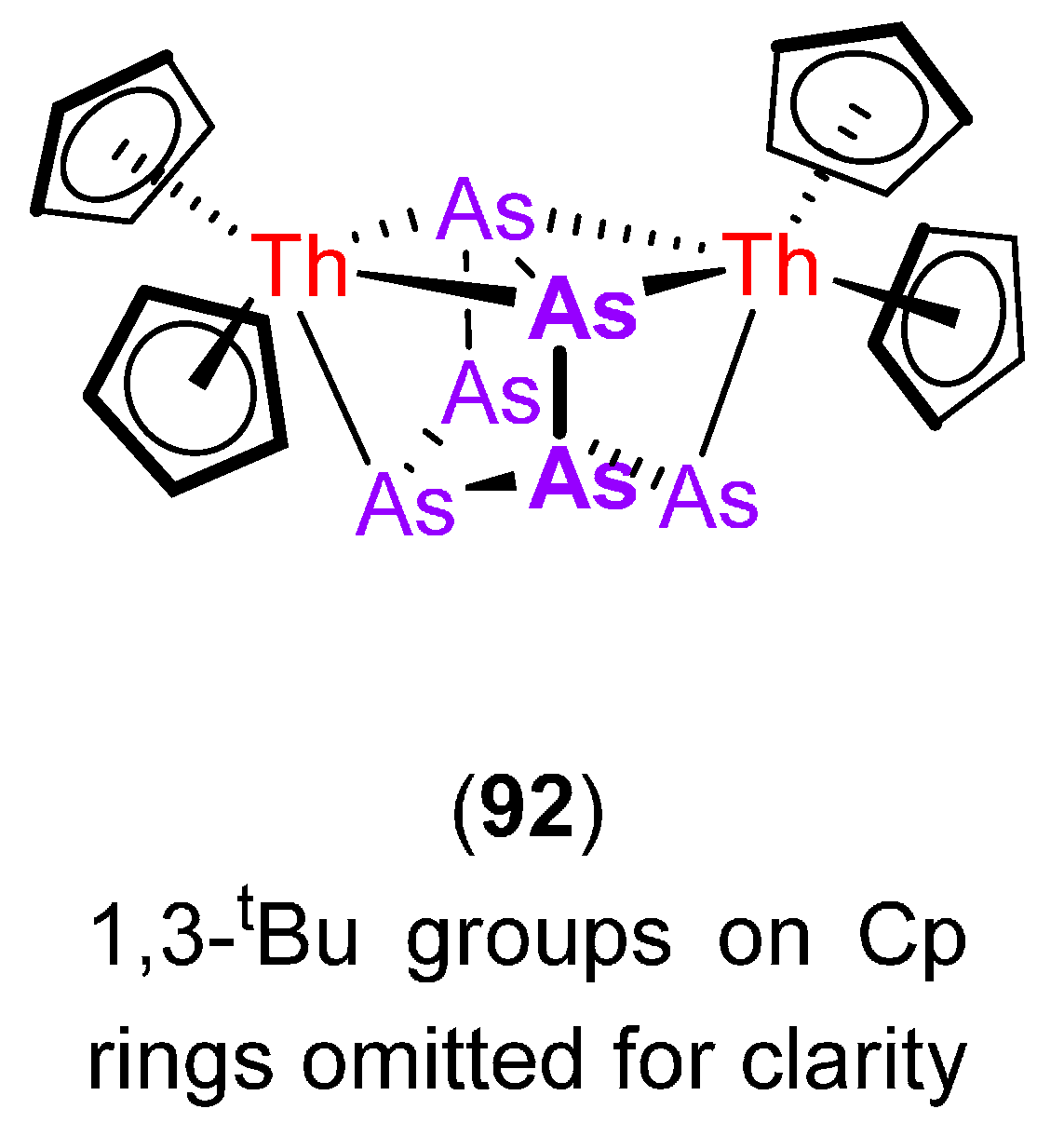
6. Arsenic, Antimony and Bismuth Activation by Rare Earth and Actinide Complexes

7. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Kaltsoyannis, N. Does covalency increase or decrease across the actinide series? Implications for minor actinide partitioning. Inorg. Chem. 2013, 52, 3407–3413. [Google Scholar] [CrossRef] [PubMed]
- Breunig, H.J.; Rösler, R.; Lork, E. Complexes with Sb2 and cyclo-Sb3 ligands: The tetrahedranes [{C5H5(CO)2Mo}2Sb2], [C5H5(CO)2MoSb3], and [C5Me5(CO)2MoSb3]. Angew. Chem. Int. Ed. 1997, 36, 2819–2821. [Google Scholar] [CrossRef]
- Evans, W.J.; Gonzales, S.L.; Ziller, J.W. The utility of (C5Me5)2Sm in isolating crystallographically characterizable zintl ions. X-ray crystal structure of a samarium complex of (Sb3)3−. J. Chem. Soc. Chem. Commun. 1992, 1138–1139. [Google Scholar] [CrossRef]
- Evans, W.J.; Gonzales, S.L.; Ziller, J.W. Organosamarium-mediated synthesis of bismuth–bismuth bonds: X-ray crystal structure of the first dibismuth complex containing a planar M2(μ-η2:η2-Bi2) unit. J. Am. Chem. Soc. 1991, 113, 9880–9882. [Google Scholar] [CrossRef]
- Hoffman, B.M.; Dean, D.R.; Seefeldt, L.C. Climbing nitrogenase: Toward a mechanism of enzymatic nitrogen fixation. Acc. Chem. Res. 2009, 42, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ribbe, M.W. Decoding the nitrogenase mechanism: The homologue approach. Acc. Chem. Res. 2010, 43, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Baerends, E.J.; Biczysko, M.; Bligaard, T.; Christensen, C.H.; Clary, D.C.; Dahl, S.; van Harrevelt, R.; Honkala, K.; Jonsson, H.; et al. Predicting catalysis: Understanding ammonia synthesis from first-principles calculations. J. Phys. Chem. B 2006, 110, 17719–17735. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Reduction of dinitrogen. Proc. Natl. Acad. Sci. USA 2006, 103, 17087. [Google Scholar] [CrossRef] [PubMed]
- Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch and the Transformation of World Food Production; Massachusetts Institute of Technology Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. GeoSci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- U.S. Geographical Survey, Mineral Commodity Surveys; U.S. Department of the Interior: Washington, DC, USA, 2015.
- Corbridge, D. Phosphorus: An Outline of its Chemistry, Biochemistry, and Technology, 5th ed.; Elsevier: New York, NY, USA, 1994. [Google Scholar]
- Quin, L.D. A Guide to Organophosphorus Chemistry; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Engel, R. Synthesis of Carbon Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Withers, P.J.A.; Elser, J.J.; Hilton, J.; Ohtake, H.; Schipper, W.J.; van Dijk, K.C. Greening the global phosphorus cycle: How green chemistry can help achieve planetary P sustainability. Green Chem. 2015, 17, 2087–2099. [Google Scholar] [CrossRef]
- Liu, H. Catalytic Ammonia Synthesis; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Schlögl, R. Catalytic synthesis of ammonia—A “Never-Ending Story”? Angew. Chem. Int. Ed. 2003, 42, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Leigh, G.J. Haber–Bosch and Other Industrial Processes. In Catalysts for Nitrogen Fixation; Smith, B., Richards, R., Newton, W., Eds.; Springer: Houten, The Netherlands, 2004; Volume 1, pp. 33–54. [Google Scholar]
- Studt, F.; Tuczek, F. Theoretical, spectroscopic, and mechanistic studies on transition-metal dinitrogen complexes: Implications to reactivity and relevance to the nitrogenase problem. J. Comput. Chem. 2006, 27, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- MacKay, B.A.; Fryzuk, M.D. Dinitrogen coordination chemistry: On the biomimetic borderlands. Chem. Rev. 2004, 104, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Barrière, F. Model Complexes of the Active Site of Nitrogenases: Recent Advances. In Bioinspired Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 225–248. [Google Scholar]
- Rolff, M.; Tuczek, F. Nitrogenase and Nitrogen Activation. In Comprehensive Inorganic Chemistry II, 2nd ed.; Poeppelmeier, J.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 593–618. [Google Scholar]
- Ribbe, M.W. Nitrogen Fixation: Methods and Protocols; Humana Press: New York, NY, USA, 2011. [Google Scholar]
- Barrière, F. Modeling of the molybdenum center in the nitrogenase FeMo-cofactor. Coord. Chem. Rev. 2003, 236, 71–89. [Google Scholar] [CrossRef]
- Smith, B.E.; Durrant, M.C.; Fairhurst, S.A.; Gormal, C.A.; Grönberg, K.L.C.; Henderson, R.A.; Ibrahim, S.K.; le Gall, T.; Pickett, C.J. Exploring the reactivity of the isolated iron-molybdenum cofactor of nitrogenase. Coord. Chem. Rev. 1999, 185–186, 669–687. [Google Scholar] [CrossRef]
- Rehder, D. Vanadium nitrogenase. J. Inorg. Biochem. 2000, 80, 133–136. [Google Scholar] [CrossRef]
- MacLachlan, E.A.; Fryzuk, M.D. Synthesis and reactivity of side-on-bound dinitrogen metal complexes. Organometallics 2006, 25, 1530–1543. [Google Scholar] [CrossRef]
- Fryzuk, M.D. Side-on end-on bound dinitrogen: An activated bonding mode that facilitates functionalizing molecular nitrogen. Acc. Chem. Res. 2009, 42, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Poveda, A.; Perilla, I.C.; Pérez, C.R. Some considerations about coordination compounds with end-on dinitrogen. J. Coord. Chem. 2001, 54, 427–440. [Google Scholar] [CrossRef]
- Gambarotta, S.; Scott, J. Multimetallic cooperative activation of N2. Angew. Chem. Int. Ed. 2004, 43, 5298–5308. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, S.F.; Holland, P.L. Dinitrogen binding and cleavage by multinuclear iron complexes. Acc. Chem. Res. 2015, 48, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, J.; Munha, R.F.; Fryzuk, M.D. The hydride route to the preparation of dinitrogen complexes. Chem. Commun. 2010, 46, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.-P.; Quadrelli, E.A. Mechanistic aspects of dinitrogen cleavage and hydrogenation to produce ammonia in catalysis and organometallic chemistry: Relevance of metal hydride bonds and dihydrogen. Chem. Soc. Rev. 2014, 43, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Sivasankar, C.; Baskaran, S.; Tamizmani, M.; Ramakrishna, K. Lessons learned and lessons to be learned for developing homogeneous transition metal complexes catalyzed reduction of N2 to ammonia. J. Organomet. Chem. 2014, 752, 44–58. [Google Scholar] [CrossRef]
- Tanabe, Y.; Nishibayashi, Y. Developing more sustainable processes for ammonia synthesis. Coord. Chem. Rev. 2013, 257, 2551–2564. [Google Scholar] [CrossRef]
- Van der Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef] [PubMed]
- Rebreyend, C.; de Bruin, B. Photolytic N2 splitting: A road to sustainable NH3 production? Angew. Chem. Int. Ed. 2015, 54, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Himmel, H.-J.; Reiher, M. Intrinsic dinitrogen activation at bare metal atoms. Angew. Chem. Int. Ed. 2006, 45, 6264–6288. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.; Taoufik, M.; Quadrelli, E.A. Ammonia and dinitrogen activation by surface organometallic chemistry on silica-grafted tantalum hydrides. Eur. J. Inorg. Chem. 2011, 2011, 1349–1359. [Google Scholar] [CrossRef]
- Ohki, Y.; Fryzuk, M.D. Dinitrogen activation by group 4 metal complexes. Angew. Chem. Int. Ed. 2007, 46, 3180–3183. [Google Scholar] [CrossRef] [PubMed]
- Chirik, P.J. Dinitrogen functionalization with bis(cyclopentadienyl) complexes of zirconium and hafnium. Dalton Trans. 2007, 1, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Green, J.C.; Himmel, H.-J. Dinitrogen fixation and activation by Ti and Zr atoms, clusters and complexes. New J. Chem. 2006, 30, 1253–1262. [Google Scholar] [CrossRef]
- Crossland, J.L.; Tyler, D.R. Iron–dinitrogen coordination chemistry: Dinitrogen activation and reactivity. Coord. Chem. Rev. 2010, 254, 1883–1894. [Google Scholar] [CrossRef]
- Hazari, N. Homogeneous iron complexes for the conversion of dinitrogen into ammonia and hydrazine. Chem. Soc. Rev. 2010, 39, 4044–4056. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Acc. Chem. Res. 2005, 38, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Nishibayashi, Y. Molybdenum-catalyzed reduction of molecular dinitrogen into ammonia under ambient reaction conditions. Comptes Rendus Chim. 2015, 18, 776–784. [Google Scholar] [CrossRef]
- Schrock, R.R. Catalytic Reduction of Dinitrogen to Ammonia by Molybdenum. In Catalysis without Precious Metals; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 25–50. [Google Scholar]
- Khoenkhoen, N.; de Bruin, B.; Reek, J.N.H.; Dzik, W.I. Reactivity of dinitrogen bound to mid- and late-transition-metal centers. Eur. J. Inorg. Chem. 2015, 2015, 567–598. [Google Scholar] [CrossRef]
- Evans, W.J.; Lee, D.S. Early developments in lanthanide-based dinitrogen reduction chemistry. Can. J. Chem. 2005, 83, 375–384. [Google Scholar] [CrossRef]
- Gardiner, M.G.; Stringer, D.N. Dinitrogen and related chemistry of the lanthanides: A review of the reductive capture of dinitrogen, as well as mono- and di-aza containing ligand chemistry of relevance to known and postulated metal mediated dinitrogen derivatives. Materials 2010, 3, 841–862. [Google Scholar] [CrossRef]
- Gardner, B.M.; Liddle, S.T. Small-molecule activation at uranium(III). Eur. J. Inorg. Chem. 2013, 2013, 3753–3770. [Google Scholar] [CrossRef]
- Liddle, S.T. The renaissance of non-aqueous uranium chemistry. Angew. Chem. Int. Ed. 2015, 54, 8604–8641. [Google Scholar] [CrossRef] [PubMed]
- Scherer, O.J. Complexes with substituent-free acyclic and cyclic phosphorus, arsenic, antimony, and bismuth ligands. Angew. Chem. Int. Ed. 1990, 29, 1104–1122. [Google Scholar] [CrossRef]
- Scherer, O.J. Pn and Asn ligands: A novel chapter in the chemistry of phosphorus and arsenic. Acc. Chem. Res. 1999, 32, 751–762. [Google Scholar] [CrossRef]
- Peruzzini, M.; Gonsalvi, L.; Romerosa, A. Coordination chemistry and functionalization of white phosphorus via transition metal complexes. Chem. Soc. Rev. 2005, 34, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Vaira, M.D.; Sacconi, L. Transition metal complexes with cyclo-triphosphorus (η3-P3) and tetrahedro-tetraphosphorus (η1-P4) ligands. Angew. Chem. Int. Ed. 1982, 21, 330–342. [Google Scholar] [CrossRef]
- Figueroa, J.S.; Cummins, C.C. A niobaziridine hydride system for white phosphorus or dinitrogen activation and N- or P-atom transfer. Dalton Trans. 2006, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Cossairt, B.M.; Piro, N.A.; Cummins, C.C. Early-transition-metal-mediated activation and transformation of white phosphorus. Chem. Rev. 2010, 110, 4164–4177. [Google Scholar] [CrossRef] [PubMed]
- Caporali, M.; Gonsalvi, L.; Rossin, A.; Peruzzini, M. P4 activation by late-transition metal complexes. Chem. Rev. 2010, 110, 4178–4235. [Google Scholar] [CrossRef] [PubMed]
- Giffin, N.A.; Masuda, J.D. Reactivity of white phosphorus with compounds of the p-block. Coord. Chem. Rev. 2011, 255, 1342–1359. [Google Scholar] [CrossRef]
- Balázs, G.; Seitz, A.; Scheer, M. Activation of White Phosphorus (P4) by Main Group Elements and Compounds. In Comprehensive Inorganic Chemistry II, 2nd ed.; Poeppelmeier, J.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1105–1132. [Google Scholar]
- Khan, S.; Sen, S.S.; Roesky, H.W. Activation of phosphorus by group 14 elements in low oxidation states. Chem. Commun. 2012, 48, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.P.; Balázs, G.; Scheer, M. Low-coordinate E1 ligand complexes of group 15 elements—A developing area. Coord. Chem. Rev. 2006, 250, 1178–1195. [Google Scholar] [CrossRef]
- Scheer, M.; Balázs, G.; Seitz, A. P4 activation by main group elements and compounds. Chem. Rev. 2010, 110, 4236–4256. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Bates, J.E.; Lorenz, S.E.; Lee, D.S.; Rego, D.B.; Ziller, J.W.; Furche, F.; Evans, W.J. (N2)3− radical chemistry via trivalent lanthanide salt/alkali metal reduction of dinitrogen: New syntheses and examples of (N2)2− and (N2)3− complexes and density functional theory comparisons of closed shell Sc3+, Y3+, and Lu3+ versus 4f9 Dy3+. Inorg. Chem. 2011, 50, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Fryzuk, M.D.; Johnson, S.A. The continuing story of dinitrogen activation. Coord. Chem. Rev. 2000, 200–202, 379–409. [Google Scholar] [CrossRef]
- Jaroschik, F.; Momin, A.; Nief, F.; LeGoff, X.-F.; Deacon, G.B.; Junk, P.C. Dinitrogen reduction and C–H activation by the divalent organoneodymium complex [(C5H2tBu3)2Nd(μ-I)K([18]crown-6)]. Angew. Chem. Int. Ed. 2009, 48, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Rego, D.B.; Ziller, J.W. Synthesis, structure, and 15N-NMR studies of paramagnetic lanthanide complexes obtained by reduction of dinitrogen. Inorg. Chem. 2006, 45, 10790–10798. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Lorenz, S.E.; Fang, M.; Furche, F.; Meyer, G.; Ziller, J.W.; Evans, W.J. Synthesis, structure, and density functional theory analysis of a scandium dinitrogen complex, [(C5Me4H)2Sc]2(μ-η2:η2-N2). J. Am. Chem. Soc. 2010, 132, 11151–11158. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Siladke, N.A.; Ziller, J.W.; Evans, W.J. Scandium and yttrium metallocene borohydride complexes: Comparisons of (BH4)1− vs. (BPh4)1− coordination and reactivity. Dalton Trans. 2012, 41, 9659–9666. [Google Scholar] [CrossRef] [PubMed]
- Schmiege, B.M.; Ziller, J.W.; Evans, W.J. Reduction of dinitrogen with an yttrium metallocene hydride precursor, [(C5Me5)2YH]2. Inorg. Chem. 2010, 49, 10506–10511. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Ulibarri, T.A.; Ziller, J.W. Isolation and X-ray crystal structure of the first dinitrogen complex of an f-element metal, [(C5Me5)2Sm]2N2. J. Am. Chem. Soc. 1988, 110, 6877–6879. [Google Scholar] [CrossRef]
- Fieser, M.E.; Bates, J.E.; Ziller, J.W.; Furche, F.; Evans, W.J. Dinitrogen reduction via photochemical activation of heteroleptic tris(cyclopentadienyl) rare-earth complexes. J. Am. Chem. Soc. 2013, 135, 3804–3807. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Allen, N.T.; Ziller, J.W. Expanding divalent organolanthanide chemistry: The first organothulium(II) complex and the in situ organodysprosium(II) reduction of dinitrogen. Angew. Chem. Int. Ed. 2002, 41, 359–361. [Google Scholar] [CrossRef]
- Evans, W.J.; Allen, N.T.; Ziller, J.W. Facile dinitrogen reduction via organometallic Tm(II) chemistry. J. Am. Chem. Soc. 2001, 123, 7927–7928. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.J.; Fieser, M.E.; Ziller, J.W.; Evans, W.J. (C5Me4H)1−-based reduction of dinitrogen by the mixed ligand tris(polyalkylcyclopentadienyl) lutetium and yttrium complexes, (C5Me5)3−x(C5Me4H)xLn. Chem. Sci. 2011, 2, 1992–1996. [Google Scholar] [CrossRef]
- Lorenz, S.E.; Schmiege, B.M.; Lee, D.S.; Ziller, J.W.; Evans, W.J. Synthesis and reactivity of bis(tetramethylcyclopentadienyl) yttrium metallocenes including the reduction of Me3SiN3 to [(Me3Si)2N]− with [(C5Me4H)2Y(THF)]2(μ-η2:η2-N2). Inorg. Chem. 2010, 49, 6655–6663. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.R.; Ziller, J.W.; Evans, W.J. Synthesis of a crystalline molecular complex of Y2+, [(18-crown-6)K][(C5H4SiMe3)3Y]. J. Am. Chem. Soc. 2011, 133, 15914–15917. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Lee, D.S.; Lie, C.; Ziller, J.W. Expanding the LnZ3/alkali-metal reduction system to organometallic and heteroleptic precursors: Formation of dinitrogen derivatives of lanthanum. Angew. Chem. Int. Ed. 2004, 43, 5517–5519. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Lee, D.S.; Johnston, M.A.; Ziller, J.W. The elusive (C5Me4H)3Lu: Its synthesis and LnZ3/K/N2 reactivity. Organometallics 2005, 24, 6393–6397. [Google Scholar] [CrossRef]
- Evans, W.J.; Lee, D.S.; Rego, D.B.; Perotti, J.M.; Kozimor, S.A.; Moore, E.K.; Ziller, J.W. Expanding dinitrogen reduction chemistry to trivalent lanthanides via the LnZ3/Alkali metal reduction system: Evaluation of the generality of forming Ln2(μ-η2:η2-N2) complexes via LnZ3/K. J. Am. Chem. Soc. 2004, 126, 14574–14582. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Zucchi, G.; Ziller, J.W. Dinitrogen reduction by Tm(II), Dy(II), and Nd(II) with simple amide and aryloxide ligands. J. Am. Chem. Soc. 2003, 125, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Lee, D.S.; Ziller, J.W. Reduction of dinitrogen to planar bimetallic M2(μ-η2:η2-N2) complexes of Y, Ho, Tm, and Lu using the K/Ln[N(SiMe3)2]3 reduction system. J. Am. Chem. Soc. 2004, 126, 454–455. [Google Scholar] [CrossRef] [PubMed]
- Corbey, J.F.; Farnaby, J.H.; Bates, J.E.; Ziller, J.W.; Furche, F.; Evans, W.J. Varying the Lewis base coordination of the Y2N2 core in the reduced dinitrogen complexes {[(Me3Si)2N]2(L)Y}2(μ-η2:η2-N2) (L = benzonitrile, pyridines, triphenylphosphine oxide, and trimethylamine N-oxide). Inorg. Chem. 2012, 51, 7867–7874. [Google Scholar] [CrossRef] [PubMed]
- Campazzi, E.; Solari, E.; Floriani, C.; Scopelliti, R. The fixation and reduction of dinitrogen using lanthanides: Praseodymium and neodymium meso-octaethylporphyrinogen-dinitrogen complexes. Chem. Commun. 1998, 2603–2604. [Google Scholar] [CrossRef]
- Cheng, J.; Takats, J.; Ferguson, M.J.; McDonald, R. Heteroleptic Tm(II) complexes: One more success for Trofimenko’s scorpionates. J. Am. Chem. Soc. 2008, 130, 1544–1545. [Google Scholar] [CrossRef] [PubMed]
- Mansell, S.M.; Farnaby, J.H.; Germeroth, A.I.; Arnold, P.L. Thermally stable uranium dinitrogen complex with siloxide supporting ligands. Organometallics 2013, 32, 4214–4222. [Google Scholar] [CrossRef]
- Mansell, S.M.; Kaltsoyannis, N.; Arnold, P.L. Small molecule activation by uranium tris(aryloxides): Experimental and computational studies of binding of N2, coupling of CO, and deoxygenation insertion of CO2 under ambient conditions. J. Am. Chem. Soc. 2011, 133, 9036–9051. [Google Scholar] [CrossRef] [PubMed]
- Perrin, L.; Maron, L.; Eisenstein, O.; Schwartz, D.J.; Burns, C.J.; Andersen, R.A. Bonding of H2, N2, ethylene, and acetylene to bivalent lanthanide metallocenes: Trends from DFT calculations on Cp2M and Cp*2M (M = Sm, Eu, Yb) and experiments with Cp*2Yb. Organometallics 2003, 22, 5447–5453. [Google Scholar] [CrossRef]
- Hamaed, H.; Lo, A.Y.; Lee, D.S.; Evans, W.J.; Schurko, R.W. Solid-state 139La- and 15N-NMR spectroscopy of lanthanum-containing metallocenes. J. Am. Chem. Soc. 2006, 128, 12638–12639. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Davis, B.L. Chemistry of tris(pentamethylcyclopentadienyl) f-element complexes, (C5Me5)3M. Chem. Rev. 2002, 102, 2119–2136. [Google Scholar] [CrossRef] [PubMed]
- Fieser, M.E.; Johnson, C.W.; Bates, J.E.; Ziller, J.W.; Furche, F.; Evans, W.J. Dinitrogen reduction, sulfur reduction, and isoprene polymerization via photochemical activation of trivalent bis(cyclopentadienyl) rare-earth-metal allyl complexes. Organometallics 2015, 34, 4387–4393. [Google Scholar] [CrossRef]
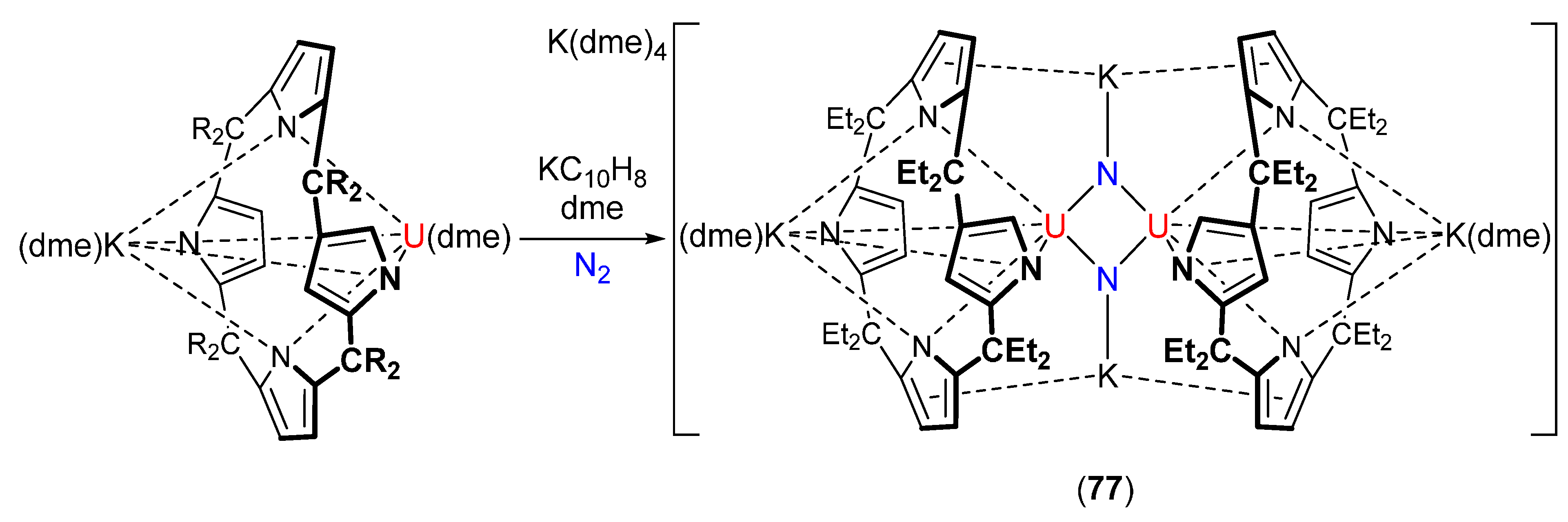
- Evans, W.J.; Fang, M.; Zucchi, G.; Furche, F.; Ziller, J.W.; Hoekstra, R.M.; Zink, J.I. Isolation of dysprosium and yttrium complexes of a three-electron reduction product in the activation of dinitrogen, the (N2)3− radical. J. Am. Chem. Soc. 2009, 131, 11195–11202. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.D.; Fang, M.; Evans, W.J.; Long, J.R. Strong exchange and magnetic blocking in N23−-radical-bridged lanthanide complexes. Nat. Chem. 2011, 3, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.D.; Fang, M.; Evans, W.J.; Long, J.R. A N23− radical-bridged terbium complex exhibiting magnetic hysteresis at 14 K. J. Am. Chem. Soc. 2011, 133, 14236–14239. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.E.; Hughbanks, T. Magnetic coupling in dinuclear Gd complexes. J. Am. Chem. Soc. 2006, 128, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Meihaus, K.R.; Corbey, J.F.; Fang, M.; Ziller, J.W.; Long, J.R.; Evans, W.J. Influence of an inner-sphere K+ ion on the magnetic behavior of N23− radical-bridged dilanthanide complexes isolated using an external magnetic field. Inorg. Chem. 2014, 53, 3099–3107. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Luo, C.-L.; Wang, B.-W.; Gao, S. Understanding the magnetic anisotropy in a family of N23− radical-bridged lanthanide complexes: Density functional theory and ab initio calculations. J. Phys. Chem. A 2013, 117, 10873–10880. [Google Scholar] [CrossRef] [PubMed]
- Rajeshkumar, T.; Rajaraman, G. Is a radical bridge a route to strong exchange interactions in lanthanide complexes? A computational examination. Chem. Commun. 2012, 48, 7856–7858. [Google Scholar] [CrossRef] [PubMed]
- Jubb, J.; Gambarotta, S. Dinitrogen reduction operated by a samarium macrocyclic complex. Encapsulation of dinitrogen into a Sm2Li4 metallic cage. J. Am. Chem. Soc. 1994, 116, 4477–4478. [Google Scholar] [CrossRef]
- Dubé, T.; Conoci, S.; Gambarotta, S.; Yap, G.P.A.; Vasapollo, G. Tetrametallic reduction of dinitrogen: Formation of a tetranuclear samarium dinitrogen complex. Angew. Chem. Int. Ed. 1999, 38, 3657–3659. [Google Scholar] [CrossRef]
- Dubé, T.; Ganesan, M.; Conoci, S.; Gambarotta, S.; Yap, G.P.A. Tetrametallic divalent samarium cluster hydride and dinitrogen complexes. Organometallics 2000, 19, 3716–3721. [Google Scholar] [CrossRef]
- Bérubé, C.D.; Yazdanbakhsh, M.; Gambarotta, S.; Yap, G.P.A. Serendipitous isolation of the first example of a mixed-valence samarium tripyrrole complex. Organometallics 2003, 22, 3742–3747. [Google Scholar] [CrossRef]
- Guan, J.; Dubé, T.; Gambarotta, S.; Yap, G.P.A. Dinitrogen labile coordination versus four-electron reduction, THF cleavage, and fragmentation promoted by a (calix-tetrapyrrole)Sm(II) complex. Organometallics 2000, 19, 4820–4827. [Google Scholar] [CrossRef]
- Ganesan, M.; Gambarotta, S.; Yap, G.P.A. Highly reactive SmII macrocyclic clusters: Precursors to N2 reduction. Angew. Chem. Int. Ed. 2001, 40, 766–769. [Google Scholar] [CrossRef]
- Fox, A.R.; Bart, S.C.; Meyer, K.; Cummins, C.C. Towards uranium catalysts. Nature 2008, 455, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.-R.; Kingham, J.R.; Kaltsoyannis, N. The strength of actinide-element bonds from the quantum theory of atoms-in-molecules. Dalton Trans. 2015, 44, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Roussel, P.; Scott, P. Complex of dinitrogen with trivalent uranium. J. Am. Chem. Soc. 1998, 120, 1070–1071. [Google Scholar] [CrossRef]
- Odom, A.L.; Arnold, P.L.; Cummins, C.C. Heterodinuclear uranium/molybdenum dinitrogen complexes. J. Am. Chem. Soc. 1998, 120, 5836–5837. [Google Scholar] [CrossRef]
- Cloke, F.G.N.; Hitchcock, P.B. Reversible binding and reduction of dinitrogen by a uranium(III) pentalene complex. J. Am. Chem. Soc. 2002, 124, 9352–9353. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Kozimor, S.A.; Ziller, J.W. A monometallic f element complex of dinitrogen: (C5Me5)3U(η1-N2). J. Am. Chem. Soc. 2003, 125, 14264–14265. [Google Scholar] [CrossRef] [PubMed]
- Laplaza, C.E.; Cummins, C.C. Dinitrogen cleavage by a three-coordinate molybdenum(III) complex. Science 1995, 268, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Mindiola, D.J.; Meyer, K.; Cherry, J.-P.F.; Baker, T.A.; Cummins, C.C. Dinitrogen cleavage stemming from a heterodinuclear niobium/molybdenum N2 complex: New nitridoniobium systems including a niobazene cyclic trimer. Organometallics 2000, 19, 1622–1624. [Google Scholar] [CrossRef]
- Curley, J.J.; Cook, T.R.; Reece, S.Y.; Müller, P.; Cummins, C.C. Shining light on dinitrogen cleavage: Structural features, redox chemistry, and photochemistry of the key intermediate bridging dinitrogen complex. J. Am. Chem. Soc. 2008, 130, 9394–9405. [Google Scholar] [CrossRef] [PubMed]
- Kaltsoyannis, N.; Scott, P. Evidence for actinide metal to ligand π backbonding. Density functional investigations of the electronic structure of [{(NH2)3(NH3)U}2(μ2-η2:η2-N2)]. Chem. Commun. 1998, 1665–1666. [Google Scholar] [CrossRef]
- Roussel, P.; Errington, W.; Kaltsoyannis, N.; Scott, P. Back bonding without σ-bonding: A unique π-complex of dinitrogen with uranium. J. Organomet. Chem. 2001, 635, 69–74. [Google Scholar] [CrossRef]
- Cloke, F.G.N.; Green, J.C.; Kaltsoyannis, N. Electronic structure of [U2(μ2-N2)(η5-C5Me5)2(η8-C8H4(SiPri3)2)2]. Organometallics 2004, 23, 832–835. [Google Scholar] [CrossRef]
- Korobkov, I.; Gambarotta, S.; Yap, G.P.A. A highly reactive uranium complex supported by the calix[4]tetrapyrrole tetraanion affording dinitrogen cleavage, solvent deoxygenation, and polysilanol depolymerization. Angew. Chem. Int. Ed. 2002, 41, 3433–3436. [Google Scholar] [CrossRef]
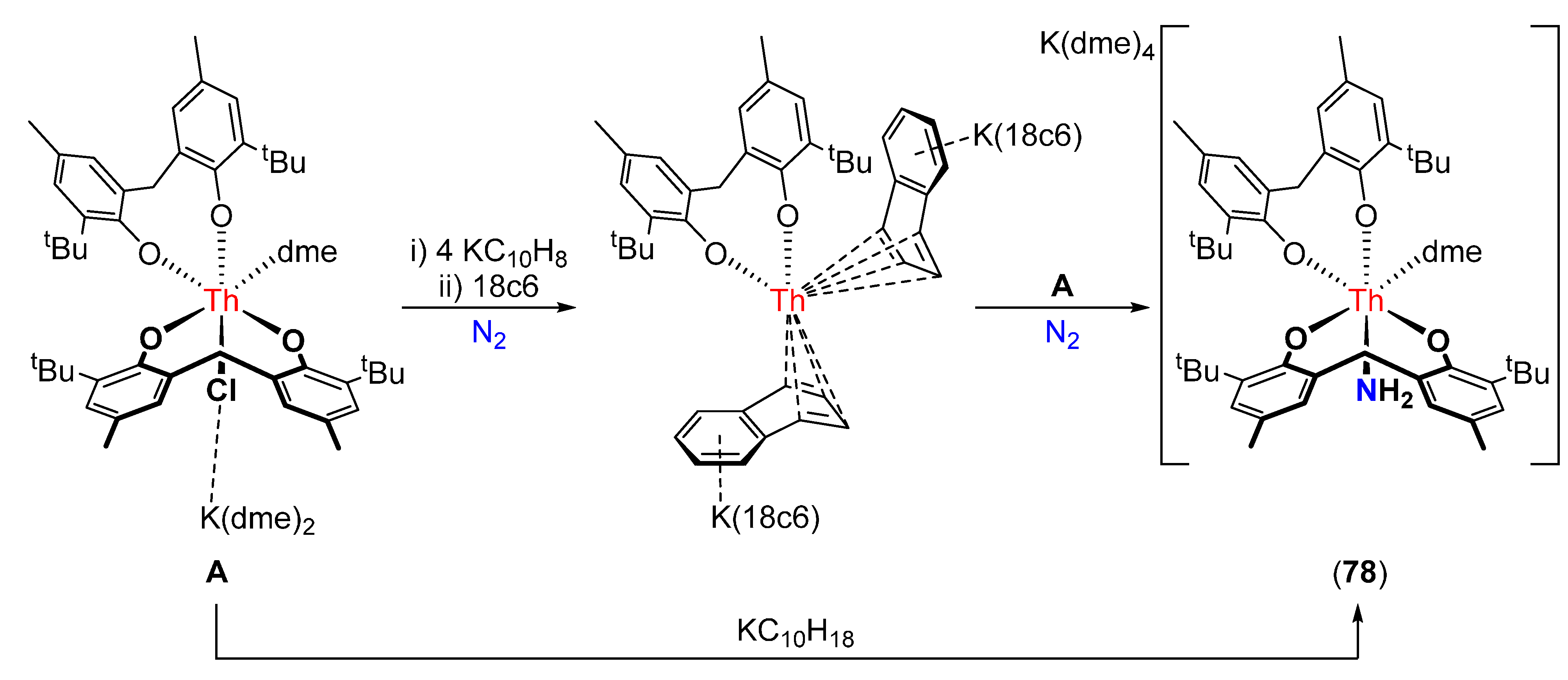
- Korobkov, I.; Gambarotta, S.; Yap, G.P.A. Amide from dinitrogen by in situ cleavage and partial hydrogenation promoted by a transient zero-valent thorium synthon. Angew. Chem. Int. Ed. 2003, 42, 4958–4961. [Google Scholar] [CrossRef] [PubMed]
- Corbridge, D.E.C.; Lowe, E.J. Structure of white phosphorus: Single crystal X-ray examination. Nature 1952, 170, 629–629. [Google Scholar] [CrossRef]
- Kühl, O. Phosphorus-31 NMR Spectroscopy: A Concise Introduction for the Synthetic Organic and Organometallic Chemist; Springer-Verlag GmbH: Berlin, Germany; Heidelberg, Germany, 2008. [Google Scholar]
- Konchenko, S.N.; Pushkarevsky, N.A.; Gamer, M.T.; Köppe, R.; Schnöckel, H.; Roesky, P.W. [{(η5-C5Me5)2Sm}4P8]: A molecular polyphosphide of the rare-earth elements. J. Am. Chem. Soc. 2009, 131, 5740–5741. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Diaconescu, P.L. P4 activation by group 3 metal arene complexes. Chem. Commun. 2012, 48, 2216–2218. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Diaconescu, P.L. P4 Activation by lanthanum and lutetium naphthalene complexes supported by a ferrocene diamide ligand. Eur. J. Inorg. Chem. 2013, 2013, 4090–4096. [Google Scholar] [CrossRef]
- Turbervill, R.S.P.; Goicoechea, J.M. From clusters to unorthodox pnictogen sources: Solution-phase reactivity of [E7]3− (E = P–Sb) anions. Chem. Rev. 2014, 114, 10807–10828. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A Found. Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Scherer, O.J.; Werner, B.; Heckmann, G.; Wolmershäuser, G. Bicyclic P6 as complex ligand. Angew. Chem. Int. Ed. 1991, 30, 553–555. [Google Scholar] [CrossRef]
- Stephens, F.H. Activation of White Phosphorus by Molybdenum and Uranium tris-Amides. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, May 2004. [Google Scholar]
- Frey, A.S.P.; Cloke, F.G.N.; Hitchcock, P.B.; Green, J.C. Activation of P4 by U(η5-C5Me5)(η8-C8H6(SiiPr3)2-1,4)(THF); the X-ray structure of [U(η5-C5Me5)(η8-C8H6(SiiPr3)2-1,4)]2(μ-η2:η2-P4). New J. Chem. 2011, 35, 2022–2026. [Google Scholar] [CrossRef]
- Gardner, B.M.; Tuna, F.; McInnes, E.J.L.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. An inverted-sandwich diuranium μ-η5:η5-cyclo-P5 complex supported by U–P5 δ-bonding. Angew. Chem. Int. Ed. 2015, 54, 7068–7072. [Google Scholar] [CrossRef] [PubMed]
- Forfar, L.C.; Clark, T.J.; Green, M.; Mansell, S.M.; Russell, C.A.; Sanguramath, R.A.; Slattery, J.M. White phosphorus as a ligand for the coinage metals. Chem. Commun. 2012, 48, 1970–1972. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, F.; Sierka, M.; Latronico, M.; Mastrorilli, P.; Virovets, A.V.; Scheer, M. Fixation and release of intact E4 tetrahedra (E = P, As). Angew. Chem. Int. Ed. 2015, 54, 4392–4396. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Tuna, F.; McInnes, E.J.L.; Lewis, W.; Blake, A.J.; Liddle, S.T. An actinide zintl cluster: A tris(triamidouranium)μ3-η2:η2:η2-heptaphosphanortricyclane and its diverse synthetic utility. Angew. Chem. Int. Ed. 2013, 52, 13334–13337. [Google Scholar] [CrossRef] [PubMed]
- Laplaza, C.E.; Davis, W.M.; Cummins, C.C. A molybdenum–phosphorus triple bond: Synthesis, structure, and reactivity of the terminal phosphido (P3−) complex [Mo(P)(NRAr)3]. Angew. Chem. Int. Ed. 1995, 34, 2042–2044. [Google Scholar] [CrossRef]
- Cherry, J.-P.F.; Stephens, F.H.; Johnson, M.J.A.; Diaconescu, P.L.; Cummins, C.C. Terminal phosphide and dinitrogen molybdenum compounds obtained from pnictide-bridged precursors. Inorg. Chem. 2001, 40, 6860–6862. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, M.H.; Folting, K.; Pasterczyk, J.W. A phosphido-capped tritungsten alkoxide cluster: W3(μ3-P)(μ-OCH2-t-Bu)3(OCH2-t-Bu)6 and speculation upon the existence of a reactive (t-BuCH2O)3W≡P intermediate. Inorg. Chem. 1988, 27, 3057–3058. [Google Scholar] [CrossRef]
- Scherer, O.J.; Sitzmann, H.; Wolmershäuser, G. Umsetzung von P4 mit (η5-C5H5)(CO)2Mo≡Mo(CO)2(η5-C5H5) zu den tetraedrischen molybdänkomplexen Pn[Mo(CO)2(η5-C5H5)]4−n (n = 2,3). J. Organomet. Chem. 1984, 268, C9–C12. [Google Scholar] [CrossRef]
- Di Vaira, M.; Ghilardi, C.A.; Midollini, S.; Sacconi, L. cyclo-Triphosphorus (δ-P3) as a ligand in cobalt and nickel complexes with 1,1,1-tris(diphenylphosphinomethyl)ethane. Formation and structures. J. Am. Chem. Soc. 1978, 100, 2550–2551. [Google Scholar] [CrossRef]
- Chirik, P.J.; Pool, J.A.; Lobkovsky, E. Functionalization of elemental phosphorus with [Zr(η5-C5Me5)(η5-C5H4tBu)H2]2. Angew. Chem. Int. Ed. 2002, 41, 3463–3465. [Google Scholar] [CrossRef]
- Gröer, T.; Baum, G.; Scheer, M. Complexes with a monohapto bound phosphorus tetrahedron and phosphaalkyne. Organometallics 1998, 17, 5916–5919. [Google Scholar] [CrossRef]
- Peruzzini, M.; Marvelli, L.; Romerosa, A.; Rossi, R.; Vizza, F.; Zanobini, F. Synthesis and characterisation of tetrahedro-tetraphosphorus complexes of rhenium—Evidence for the first bridging complex of white phosphorus. Eur. J. Inorg. Chem. 1999, 1999, 931–933. [Google Scholar] [CrossRef]
- Urnėžius, E.; Brennessel, W.W.; Cramer, C.J.; Ellis, J.E.; Schleyer, P.V.R. A carbon-free sandwich complex [(P5)2Ti]2−. Science 2002, 295, 832–834. [Google Scholar]
- Scherer, O.J.; Sitzmann, H.; Wolmershäuser, G. Hexaphosphabenzene as complex ligand. Angew. Chem. Int. Ed. 1985, 24, 351–353. [Google Scholar] [CrossRef]
- Scherer, O.J.; Berg, G.; Wolmershäuser, G. P8 and P12 as complex ligands. Chem. Ber. 1996, 129, 53–58. [Google Scholar] [CrossRef]
- Scheer, M.; Deng, S.; Scherer, O.J.; Sierka, M. Tetraphosphacyclopentadienyl and triphosphaallyl ligands in iron complexes. Angew. Chem. Int. Ed. 2005, 44, 3755–3758. [Google Scholar] [CrossRef] [PubMed]
- Scherer, O.J.; Schulze, J.; Wolmershäuser, G. Bicyclisches As6 als komplexligand. J. Organomet. Chem. 1994, 484, C5–C7. [Google Scholar] [CrossRef]
- Di Vaira, M.; Midollini, S.; Sacconi, L.; Zanobini, F. cyclo-Triarsenic as μ,η-ligand in transition-metal complexes. Angew. Chem. Int. Ed. 1978, 17, 676–677. [Google Scholar] [CrossRef]
- Scherer, O.J.; Kemény, G.; Wolmershäuser, G. [Cp4Fe4(E2)2] clusters with triangulated dodecahedral Fe4E4 skeletons (E = P, As). Chem. Ber. 1995, 128, 1145–1148. [Google Scholar] [CrossRef]
- Schwarzmaier, C.; Timoshkin, A.Y.; Scheer, M. An end-on-coordinated As4 tetrahedron. Angew. Chem. Int. Ed. 2013, 52, 7600–7603. [Google Scholar] [CrossRef] [PubMed]
- Schwarzmaier, C.; Sierka, M.; Scheer, M. Intact As4 tetrahedra coordinated side-on to metal cations. Angew. Chem. Int. Ed. 2013, 52, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Scherer, O.J.; Sitzmann, H.; Wolmershäuser, G. (E2)2-einheiten (E = P, As) als clusterbausteine. J. Organomet. Chem. 1986, 309, 77–86. [Google Scholar] [CrossRef]
- Spinney, H.A.; Piro, N.A.; Cummins, C.C. Triple-bond reactivity of an AsP complex intermediate: Synthesis stemming from molecular arsenic, As4. J. Am. Chem. Soc. 2009, 131, 16233–16243. [Google Scholar] [CrossRef] [PubMed]
- Heinl, S.; Scheer, M. Activation of group 15 based cage compounds by [CpBIGFe(CO)2] radicals. Chem. Sci. 2014, 5, 3221–3225. [Google Scholar] [CrossRef]
- Gra; Bodensteiner, M.; Zabel, M.; Scheer, M. Synthesis of arsenic-rich Asn ligand complexes from yellow arsenic. Chem. Sci. 2015, 6, 1379–1382. [Google Scholar]
- Schwarzmaier, C.; Bodensteiner, M.; Timoshkin, A.Y.; Scheer, M. An approach to mixed PnAsm ligand complexes. Angew. Chem. Int. Ed. 2014, 53, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Curley, J.J.; Piro, N.A.; Cummins, C.C. A terminal molybdenum arsenide complex synthesized from yellow arsenic. Inorg. Chem. 2009, 48, 9599–9601. [Google Scholar] [CrossRef] [PubMed]
- Balázs, G.; Sierka, M.; Scheer, M. Antimony–tungsten triple bond: A stable complex with a terminal antimony ligand. Angew. Chem. Int. Ed. 2005, 44, 4920–4924. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Fang, M.; Bates, J.E.; Furche, F.; Ziller, J.W.; Kiesz, M.D.; Zink, J.I. Isolation of a radical dianion of nitrogen oxide (NO)2−. Nat. Chem. 2010, 2, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Corbey, J.F.; Fang, M.; Ziller, J.W.; Evans, W.J. Cocrystallization of (μ-S2)2− and (μ-S)2− and formation of an [η2-S3N(SiMe3)2] ligand from chalcogen reduction by (N2)2− in a bimetallic yttrium amide complex. Inorg. Chem. 2015, 54, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Lee, D.S.; Ziller, J.W.; Kaltsoyannis, N. Trivalent [(C5Me5)2(THF)Ln]2(μ-η2:η2-N2) complexes as reducing agents including the reductive homologation of CO to a ketene carboxylate, (μ-η4-O2CCCO)2. J. Am. Chem. Soc. 2006, 128, 14176–14184. [Google Scholar] [CrossRef] [PubMed]
- Lu, E.; Li, Y.; Chen, Y. A scandium terminal imido complex: Synthesis, structure and DFT studies. Chem. Commun. 2010, 46, 4469–4471. [Google Scholar] [CrossRef] [PubMed]
- Rong, W.; Cheng, J.; Mou, Z.; Xie, H.; Cui, D. Facile preparation of a scandium terminal imido complex supported by a phosphazene ligand. Organometallics 2013, 32, 5523–5529. [Google Scholar] [CrossRef]
- Schädle, D.; Meermann-Zimmermann, M.; Schädle, C.; Maichle-Mössmer, C.; Anwander, R. Rare-earth metal complexes with terminal imido ligands. Eur. J. Inorg. Chem. 2015, 2015, 1334–1339. [Google Scholar] [CrossRef]
- Sobota, P.; Janas, Z. Formation of a nitrogen–carbon bond from N2 and CO. Influence of MgCl2 on the N2 reduction process in the system TiCl4/Mg. J. Organomet. Chem. 1984, 276, 171–176. [Google Scholar] [CrossRef]
- Semproni, S.P.; Margulieux, G.W.; Chirik, P.J. Di- and tetrametallic hafnocene oxamidides prepared from CO-induced N2 bond cleavage and thermal rearrangement to hafnocene cyanide derivatives. Organometallics 2012, 31, 6278–6287. [Google Scholar] [CrossRef]
- Knobloch, D.J.; Lobkovsky, E.; Chirik, P.J. Carbon monoxide-induced dinitrogen cleavage with group 4 metallocenes: Reaction scope and coupling to N–H bond formation and CO deoxygenation. J. Am. Chem. Soc. 2010, 132, 10553–10564. [Google Scholar] [CrossRef] [PubMed]
- Semproni, S.P.; Milsmann, C.; Chirik, P.J. Structure and reactivity of a hafnocene μ-nitrido prepared from dinitrogen cleavage. Angew. Chem. Int. Ed. 2012, 51, 5213–5216. [Google Scholar] [CrossRef] [PubMed]
- Semproni, S.P.; Chirik, P.J. Synthesis of a base-free hafnium nitride from N2 cleavage: A versatile platform for dinitrogen functionalization. J. Am. Chem. Soc. 2013, 135, 11373–11383. [Google Scholar] [CrossRef] [PubMed]
- MacKay, B.A.; Johnson, S.A.; Patrick, B.O.; Fryzuk, M.D. Functionalization and cleavage of coordinated dinitrogen via hydroboration using primary and secondary boranes. Can. J. Chem. 2005, 83, 315–323. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; MacKay, B.A.; Johnson, S.A.; Patrick, B.O. Hydroboration of coordinated dinitrogen: A new reaction for the N2 ligand that results in its functionalization and cleavage. Angew. Chem. Int. Ed. 2002, 41, 3709–3712. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; MacKay, B.A.; Patrick, B.O. Hydrosilylation of a dinuclear tantalum dinitrogen complex: Cleavage of N2 and functionalization of both nitrogen atoms. J. Am. Chem. Soc. 2003, 125, 3234–3235. [Google Scholar] [CrossRef] [PubMed]
- Spencer, L.P.; MacKay, B.A.; Patrick, B.O.; Fryzuk, M.D. Inner-sphere two-electron reduction leads to cleavage and functionalization of coordinated dinitrogen. Proc. Natl. Acad. Sci. USA 2006, 103, 17094–17098. [Google Scholar] [CrossRef] [PubMed]
- Margulieux, G.W.; Turner, Z.R.; Chirik, P.J. Synthesis and ligand modification chemistry of a molybdenum dinitrogen complex: Redox and chemical activity of a bis(imino)pyridine ligand. Angew. Chem. Int. Ed. 2014, 53, 14211–14215. [Google Scholar] [CrossRef] [PubMed]
- Milsmann, C.; Turner, Z.R.; Semproni, S.P.; Chirik, P.J. Azo N=N bond cleavage with a redox-active vanadium compound involving metal–ligand cooperativity. Angew. Chem. Int. Ed. 2012, 51, 5386–5390. [Google Scholar] [CrossRef] [PubMed]
- King, D.M.; Tuna, F.; McInnes, E.J.L.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. Synthesis and structure of a terminal uranium nitride complex. Science 2012, 337, 717–720. [Google Scholar] [CrossRef] [PubMed]
- King, D.M.; Tuna, F.; McInnes, E.J.L.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. Isolation and characterization of a uranium(VI)–nitride triple bond. Nat. Chem. 2013, 5, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Cleaves, P.A.; King, D.M.; Kefalidis, C.E.; Maron, L.; Tuna, F.; McInnes, E.J.L.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. Two-electron reductive carbonylation of terminal uranium(V) and uranium(VI) nitrides to cyanate by carbon monoxide. Angew. Chem. Int. Ed. 2014, 53, 10412–10415. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Balázs, G.; Scheer, M.; Tuna, F.; McInnes, E.J.L.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. Triamidoamine uranium(IV)–arsenic complexes containing one-, two- and threefold U–As bonding interactions. Nat. Chem. 2015, 7, 582–590. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turner, Z.R. Molecular Pnictogen Activation by Rare Earth and Actinide Complexes. Inorganics 2015, 3, 597-635. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040597
Turner ZR. Molecular Pnictogen Activation by Rare Earth and Actinide Complexes. Inorganics. 2015; 3(4):597-635. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040597
Chicago/Turabian StyleTurner, Zoë R. 2015. "Molecular Pnictogen Activation by Rare Earth and Actinide Complexes" Inorganics 3, no. 4: 597-635. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040597