
Formation of Micro and Mesoporous Amorphous Silica-Based Materials from Single Source Precursors

 ,
,
Abstract
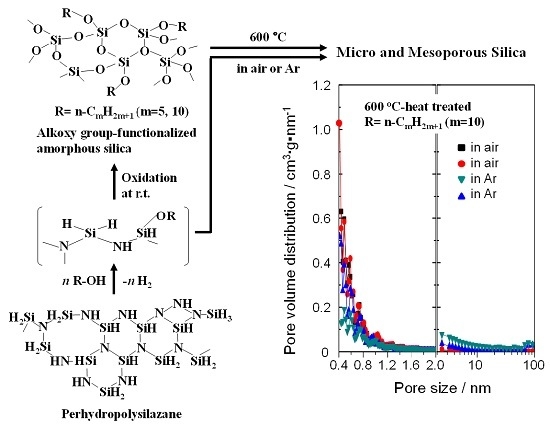
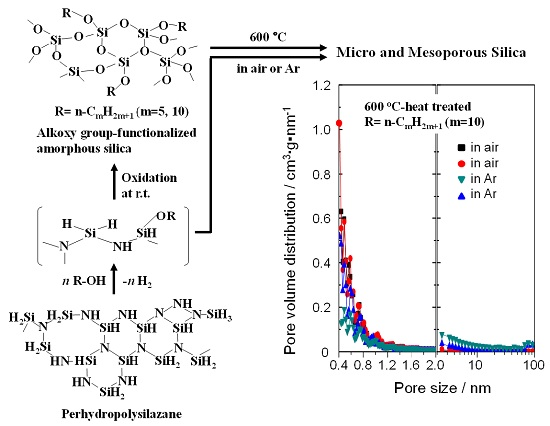
:
1. Introduction
2. Results and Discussion
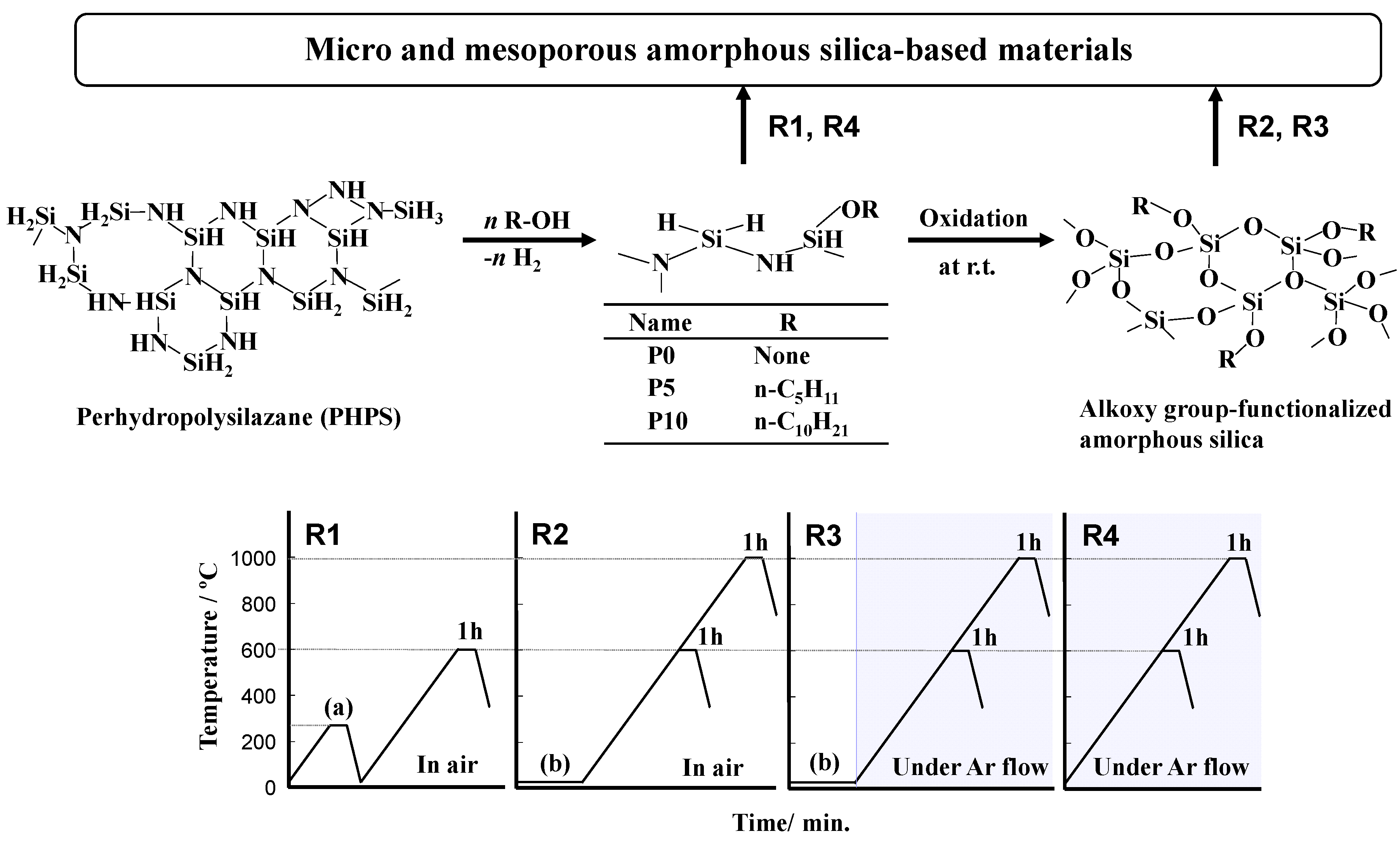
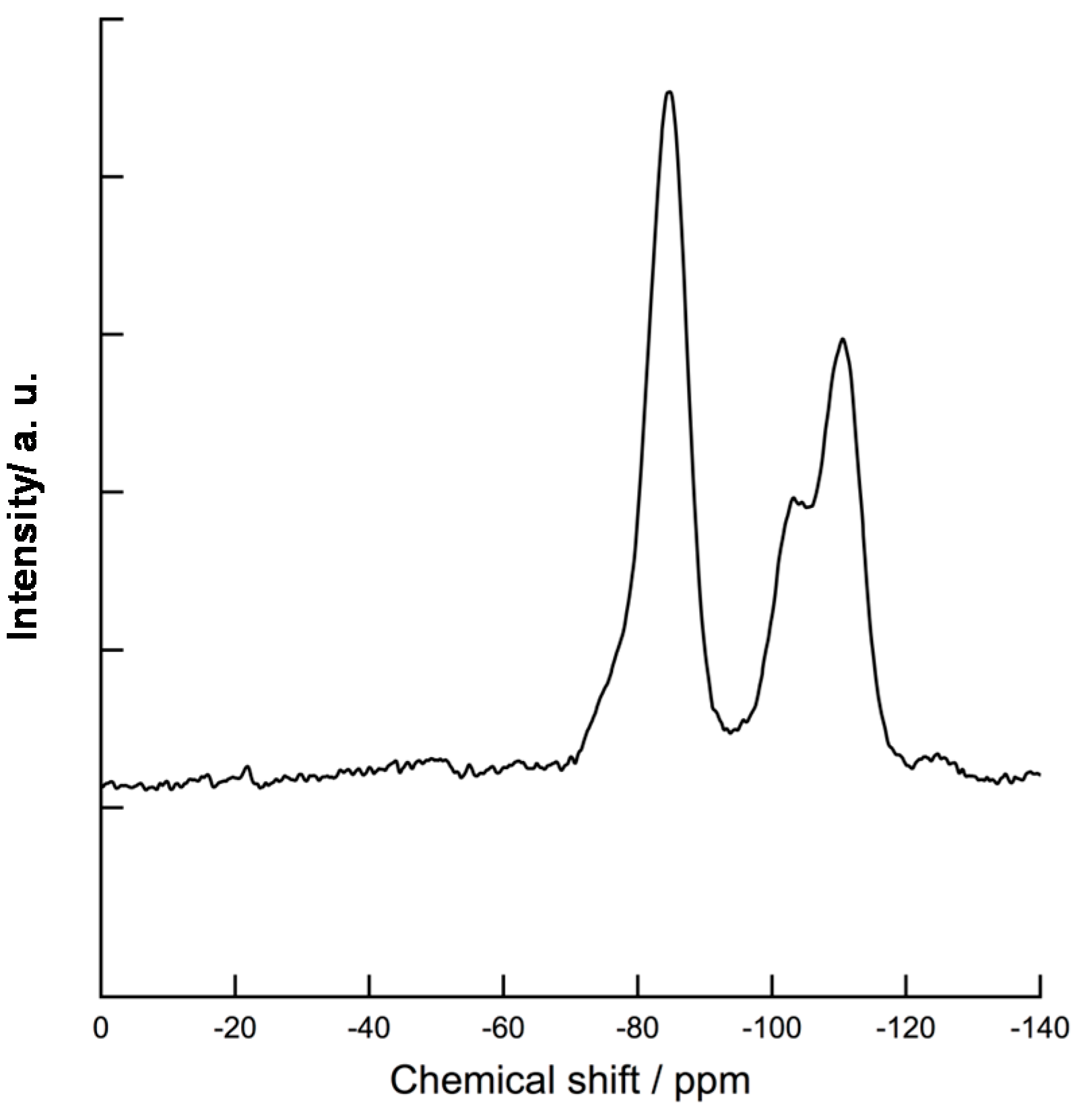
2.1. Polymer Precursors and Alkoxy Group-Functionalized Amorphous Silica
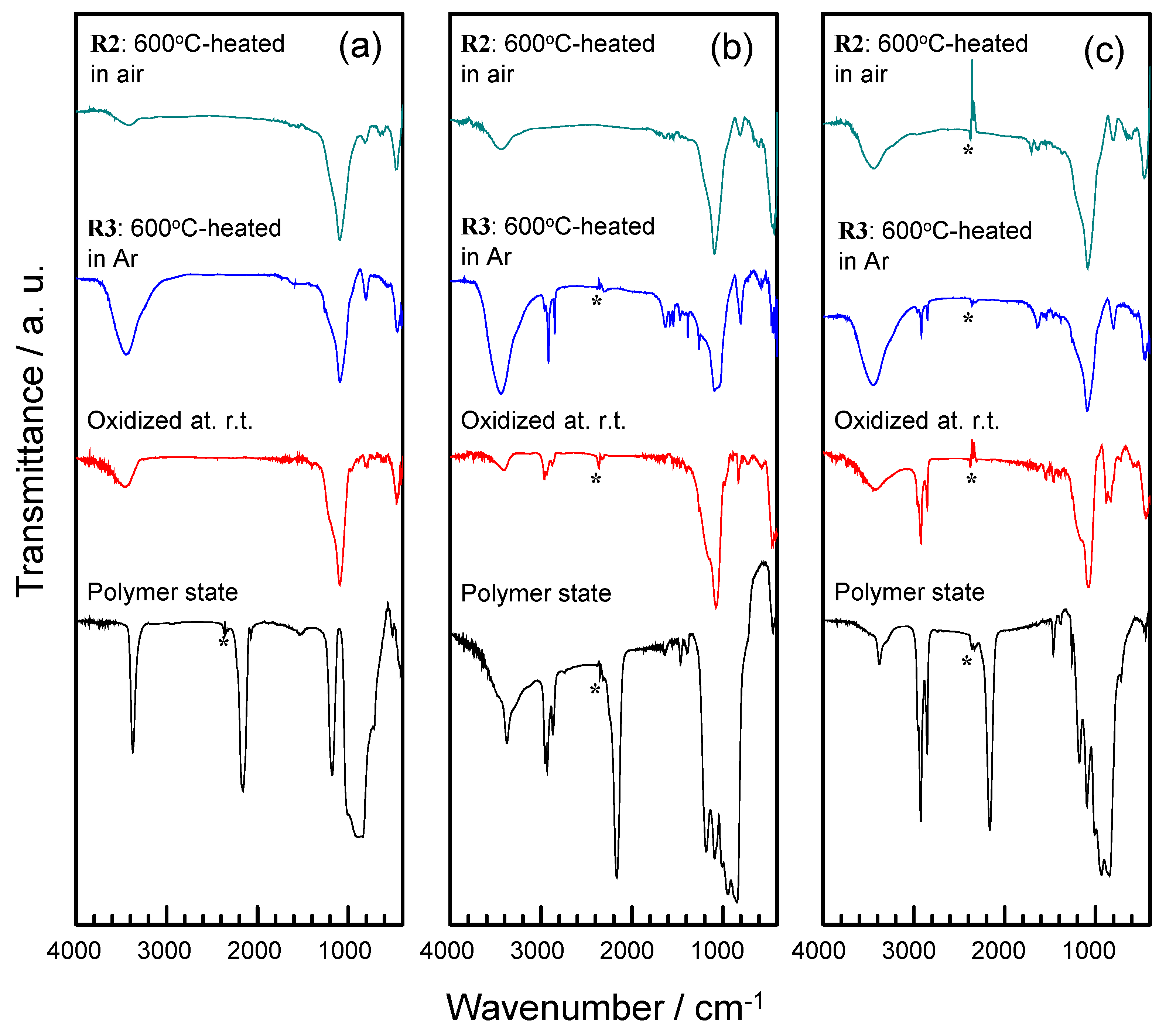
2.2. Thermal Conversion to Amorphous Silica-Based Materials
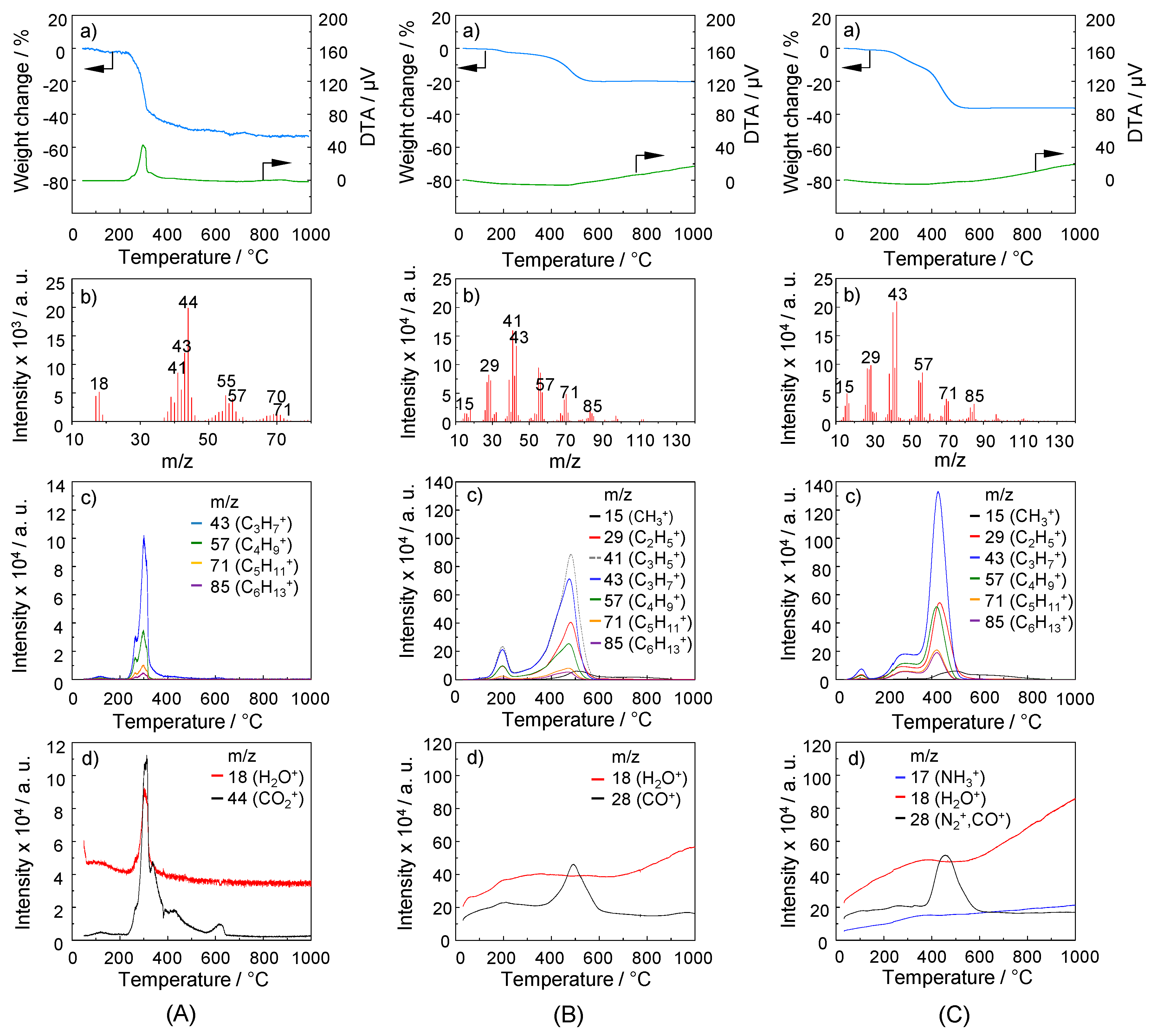
2.3. TG-MS Analysis
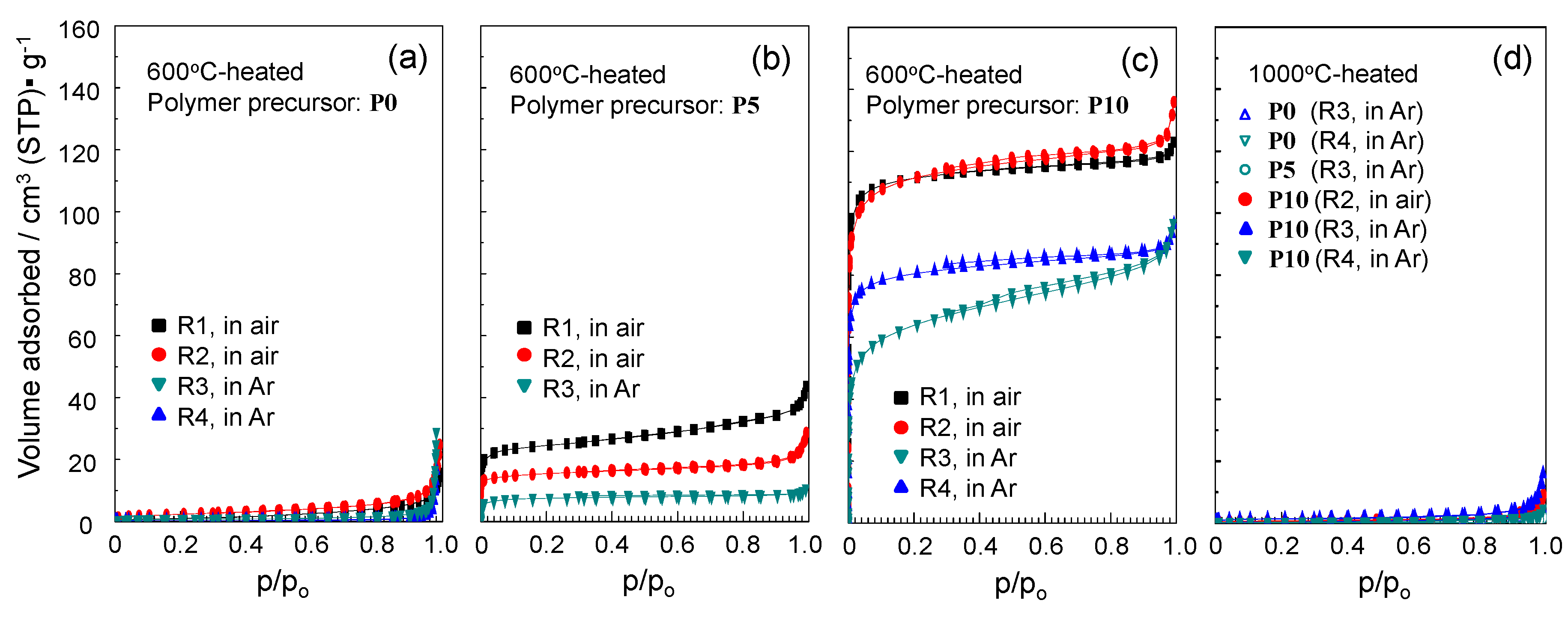
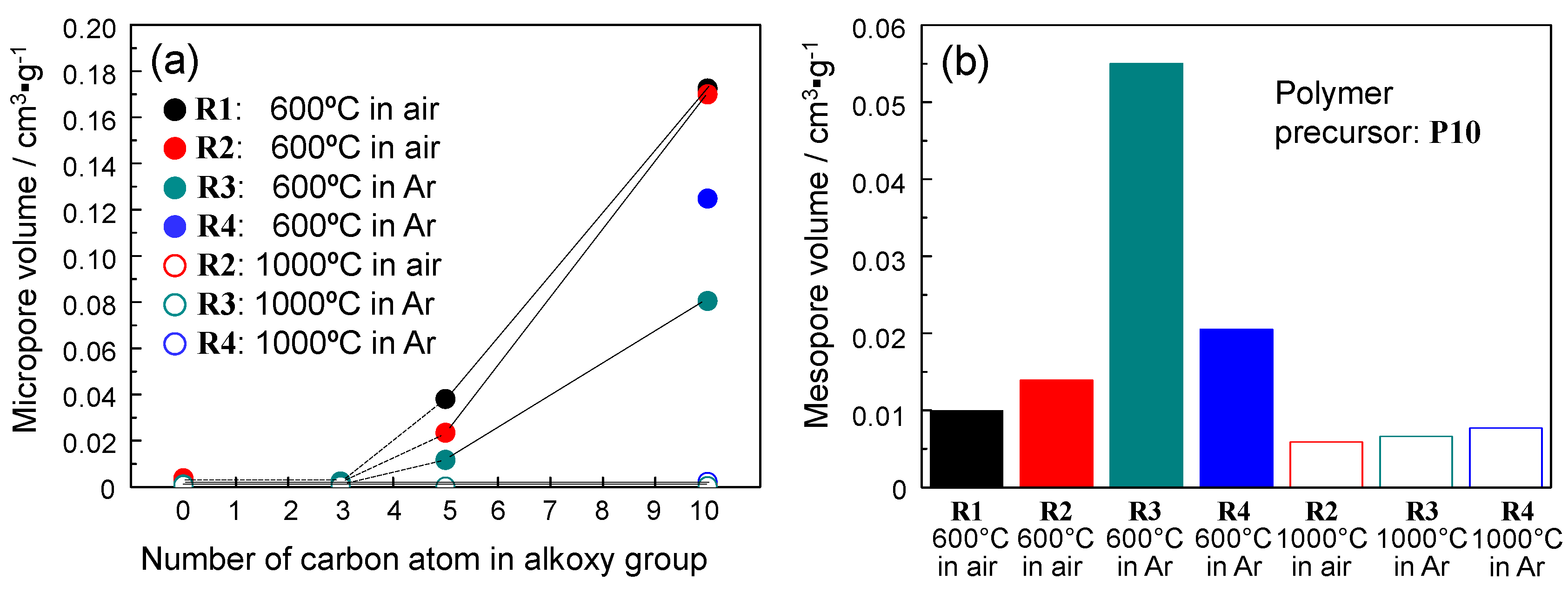
2.4. N2 Adsorption/Desorption Isotherm and Porosities Evaluated for the Polymer-Derived Amorphous Silica-Based Materials
3. Experimental Procedures
3.1. Precursor Synthesis
3.2. Conversion of Polymer Precursor to Amorphous Silica-Based Materials
R1: Oxidative crosslinking and subsequent heat treatment in air [31]
R2: Room temperature oxidation followed by heat treatment in air [32]
R3: Room temperature oxidation followed by heat treatment in Ar
R4: Heat treatment in Ar
3.3. Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rice, R.W. Ceramics from polymer pyrolysis, opportunities and needs. A materials perspective. Am. Ceram. Soc. Bull. 1983, 62, 889–892. [Google Scholar]
- Wynne, K.J.; Rice, R.W. Ceramics via polymer pyrolysis. Annu. Rev. Mater. Sci. 1984, 14, 297–334. [Google Scholar] [CrossRef]
- Riedel, R.; Dressler, W. Chemical formation of ceramics. Ceram. Int. 1996, 22, 233–239. [Google Scholar] [CrossRef]
- Seyferth, D.; Strohmann, C.; Dando, N.R.; Perrotta, A. Poly(ureidosilazanes): Preceramic polymeric precursors for silicon carbonitride and silicon nitride. Synthesis, characterization and pyrolytic conversion to Si3N4/SiC ceramics. J. Chem. Mater. 1995, 7, 2058–2066. [Google Scholar] [CrossRef]
- Kroke, E.; Li, Y-L.; Konetschny, C.; Lecomte, E.; Fasel, C.; Riedel, R. Silazane derived ceramics and related materials. Mater. Sci. Eng. R 2000, 26, 97–199. [Google Scholar] [CrossRef]
- Funayama, O.; Arai, M.; Tashiro, Y.; Aoki, H.; Suzuki, T.; Tamura, K.; Kaya, H.; Nishii, H.; Isoda, T. Tensile strength of silicon nitride fibers produced from perhydropolysilazane. J. Ceram. Soc. Jpn. 1990, 98, 104–107. [Google Scholar] [CrossRef]
- Miyajima, K.; Eda, T.; Ohta, H.; Ando, Y.; Nagaya, S.; Ohba, T.; Iwamoto, Y. Development of Si–N based hydrogen separation membrane. Ceram. Trans. 2010, 213, 87–94. [Google Scholar]
- Schitco, C.; Bazarjani, M.S.; Riedel, R.; Gurlo, A. NH3-assisted synthesis of microporous silicon oxycarbonitride ceramics from preceramic polymers: A combined N2 and CO2 adsorption and small angle X-ray scattering study. J. Mater. Chem. A 2015, 3, 805–818. [Google Scholar] [CrossRef]
- Kusakabe, K.; Li, Z.Y.; Maeda, H.; Morooka, S. Preparation of supported composite membrane by pyrolysis of polycarbosilane for gas separation at high temperature. J. Membr. Sci. 1995, 103, 175–180. [Google Scholar] [CrossRef]
- Li, Z.Y.; Kusakabe, K.; Morooka, S. Preparation of thermostable amorphous Si–C–O membrane and its application to gas separation at elevated temperature. J. Membr. Sci. 1996, 118, 159–168. [Google Scholar]
- Nagano, T.; Sato, K.; Saito, T.; Iwamoto, Y. Gas permeation properties of amorphous SiC membranes synthesized from polycarbosilane without oxygen-curing process. J. Ceram. Soc. Jpn. 2006, 114, 533–538. [Google Scholar] [CrossRef]
- Suda, H.; Yamauchi, H.; Uchimaru, Y.; Fujiwara, I.; Haraya, K. Preparation and gas permeation properties of silicon carbide-based inorganic membranes for hydrogen separation. Desalination 2006, 193, 252–255. [Google Scholar] [CrossRef]
- Mourhatch, R.; Tsotsis, T.T.; Sahimi, M. Network model for the evolution of the pore structure of silicon-carbide membranes during their fabrication. J. Membr. Sci. 2010, 356, 138–146. [Google Scholar] [CrossRef]
- Takeyama, A.; Sugimoto, M.; Yoshikawa, M. Gas permeation property of SiC membrane using curing of polymer precursor film by electron beam irradiation in helium atmosphere. Mater. Trans. 2011, 52, 1276–1280. [Google Scholar] [CrossRef]
- Völger, K.W.; Hauser, R.; Kroke, E.; Riedel, R.; Ikuhara, Y.H.; Iwamoto, Y. Synthesis and characterization of novel non-oxide sol-gel derived mesoporous amorphous Si–C–N membranes. J. Ceram. Soc. Jpn. 2006, 114, 567–570. [Google Scholar] [CrossRef]
- Soraru, G.D.; Liu, Q.; Interrante, L.V.; Apple, T. Role of precursor molecular structure on the microstructure and high temperature stability of silicon oxycarbide glasses derived from methylene-bridged polycarbosilanes. Chem. Mater. 1998, 10, 4047–4054. [Google Scholar] [CrossRef]
- Liu, Q.; Shi, W.; Babonneau, F.; Interrante, L.V. Synthesis of polycarbosilane/siloxane hybrid polymers and their pyrolytic conversion to silicon oxycarbide ceramics. Chem. Mater. 1997, 9, 2434–2441. [Google Scholar] [CrossRef]
- Lee, L.; Tsai, D.S. A hydrogen-permselective silicon oxycarbide membrane derived from polydimethylsilane. J. Am. Ceram. Soc. 1999, 82, 2796–2800. [Google Scholar] [CrossRef]
- Hauser, R.; Nahar-Borchard, S.; Riedel, R.; Ikuhara, Y.H.; Iwamoto, Y. Polymer-derived SiBCN ceramic and their potential application for high temperature membranes. J. Ceram. Soc. Jpn. 2006, 114, 524–528. [Google Scholar] [CrossRef]
- Prasad, R.M.; Iwamoto, Y.; Riedel, R.; Gurlo, A. Multilayer amorphous Si–B–C–N/γ-Al2O3/α-Al2O3 membranes for hydrogen purification. Adv. Eng. Mater. 2010, 12, 522–528. [Google Scholar] [CrossRef]
- Bazarjani, M.S.; Müller, M.M.; Kleebe, H.-J.; Jüttke, Y.; Voigt, I.; Yazdi, M.B.; Alff, L.; Riedel, R.; Gurlo, A. High-temperature stability and saturation magnetization of superparamagnetic nickel nanoparticles in microporous polysilazane-derived ceramics and their gas permeation properties. Appl. Mater. Interfaces 2014, 6, 12270–12278. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, M. Chapter 7 Polysilazanes. In Inorganic Materials; De Jaeger, R., Gleria, M., Eds.; Nova Science Publishers Inc.: New York, NY, USA, 2007; pp. 371–413. [Google Scholar]
- Iwamoto, Y.; Sato, K.; Kato, T.; Inada, T.; Kubo, Y. A hydrogen-permselective amorphous silica membrane derived from polysilazane. J. Eur. Ceram. Soc. 2005, 25, 257–264. [Google Scholar] [CrossRef]
- Funayama, O.; Kato, T.; Tashiro, Y.; Isoda, T. Synthesis of a polyborosilazane and its conversion into inorganic compounds. J. Am. Ceram. Soc. 1993, 76, 717–723. [Google Scholar] [CrossRef]
- Funayama, O.; Tashiro, Y.; Aoki, T.; Isoda, T. Synthesis and pyrolysis of polyaluminosilazane. J. Ceram. Soc. Jpn. 1994, 102, 908–912. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Kikuta, K.; Hirano, S. Microstructural development of Si3N4–SiC–Y2O3 ceramics derived from polymeric precursors. J. Mater. Res. 1998, 13, 353–361. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Kikuta, K.; Hirano, S. Synthesis of poly-titanosilazanes and conversion into Si3N4–TiN ceramics. J. Ceram. Soc. Jpn. 2000, 108, 350–356. [Google Scholar] [CrossRef]
- Kubo, T.; Tadaoka, E.; Kozuka, H. Formation of silica coating films from spin-on polysilazane at room temperature and their stability in hot water. J. Mater. Res. 2004, 19, 635–642. [Google Scholar] [CrossRef]
- Kubo, T.; Kozuka, H. Conversion of perhydropolysilazane-to-silica thin films by exposure to vapor from aqueous ammonia at room temperature. J. Ceram. Soc. Jpn. 2006, 114, 517–523. [Google Scholar] [CrossRef]
- Miyajima, K.; Eda, T.; Nair, B.N.; Iwamoto, Y. Organic-inorganic layered membrane for selective hydrogen permeation together with dehydration. J. Membr. Sci. 2012, 421–422, 124–130. [Google Scholar] [CrossRef]
- Sokri, M.N.M.; Daiko, Y.; Honda, S.; Iwamoto, Y. Synthesis of microporous amorphous silica from perhydropolysilazane chemically modified with alcohol derivatives. J. Ceram. Soc. Jpn. 2015, 123, 292–297. [Google Scholar] [CrossRef]
- Sokri, M.N.M.; Onishi, T.; Mouline, Z.; Daiko, Y.; Honda, S.; Iwamoto, Y. Polymer-derived amorphous silica-based inorganic-organic hybrids having alkoxy groups: Intermediates for synthesizing microporous amorphous silica materials. J. Ceram. Soc. Jpn. 2015, 123, 732–738. [Google Scholar] [CrossRef]
- Seyferth, D.; Wiseman, G.; Prud’homme, C. A liquid silazane precursor to silicon nitride. J. Am. Ceram. Soc. 1983, 66, C-13–C-14. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C.; Morrill, T.C. Spectrometric Identification of Organic Compounds, 5th ed.; John Wiley and Sons, Inc.: New York, NY, USA, 1991. [Google Scholar]
- Iwamoto, Y.; Matsunaga, K.; Saito, T.; Völger, W.; Kroke, E.; Riedel, R. Crystallization behaviors of amorphous Si–C–N ceramics derived from organometallic precursors. J. Am. Ceram. Soc. 2001, 84, 2170–2178. [Google Scholar] [CrossRef]
- Stein, S.E. Mass Spectra in NIST Chemistry WebBook, NIST Standard Reference Database Number 69. Available online: http://webbook.nist.gov (accessed on 2 April 2015).
- Cruz, J.M.D.; Lozovoy, V.V.; Dantus, M. Quantitative mass spectrometric identification of isomers applying coherent laser control. J. Phys. Chem. Lett. A 2005, 109, 8447–8450. [Google Scholar]
- Johnstone, R.A.W. Mass Spectrometry for Organic Chemists; Cambridge Chemistry Texts; Cambridge University Press: Cambridge, UK, 1972; pp. 64–67. [Google Scholar]
- Watson, J.T.; Sparkman, D. Introduction to Mass Spectrometry: Instrumentation, Applications, and Strategies for Data Interpretation, 4th ed.; John Wiley and Sons, Inc.: New York, NY, USA, 2013; pp. 368–376. [Google Scholar]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solid and Powders; Springer: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Breck, D.W. Zeolite Molecular Sieves; John Wiley and Sons, Inc.: New York, NY, USA, 1974; p. 636. [Google Scholar]
- Li, Y.; Feng, Z.; Lian, Y.; Sun, K.; Zhang, L.; Jia, G.; Yang, Q.; Li, C. Direct synthesis of highly ordered Fe-SBA-15 mesoporous materials under weak acidic conditions. Microporous Mesoporous Mater. 2005, 84, 41–49. [Google Scholar] [CrossRef]
- Apopei, A.I.; Buzgar, N.; Buzatu, A. Raman and infrared spectroscopy of kaersutite and certain common amphiboles. AUI Geol. 2011, 57, 35–58. [Google Scholar]
- Makreski, P.; Jovanovski, G.; Gajovic, A. Minerals from Macedonia XVII. Vibrational spectra of some common appearing amphiboles. Vib. Spectrosc. 2006, 40, 98–109. [Google Scholar] [CrossRef]
- Ventura, G.D.; Robert, J.-L.; Beny, J.-M. Tetrahedrally coordinated Ti4+ in synthetic Ti-rich potassic richterite: Evidence from XRD, FTIR, and Raman studies. Am. Miner. 1991, 76, 1134–1140. [Google Scholar]
- Uchino, T.; Kitagawa, Y.; Yoko, T. Structure, energies, and vibrational properties of silica rings in SiO2 glass. Phys. Rev. B 2000, 61, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Matson, D.W.; Sharma, S.K.; Philpotts, J.A. Raman spectra of some tectosilicates and of glasses along the orthoclase-anorthite and nepheline-anorthite joins. Am. Miner. 1986, 71, 694–704. [Google Scholar]
- Kingma, K.J.; Hemley, R.J. Raman spectroscopic study of microcrystalline silica. Am. Miner. 1994, 79, 269–273. [Google Scholar]
- Oyama, S.; Lee, D.; Hacarlioglu, P.; Saraf, R.F. Theory of hydrogen permeability in nonporous silica membranes. J. Membr. Sci. 2004, 244, 45–53. [Google Scholar] [CrossRef]
- Hacarlioglu, P.; Lee, D.; Gibbs, G.V.; Oyama, S. Activation energies for permeation of He and H2 through silica membranes: An ab initio calculation study. J. Membr. Sci. 2008, 313, 277–283. [Google Scholar] [CrossRef]
- Saito, A.; Foley, H. Curvature and parametric sensitivity in models for adsorption in micropores. AIChE J. 1991, 37, 429–436. [Google Scholar] [CrossRef]
- Barrett, P.; Joyner, G.; Halenda, P.H. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
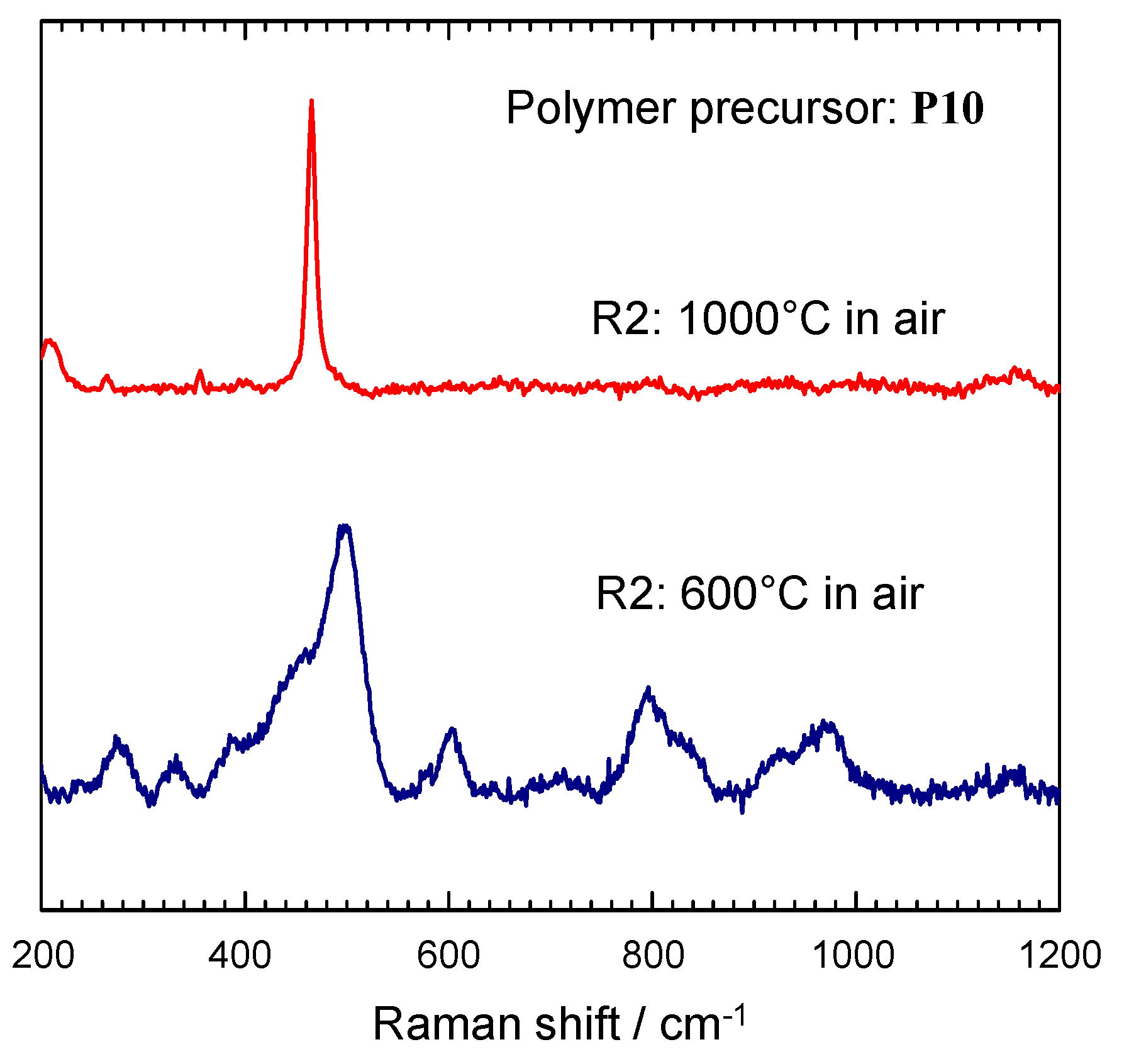{kind=link}
| Polymer | Conversion Route | Flow Gas | Temp./°C | Composition/wt % | Empirical Ratio | ||||
|---|---|---|---|---|---|---|---|---|---|
| Si | C | O | N | H | |||||
| P0 | R1 | air | 600 | 68.1 | 0.0 | 30.2 | 0.7 | 0.0 | Si1.0C0.00O0.78N0.02H0.00 |
| R2 | air | 600 | 59.1 | 0.0 | 39.3 | 0.6 | 0.0 | Si1.0C0.00O1.17N0.02H0.00 | |
| R3 | Ar | 600 | 54.5 | 0.0 | 42.4 | 1.9 | 0.2 | Si1.0C0.00O1.37N0.07H0.10 | |
| R4 | Ar | 600 | 65.8 | 0.9 | 4.8 | 26.9 | 0.6 | Si1.0C0.03O0.13N0.82H0.25 | |
| R3 | Ar | 1000 | 58.1 | 0.0 | 40.2 | 0.5 | 0.2 | Si1.0C0.00O1.21N0.02H0.10 | |
| R4 | Ar | 1000 | 63.4 | 0.9 | 5.0 | 29.1 | 0.6 | Si1.0C0.03O0.14N0.92H0.26 | |
| P5 | R1 | air | 600 | 64.7 | 0.0 | 34.2 | 0.1 | 0.0 | Si1.0C0.00O0.93N0.003H0.00 |
| R2 | air | 600 | 60.7 | 0.0 | 38.2 | 0.1 | 0.0 | Si1.0C0.00O1.10N0.003H0.00 | |
| R3 | Ar | 600 | 55.6 | 6.2 | 36.2 | 0.6 | 0.4 | Si1.0C0.26O1.13N0.02H0.20 | |
| R3 | Ar | 1000 | 52.7 | 4.1 | 41.8 | 0.3 | 0.1 | Si1.0C0.18O1.39N0.01H0.05 | |
| P10 | R1 | air | 600 | 64.7 | 0.0 | 34.3 | 0.0 | 0.0 | Si1.0C0.00O0.93N0.00H0.00 |
| R2 | air | 600 | 63.1 | 0.0 | 35.9 | 0.0 | 0.0 | Si1.0C0.00O1.00N0.00H0.00 | |
| R3 | Ar | 600 | 51.9 | 9.5 | 36.6 | 0.1 | 0.9 | Si1.0C0.43O1.24N0.004H0.48 | |
| R4 | Ar | 600 | 57.7 | 6.7 | 30.2 | 3.0 | 1.4 | Si1.0C0.27O0.92N0.10H0.68 | |
| R2 | air | 1000 | 63.6 | 0.0 | 35.4 | 0.0 | 0.0 | Si1.0C0.00O0.98N0.00H0.00 | |
| R3 | Ar | 1000 | 49.4 | 6.1 | 42.3 | 0.0 | 1.2 | Si1.0C0.29O1.50N0.00H0.68 | |
| R4 | Ar | 1000 | 59.6 | 8.4 | 14.9 | 14.4 | 1.7 | Si1.0C0.33O0.44N0.48H0.79 | |
| ν, (Si–O–Si)/cm−1 | Number of SiO4 in Rings | Notes | Reference |
|---|---|---|---|
| 381 (νs) | 6 | Observe in α-Carnegieite | Matson et al. [48] |
| 463 (νs) | 6 | Observe in moganite | Kingma and Hemley [49] |
| 465 (νs) | 6 | Observe in α-quartz | Kingma and Hemley [49] |
| 498 (νs) | 4 + 6 | Observe in Leucite | Matson et al. [48] |
| 606 (νs) | 3 | - | Uchino et al. [48] |
| 800 (νas) | - | O–Si–O vibration (asymmetric stretching mode) | Li et al. [43] |
| 978 (νas) | - | Si–O–Si bond due to defect by surface silanol group (asymmetric stretching mode) | Li et al. [43] |
| 1000–1200 (νas) | - | Asymmetric stretching SiO4 vibrations | Apopei et al. [44] |
| Makreski et al. [45] | |||
| Ventura et al. [46] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohd Sokri, M.N.; Daiko, Y.; Mouline, Z.; Honda, S.; Iwamoto, Y. Formation of Micro and Mesoporous Amorphous Silica-Based Materials from Single Source Precursors. Inorganics 2016, 4, 5. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics4010005
Mohd Sokri MN, Daiko Y, Mouline Z, Honda S, Iwamoto Y. Formation of Micro and Mesoporous Amorphous Silica-Based Materials from Single Source Precursors. Inorganics. 2016; 4(1):5. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics4010005
Chicago/Turabian StyleMohd Sokri, Mohd Nazri, Yusuke Daiko, Zineb Mouline, Sawao Honda, and Yuji Iwamoto. 2016. "Formation of Micro and Mesoporous Amorphous Silica-Based Materials from Single Source Precursors" Inorganics 4, no. 1: 5. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics4010005