Synthesis and Reactivity of Mn–CF3 Complexes

Department of Chemistry and Biomolecular Sciences and Centre for Catalysis Research and Innovation, University of Ottawa, 30 Marie Curie, Ottawa, ON K1N 6N5, Canada
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(1), 3; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7010003
Submission received: 29 October 2018
/
Revised: 19 December 2018
/
Accepted: 25 December 2018
/
Published: 6 January 2019
(This article belongs to the Special Issue First-Row Transition Metal Complexes)
Abstract
:The synthesis, characterization and reactivity of several bi- and tridentate, N-ligated manganese carbonyl trifluoromethyl complexes are presented. These complexes exhibit elongated Mn–CCF3 bonds (versus Mn(CF3)(CO)5), suggesting a lability that could be utilized for the transfer or insertion of the CF3 functional group into organic substrates. Unlike their Mn–X congeners (X = Cl, Br), these Mn–CF3 complexes exhibit a preference for hard donor ancillary ligands, thus enabling the synthesis of 4 N-ligated Mn–CF3 complexes including a mixed-donor tridentate complex using an NNS Schiff base ([2-(methylthio)-N-(1-(pyridin-2-yl)ethylidene)aniline]). Although we have not yet identified efficient CF3 transfer reactions, fluoride abstraction from the Mn–CF3 complexes using trimethylsilyl triflate affords the first stable Mn fluorocarbenes as evidenced by 19F NMR spectroscopy.
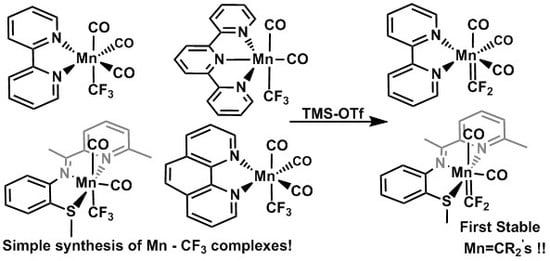
1. Introduction
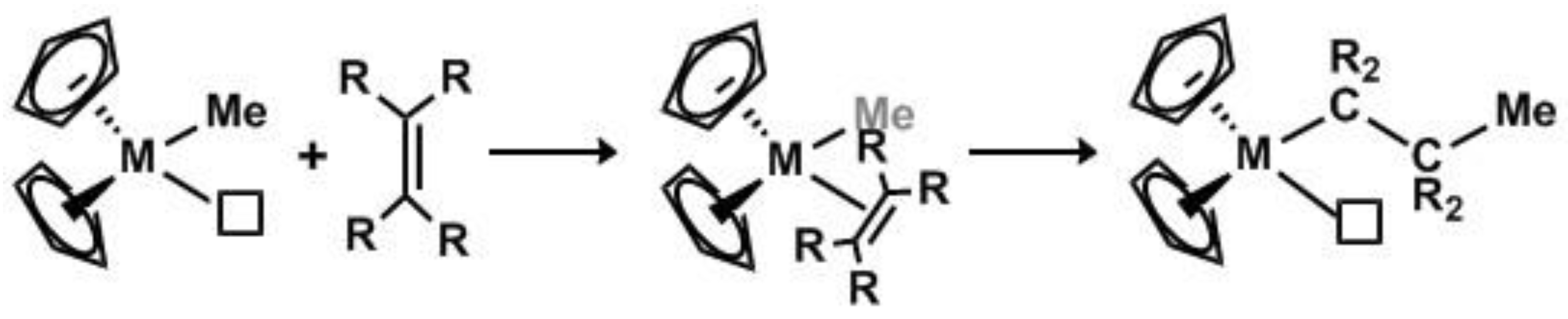
Organometallic compounds and especially metal alkyls (M–R) are immensely important players in catalysis [1,2]. Catalysis utilizing metal fluoroalkyl complexes, however, is less common due to the inherent stability of metal perfluoroalkyl bonds (M–RF) [3,4,5]. Nonetheless, these compounds are useful to the increasingly important field of fluoro-organic synthesis [6,7,8,9,10,11,12,13,14]. Prominent examples include [Cu]−RF reagents for stoichiometric perfluoroalkyl transfer to organic substrates [15,16,17,18,19,20] and increasing numbers of transition metal (e.g., Cu, Ni, Pd) catalyzed C−RF (where RF is usually CF3) bond-forming processes [21,22], which can be used to obtain high-value fluorinated pharmaceuticals and agrochemicals [4,6,8]. One unsolved challenge, however, involves metal-catalyzed polymerization of fluoroalkenes via the Cossee–Arlman mechanism as commonly practiced with metallocene or Ziegler–Natta type catalysts (Scheme 1).
Early reports by Ziegler and coworkers of metal-catalyzed alkene polymerization promoted by Et2AlCl have evolved 5 decades later to a tour de force of organometallic chemistry with molecular control of polypropylene tacticity, living catalysts for block co-polymer formation, and late metal chain-walking as only three of many highlights [23,24,25]. In contrast, polymerization of fluoroalkenes traditionally utilizes radical processes, either in the gas phase or in aqueous emulsions [26,27,28,29]. As a result, while the properties of fluoropolymers can be tuned by altering reaction conditions or changing the relative amounts of co-monomers, attempts at molecular control have met with little success. Early work by Sianesi and Caporiccio reported the polymerization of hexafluoropropene (HFP) using a traditional Ziegler-Natta catalytic system [(Ti(OiPr)4 + Al(iBu)3] over 15 days to produce a material thought to be low mol. wt. poly-HFP, but no follow-up investigations have appeared [30]. Similar work utilizing a chromium metallocene catalyst [(Cr(C6H6)2] to produce dimers and trimers of HFP [31] purportedly through a metal-mediated process was later suggested to proceed via a soluble source of fluoride ion [32]. Finally, research by Kiplinger, Hughes, and co-workers demonstrated that decomposition of the shock-sensitive Cp2TiF(CF3) complex produced various insoluble oligomers in which both –(CF2CF2)– and –(CF2CFH)– units were identified. The precise mechanism for the formation of these fluoro-oligomers, including whether Ti nanoparticles were involved, was never determined [3].
As can be seen from this previous research, metal-mediated formation of fluoro-oligomers and polymers only occurs under unusual conditions. This is certainly not unexpected as the best alkene polymerization catalysts are typically electrophilic. Moreover, attempts to use nucleophilic metal alkyl complexes typically result in stable fluoroalkene complexes that in some cases are better thought of as metallacyclopropanes due to extensive metal to alkene back-bonding. A rare well-characterized example involving insertion of a fluoroalkene into a metal alkyl was reported by Wilford and Stone (Equation (1)) [33]. Notably, they also observed that further insertion of TFE into the M-CRF bond did not occur.
M(CH3)(CO)5 + CF2=CF2 → M(CF2CF2CH3)(CO)5 (M = Mn, Re)
Inspired by this work, we hypothesized that the half-filled d shell of a Mn(II) fluoroalkyl complex may contain a weak enough M–CRF bond to allow for multiple alkene insertions. Indeed, Fujisawa, Nubika, and co-workers showed that several tris(pyrazolyl)-borate and -methane-ligated Mn(II) halide complexes activated by Al(i-Bu)3 and [Ph3C][B(C6F5)4] are effective propylene polymerization catalysts [34]. For insertion of electron-poor fluoroalkenes, however, we would need a neutral Mn(II) complex with strongly electron-donating ligands. Computational studies of M–CF3 versus M–CH3 show that CF3 groups are significant σ-donors despite being considered a strong electron withdrawing group in organic chemistry. This, combined with their weak π-acceptor attributes, leads to significantly increased electron density on metal centers. [35] This research goes on to mention that while CF3 groups tend to stabilize the metal d-orbitals, making them less reactive towards electrophiles, only group 7 complexes showed an increase in the overall negative natural charge on the metal center. For this reason we pursued the formation of Mn–CF3 complexes to determine their reactivity towards fluoroalkenes. As Mn(II) prefers ”hard” ligands we initiated our study with typical N-donor ligands. After repeated unsuccessful attempts to install the CF3 ligand on Mn(II) precursors, we prepared a variety of monovalent Ln(CO)5−nMn(I)–CF3 (n = 1–3; L = bi- and tridentate N-donors) complexes with a view to eventual oxidation to Mn(II).
The few examples of Mn–CF3 complexes are either derived from the original synthesis of (CO)5MnCF3, (1), by McClellan in the 1960s [36] or from a more recent route involving reactions of Mn(CO)5Br with AgPF6 and then Cd(RF)2 reagents [37]. The first synthesis of (1) involved treatment of Na[Mn(CO)5] with trifluoroacetic anhydride (TFAA) forming Mn(CO)5COCF3 [38]. Sublimation of this compound at 100 °C not only separates it from the Na[OCOCF3] salt but also partially decarbonylates the Mn–COCF3 unit, yielding a mixture of Mn(CO)5COCF3 and (1). Intriguingly, further workup was not necessary to form several N-ligated Mn–CF3 complexes (vide infra). Here we report the synthesis, characterization and reactivity of these complexes.
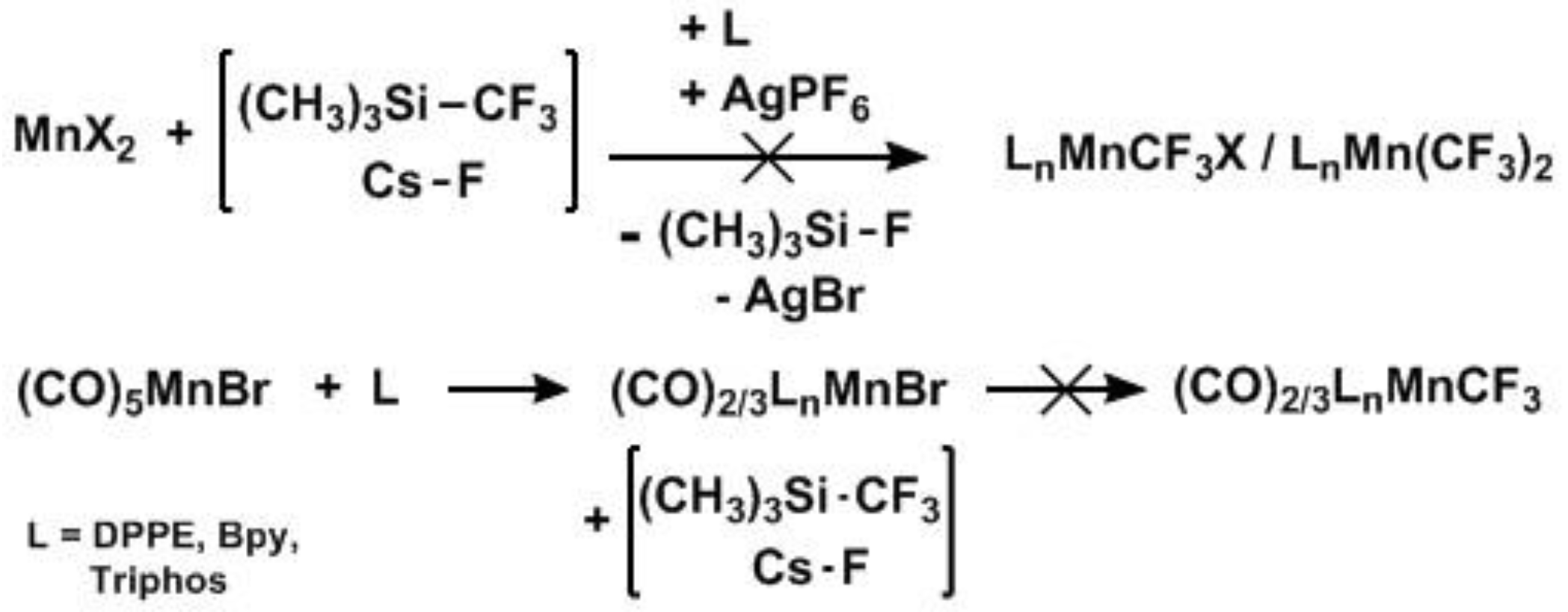
2. Results
Synthesis of Mn(CO)5CF3, (1): Initial attempts at the formation of Mn–CF3 complexes began with reactions of MnX2 complexes (X = OAc, Br, Cl) with the Ruppert-Prakash reagent (trifluoro-methyltrimethylsilane [TMS–CF3] + F− source) in an attempt to synthesize Mn–CF3 complexes directly. (Scheme 2, top) Unfortunately displacement of the X group by CF3 proved difficult. Even after halide abstraction with Lewis acids such as AgPF6, addition of CF3 anions was unsuccessful. Moving to a Mn(I) source, (CO)5MnBr, allowed for an easy exchange of CO groups with various ligands [39,40,41,42] but displacement of Br with CF3 again was problematic, even after abstraction with AgPF6. While we chose to avoid toxic and difficult to prepare Cd(CF3)2, this may offer an alternate route to Mn–CF3 complexes with soft donors such as phosphines. Instead, we utilized the carbonyl Mn–CF3 source, following the report of McClellan [36]. The initially obtained manganese perfluoroacyl/salt mixture was sublimed at 100 °C in a static vacuum affording light yellow crystals of Mn(CO)5COCF3 and 1 in a 4:6 ratio (by 19F NMR) in 60% yield based on TFAA. Further workup was unnecessary as we found that ligand substitution on the acyl complex was accompanied by rapid decarbonylation at room temperature.
2.1. Substitution Reactions of Mn(CO)5COCF3 to form New Mn(I)–CF3 Complexes
Attempted reactions of the Mn(CO)5CF3/Mn(CO)5COCF3 mixture with various bi- and tridentate P-donor ligands, including: PPh3, PMe3, DPPE, DPPF, DMPE, Tripod, Triphos, P(OiPr)3, P(OPh)3 were unsuccessful as soft donor ligands appeared ineffective at displacing the strongly held carbonyls unlike the similar complexes (CO)5Mn–X complexes (X = Br, I, Me) which have a rich coordination chemistry with soft ligands [39,40,41,42]. Hard donor ligands gave moderate success but reactions required harsh conditions (100 °C, 48 h reaction time) to afford mixtures of starting material and the desired products. As a result, we utilized a known method for the displacement of CO ligands via decarboxylation using trimethylamine N-oxide [43] which yielded relatively pure (≥90%) trimethylamine complexes, [(NMe3)n(CO)5−nMn–CF3], (2), (5) (Scheme 3) which were utilized directly for further syntheses. Reaction of 2 with bipyridine (Bpy) and phenanthroline (Phen) furnished complexes (3) and (4) respectively (50 °C, 12 h, Scheme 4).
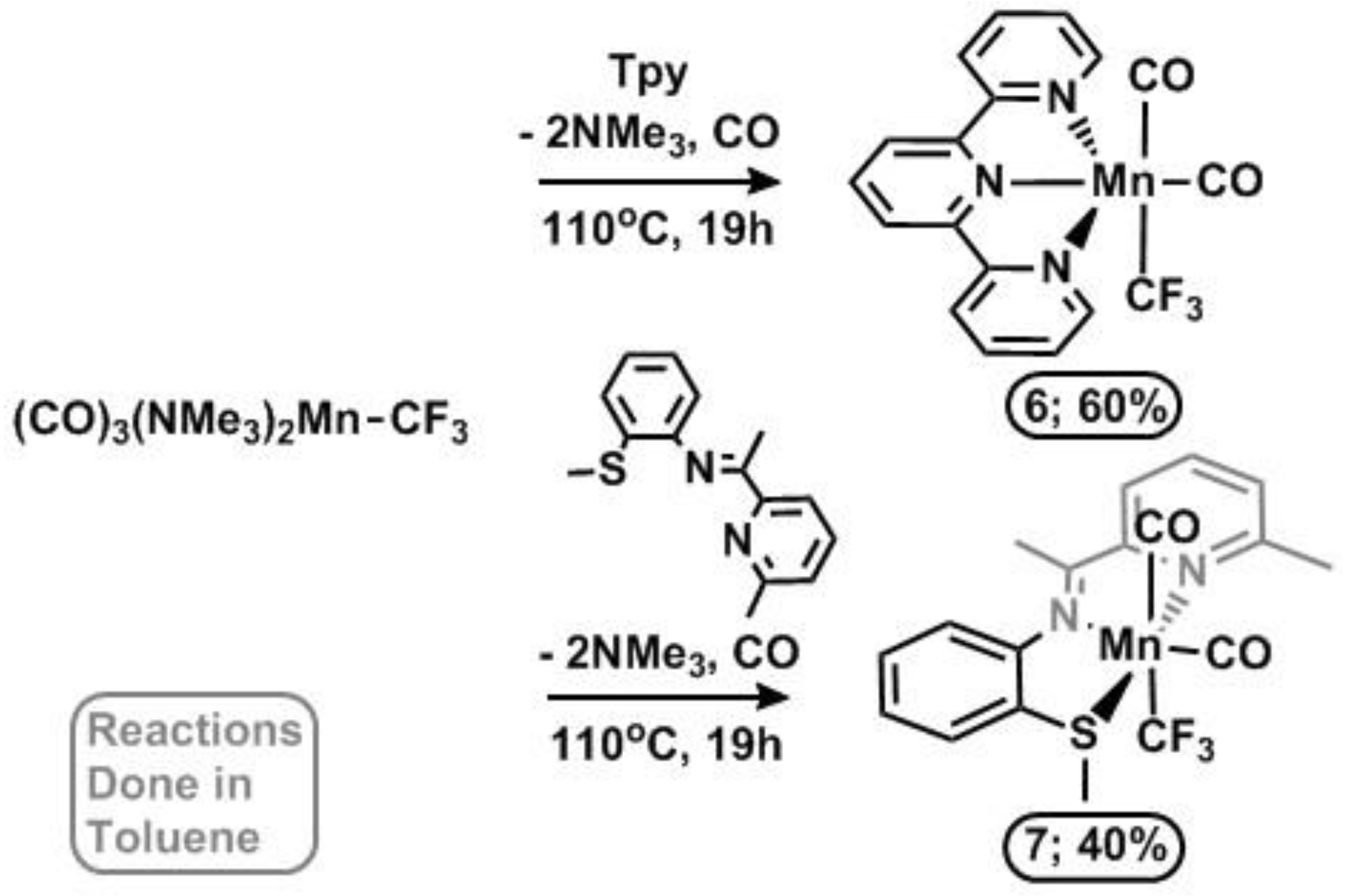
Formation of a tridentate N-ligated Mn–CF3 complex required a modified synthesis. Three equivalents of Me3NO were utilized to form [(Me3N)3(CO)2Mn–CF3], (5), in toluene which was then refluxed with terpyridine (Tpy) (19 h, 100 °C) giving (6) in 60% yield. This method was also utilized with a tridentate mixed-donor NNS Schiff Base [2-(methylthio)-N-(1-(pyridin-2-yl)ethylidene)aniline] [39] to give (7) in 40% yield (Scheme 5). In contrast, reactions of (2) and (5) with soft phosphine ligands gave mixtures of products, again showing the significant effect of the CF3 group given that Mn–X complexes (X = Br, Cl) readily coordinate soft donors [39,40,41,42].
2.2. Solid State Structures of New Mn–CF3 Complexes
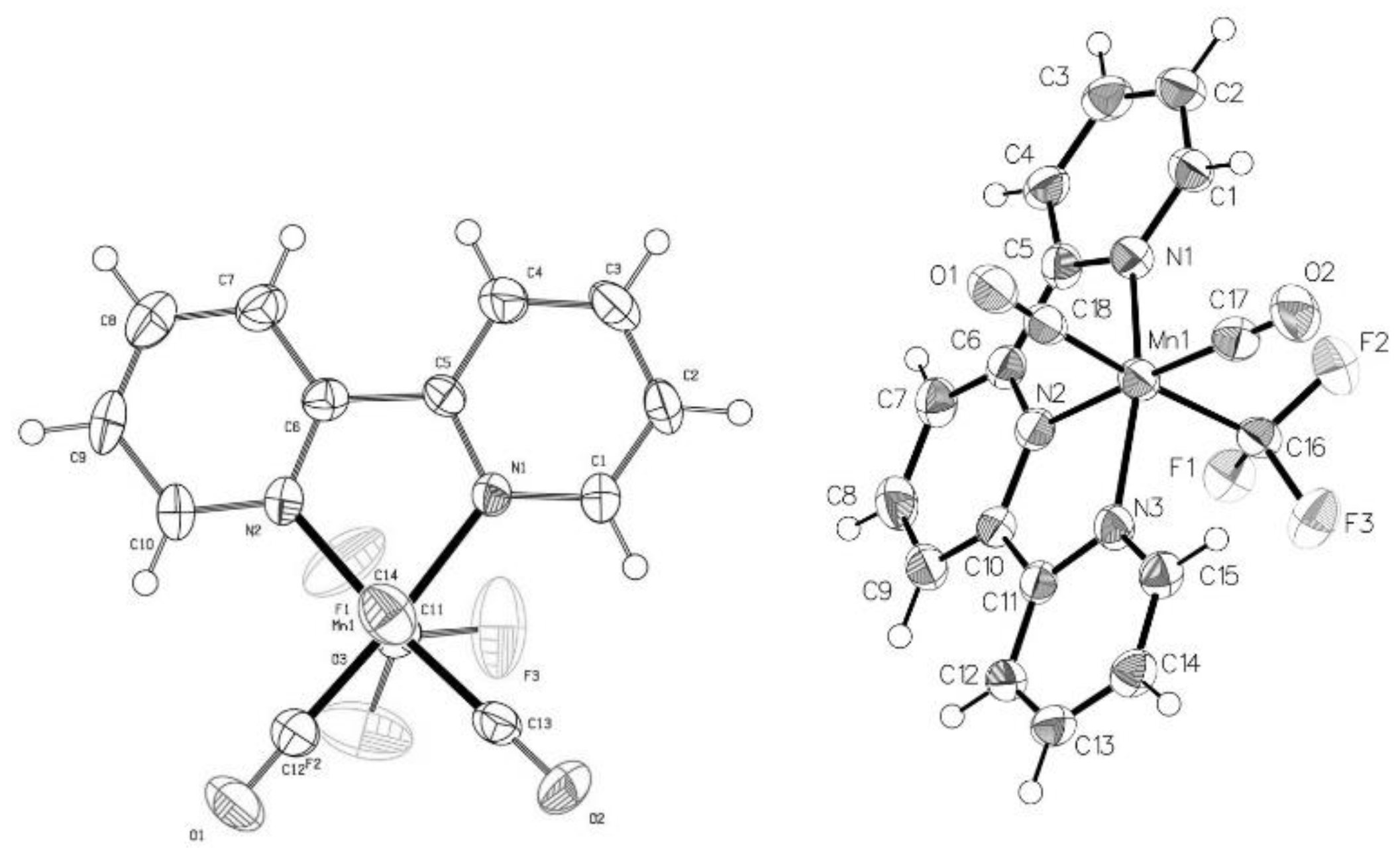
The molecular structures of (3) and (6) determined by single crystal X-ray diffraction are shown in Figure 1 and selected bond lengths are compared to the known Mn(CO)5CF3 complex in Table 1.
In both pseudo-octahedral structures the CF3 ligand is trans to CO. The elongation of the Mn–CCO bond lengths trans to CF3 (versus those that are cis) is consistent with the strong trans influence of the CF3 group [4,32,44]. The Mn–CCF3 bond distance in (3), however, is significantly shorter than that in (1). The reasoning for this observation may be due to more significant π-backbonding into the low-lying C–F σ*orbitals as the N-donor ligand adds more electron density to the metal center [3]. Research by Grushin and Macgregor, however, suggests that M–CF3 bonding has little to no (<8%) π-backbonding character [35]. If significant ionic character is invoked for the Mn–CCF3 bond then the replacement of 2 CO ligands with 2 hard N donors would decrease the Lewis acidity at Mn, increasing its interaction with the partial positive charge of the CF3 carbon [3,4]. In contrast, the Mn–CCF3 bond distance in (6) is significantly longer than that in (1) in spite of the additional N-donors and one less CO ligand competing for the metal’s π-back-donation. This may be due to more significant π-backbonding to the Tpy [45] and CO ligands which all display shorter bond lengths to Mn than those in (3) (Table 1). This would support the ionic interpretation of the bonding between manganese and the trifluoromethyl ligand as electron density is removed from the positive metal center, thereby increasing repulsion with the partially positive CF3 carbon.
2.3. NMR Data for Mn–CF3
The solution phase 19F NMR data in CD3CN for (2)–(4) are consistent with other M–CF3 complexes (M = Fe, Co, Ni, Mn) with the CF3 resonance between −15 and −30 ppm. The perfluoroacyl signal at ca. −90 ppm observed for Mn(CO)5(COCF3) is not present after addition of any donor ligand that causes room temperature decarbonylation to form Mn–CF3 complexes. The 19F NMR spectrum of (7) shows two peaks at −19.88 and −20.43 ppm suggesting the presence of two different coordination isomers (presumably with the CF3 cis- versus trans- to CO; Scheme 6). The alternate possibility of a hemilabile thioether donor [39] was discounted on the basis of variable temperature 19F and 1H NMR experiments that showed no changes in the intensities of the Mn–CF3 or S–Me and N=C(CH3) peaks, respectively. Addition of a coordinating solvent such as CD3CN also had no effect on the integration of these resonances. The 19F and 1H NMR spectra for all Mn–CF3 complexes are characteristically broad due to the 100% abundance of the quadrupolar 55Mn nucleus [46] and 55Mn NMR signals were not observed, presumably due to excessive quadruploar broadening.
2.4. IR data
Selected FT-IR data are listed in Table 2. As expected, the CO stretching frequencies shift to lower energies with increasing σ-donor strength of the ancillary ligands/electron density on the metal centers, reaching a maximum electron density for the Mn(Tpy)(CO)2CF3 and Mn(NNS)–(CO)2CF3 complexes. Again, ionic bonding in complex (6) could explain the longer M–CRF bond distance despite having more electron density on the metal as discussed by Hughes [4].
2.5. Cyclic Voltammetry Data
Cyclic voltammetry (CV) was employed to determine if the Mn–CF3 complexes could be successfully oxidized or reduced without decomposition. Complexes (3), (4), and (7) were all subjected to CV between −2.5 and 2.5 V in a THF/electrolyte solution (0.1 M [(Bu)4N][BF4] supporting electrolyte) with ferrocene as a reference. The cyclic voltammograms of (3), (4), and (7) exhibited irreversible oxidation waves at 0.64, 0.66, and 0.53 V (versus ferrocene) respectively even when faster sweep rates (200 mV/s) were utilized. This suggests that the complexes decompose when oxidized from Mn(I) to Mn(II), most likely due to loss of CO ligands by the coordinatively labile high spin d5 complex due to the absence of ligand field stabilization. However, each of these complexes showed a quasi-reversible reductions at: −2.3 (complex (3)), −2.4 (complex (4)) and −2.2 V (complex (7)). Slower sweep rates of 50 mV/s were used to probe the stability of these reduced complexes and the quasi-reversible waves remained, suggesting the formation of stable Mn(0) species.
2.6. Mass Spectrometry
Electron impact mass spectrometry (EI-MS) was attempted on complexes (3), (4), (6), and (7). Electrospray ionization mass spectroscopy (ESI-MS) was initially performed; however this led to significant fragmentation of the parent ions due to a presumed oxidation to unstable Mn(II) species for all Mn–CF3 complexes. For this reason we turned to EI-MS. Complexes (6) and (7) were too unstable to provide a useful MS spectrum but complexes (3) and (4) gave consistent fragmentation patterns suggesting the presence of the parent ions. The fragment observed for (3) was [(N–N)(F)Mn=CF2]+ (280.00375 Da, 0.2% int.; N–N = Bipy) derived from loss of all CO ligands followed by intramolecular α-F abstraction by the now electron-rich Mn center. This was corroborated by the presence of both [(N–N)Mn–CF3]+ (303.99905 Da, 0.1% int.) and [(N–N)Mn–F]+ (254.00625 Da, 2.25% int.){N–N = Phen} fragment ions in the EI-MS of (4). EI-MS spectral data and proposed reaction pathway for gas-phase formation of [(N–N)MnF]+ and [N–N](F)Mn=CF2]+ are available in the Supplementary Materials.
2.7. Reactivity
Due to the successful insertion of tetrafluoroethylene (CF2=CF2; TFE) into Mn–H and Mn–CH3 [33] and encouraged by the elongated M–CF3 bonds possessed by our new complexes, we pursued reactions with fluoroalkenes such as vinylidene fluoride (CF2=CH2; VDF) and TFE. Unfortunately, even under 5 bar of VDF and higher temperatures (80 °C), these reactions failed to produce the desired insertion products. At first it was suspected that this may have been due to the inability to dissociate one of the remaining CO ligands, thus preventing coordination of the fluoroalkenes. For this reason we moved to the NNS complex (7) where displacement of the soft donor thiol group may allow for the coordination of olefins. This complex was still unable to coordinate the fluoroalkenes (TFE, VDF) even under forcing conditions. Attempts to labilize the CO ligands by oxidizing (3) or (4) to Mn(II) complexes utilizing [Fe(Cp)2][BF4] was successful, but without crystal field stabilization, the newly formed high-spin d5 Mn complexes decomposed to form [(N–N)3Mn][BF4]2 and other unidentified Mn(II) complexes as confirmed by cyclic voltammetry (vide supra). Additionally, as M–CF3 complexes are known to stabilize higher oxidation states, we attempted to form Ar–CF3 compounds through oxidative addition of Ar–I and subsequent reductive elimination of the desired compounds. However, reactions of aryl halides with complexes (3), (4), (6), and (7) showed no change by 19F NMR regardless of reaction temperature or solvent.
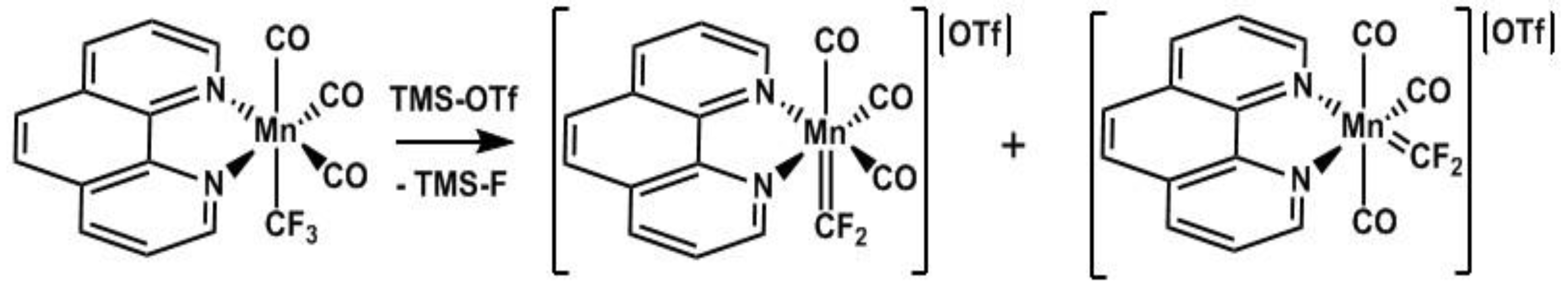
Finally, we investigated fluoride abstraction with a Lewis acid. Previous research invoked the formation of Mn=CF2 carbenes as intermediates in C-halide exchange reactions but the Mn(CO)5 unit was unable to stabilize the electron deficient CF2 group [47,48]. In contrast, addition of trimethylsilyl triflate (TMS-OTf) to complex (4) gave a color change within minutes and the 19F NMR spectrum revealed new resonances at 155.5 and 156.3 ppm suggesting formation of the first stable Mn=CF2 carbene complex, cis/trans-{[(Phen)(CO)3Mn=CF2][OTf]}, 8 (Scheme 7). Given that this new carbene is cationic it is assumed that it will be strongly electrophilic unlike previous examples that our group has reported [49,50,51].
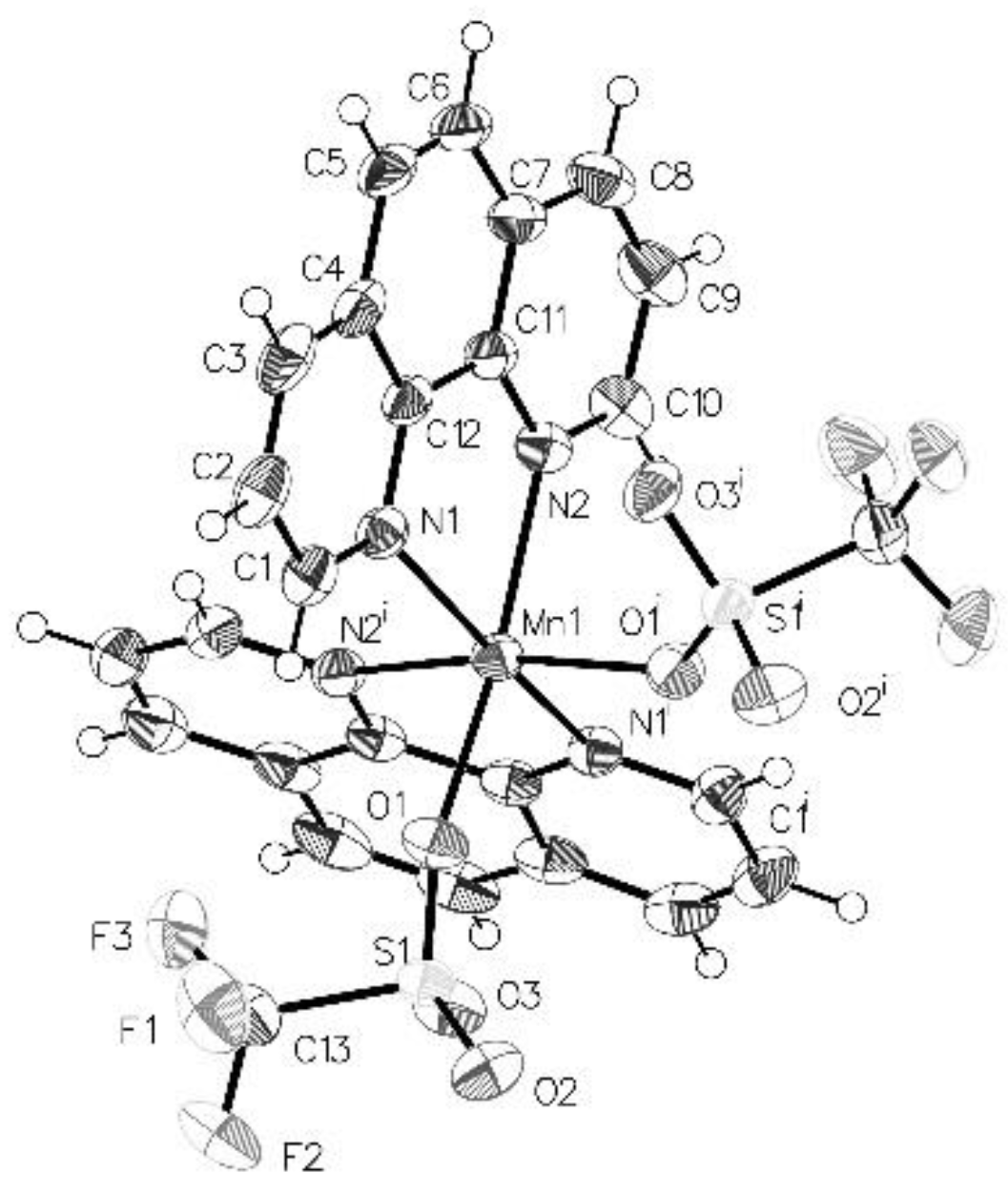
The above transformation serves as a general preparative method using various Mn–CF3 complexes as 19F NMR spectra revealed the formation of new carbenes (as isomeric mixtures) when complexes (3), (4), and (7) were subjected to fluoride abstraction using TMS-OTf . Unfortunately, even with multiple N donors the carbene complexes tended to decompose over 24 h at room temperature, precluding us from obtaining elemental analyses. Given the rarity of first-row metal fluorocarbenes, preliminary reactivity studies were undertaken with ethylene. Monitoring the reaction of complex 8 with ethylene by 19F NMR showed complete consumption of the carbene after 12 h at room temperature. Although several new resonances were observed, none could be assigned to the expected cyclopropanation product [52]. X-ray quality crystals obtained from the reaction solution revealed a new divalent product, Mn(Phen)2(OTf)2, 9 (Figure 2). Details of this redox reaction are as yet unclear and further reactivity investigations of 8 are underway.
3. Materials and Methods
General Procedures: Experiments were conducted under nitrogen, using Schlenk techniques or an MBraun glovebox. All solvents were deoxygenated by purging with nitrogen. Toluene, hexanes, diethyl ether (DEE), and tetrahydrofuran (THF) were dried on columns of activated alumina using a J. C. Meyer (formerly Glass Contours) solvent purification system. Dichloromethane (DCM) and CDCl3 were dried by refluxing solution over calcium hydride (CaH2) followed by distillation. C6D6 was dried over activated alumina (heated at 300 °C >8 h under vacuum) (~15 wt %). All solvents were stored over activated (heated at 250 °C for >6 h under vacuum) 4 Å molecular sieves. Glassware was oven-dried at 150 °C for >2 h. The following chemicals were obtained commercially: Mn2(CO)10 (Strem, 98%, Newburyport, MA, USA),trifluoroacetic anhydride (Aldrich, >99%), 2,2′-bipyridine (Strem, 98+%), 1,10-phenanthroline (Strem, anhydrous 99%), 2,2′:6′,2′′-terpyridine (Aldrich, ≥98.5, trimethylsilyl triflate (Aldrich, 98%, Oakville, ON, Canada), C6D6/CDCl3/CDCN (Cambridge Isotope Laboratories, d-99.5%). Mn(CO)5CF3 and the NNS ligand were prepared following literature procedures [36,39]. 1H, 19F, and 31P{1H} NMR spectra were recorded on 300 MHz Bruker Avance or AvanceII instruments (Bruker, Billerica, MA, USA) at RT (21–23 °C). 1H NMR spectra were referenced to the residual proton peaks (C6D6: 7.16 ppm; CDCl3: 7.26 ppm). 19F NMR spectra were referenced to internal 1,3-bis(trifluoromethyl)-benzene (BTB) (Aldrich, 99%), set to −63.5 ppm. 19F NMR yields were calculated from product integration relative to a known quantity of BTB using 9 s delay times. 31P{1H} NMR data were referenced to external H3PO4, set to 0.0 ppm. IR data were obtained on a Nicolet NEXUS 670 FT-IR spectrometer using neat/solid samples by allowing a DCM solution of compounds (3), (4), (6), and (7) to evaporate on a NaCl plate under a stream of nitrogen. Elemental analyses were performed at the University of Ottawa. Electrochemical measurements were performed using a Princeton Applied Research (PAR) VersaSTAT 3 potentiostat/galvanostat/frequency response analyzer (Ametek Scientific Instruments, Mississauga, ON, Canada) and V3-Studio electrochemical software version 1.0.281 (Ametek Scientific Instruments, Mississauga, ON, Canada, 2008) (PAR) employing a three compartment glass cell containing a 5 mmol THF/electrolyte solution of each complex (0.1 M [(Bu)4N][BF4]). Mass spectroscopy was performed on a Kratos Analytical–Concept Magnetic sector Electron impact mass spectrometer (Kratos Analytical, Wharfside, Manchester, UK).
Modified Synthesis of Mn(CO)5CF3/Mn(CO)5COCF3 (1). Synthesis followed the procedure of McClellan and co-workers [36] but the complex was not further purified. All following preps utilized this starting material as a mixture of Mn(CO)5CF3 and Mn(CO)5COCF3 after sublimation. The complexes decarbonylated spontaneously following association of the N-donor ligand.
Synthesis of Mn(CO)5−n(NMe3)nCF3 intermediates, n = 1 (2) and 3 (5). Me3NO ([34 mg × n]; n = 1 or 3) was combined with a THF solution [3 mL; Preps (3) and (4)] or toluene [3 mL; Preps (6) and (7)] of Mn(CO)5CF3/Mn(CO)5COCF3 (100 mg, 0.38 mmol) (Note: if the solids are combined without solvent a reaction occurs decomposing the two starting materials) once combined the solution changed color from light yellow to orange and significant gas release was observed. The solution was stirred for 3 h at room temperature forming impure Mn(CO)5−n(NMe3)nCF3 intermediates; Yield: 75% based on 19F NMR. The preps for complexes (3), (4), (6), and (7) utilized these products directly without further workup.
Synthesis of (3). Bipy (59.4 mg, 0.38 mmol) was added to a THF solution (ca. 3 mL) of Mn(CO)4(NMe3)CF3 (2) and then heated at 50 °C for 24 h. The suspension was cooled to −34 °C overnight before the solid was collected by filtration and washed with hexanes (3 × 1.0 mL) followed by cold Et2O (2 × 0.2 mL). The solid was then dried under reduced pressure giving a yellow solid. Yield: 73 mg, 53% based on Mn(CO)5CF3/Mn(CO)5COCF3. IR (neat): 2360 (w), 2330 (w), 2020 (s), 1920 (s), 1620 (w), 1600 (w), 1470 (w), 1450 (w), 1320 (w), 1240(w), 1230(w), 1170 (w), 1130 (w), 1050 (m), 953 (m), 889(w), 852 (w), and 768(m) cm−1. 1H NMR (300 MHz, CDCl3): 9 (unresolved multiplet, 2H), 8.3 (unresolved multiplet, 2H), 8.2 (unresolved multiplet, 2H), 7.6 (unresolved multiplet, 2H). 19F NMR (282 MHz, CDCl3): −21.1 ppm (br s, CF3).
Synthesis of (4). Phen (68.5 mg, 0.38 mmol) was added to a THF solution (ca. 3 mL) of Mn(CO)4(NMe3)CF3 (2) and the solution was heated to 50 °C for 19 h. Hexanes (8 mL) was added to the now yellow solution to induce precipitation. The suspension was cooled to −34 °C overnight before the solid was collected by filtration and washed with hexanes (3 × 1.0 mL) followed by cold Et2O (2 × 0.2 mL). The solid was then dried under reduced pressure giving a yellow solid. Yield: 85 mg, 60% based on Phen. IR (neat): 2960 (w), 2920 (w), 2850 (w), 2010 (s), 1930 (s), 1650 (w), 1430 (m), 1350 (m), 1050 (m), 949 (m), and 849 (w). 1H NMR (300 MHz, CDCl3): δ 9.42 (unresolved d, 2H), 8.43 (unresolved dd, 2H), 8.0 (multiplet, 2H), 7.8 ppm (unresolved d, 2H). 19F NMR (282 MHz, CDCl3): −20.9 ppm (br s, CF3).
Synthesis of (6). Me3NO (85.6 mg, 1.14 mmol; 3 equiv.) was added to a toluene solution (ca. 6 mL) of Mn(CO)5CF3/Mn(CO)5COCF3 (100 mg, 0.38 mmol) and the solution was stirred at RT for 2 h until the solution was light orange. Tpy (88.7 mg, 0.38 mmol) was added to the solution which was then refluxed under nitrogen at 110 °C for 4 h. The final solution was red. Hexanes was added to the solution to precipitate red crystals. The solution was cooled to −32 °C for 3 h before the solid was collected on a glass frit and washed with hexanes (3 × 1.5 mL) followed by Et2O (3 × 1.0 mL) then dried under reduced pressure. Yield: 105 mg, 67% based on Tpy. IR (neat): 2970 (w), 2930 (w), 2850 (w), 2360 (w), 2330 (w), 2020 (s), 1900 (s), 1850 (s), 1600 (w), 1590 (w), 1560 (w), 1460 (w), 1430 (w), 1260 (w), 1050 (s), 953 (m), and 769 cm−1 (m). 1H NMR (300 MHz, CDCN): 9.3–8.5 (broad multiplets; 2H), 8.5–7.8 (broad multiplets, 6H), 7.8–7 ppm (broad multiplets, 3H). 19F NMR (282 MHz, CDCl3): −20.9 ppm (br s, CF3).
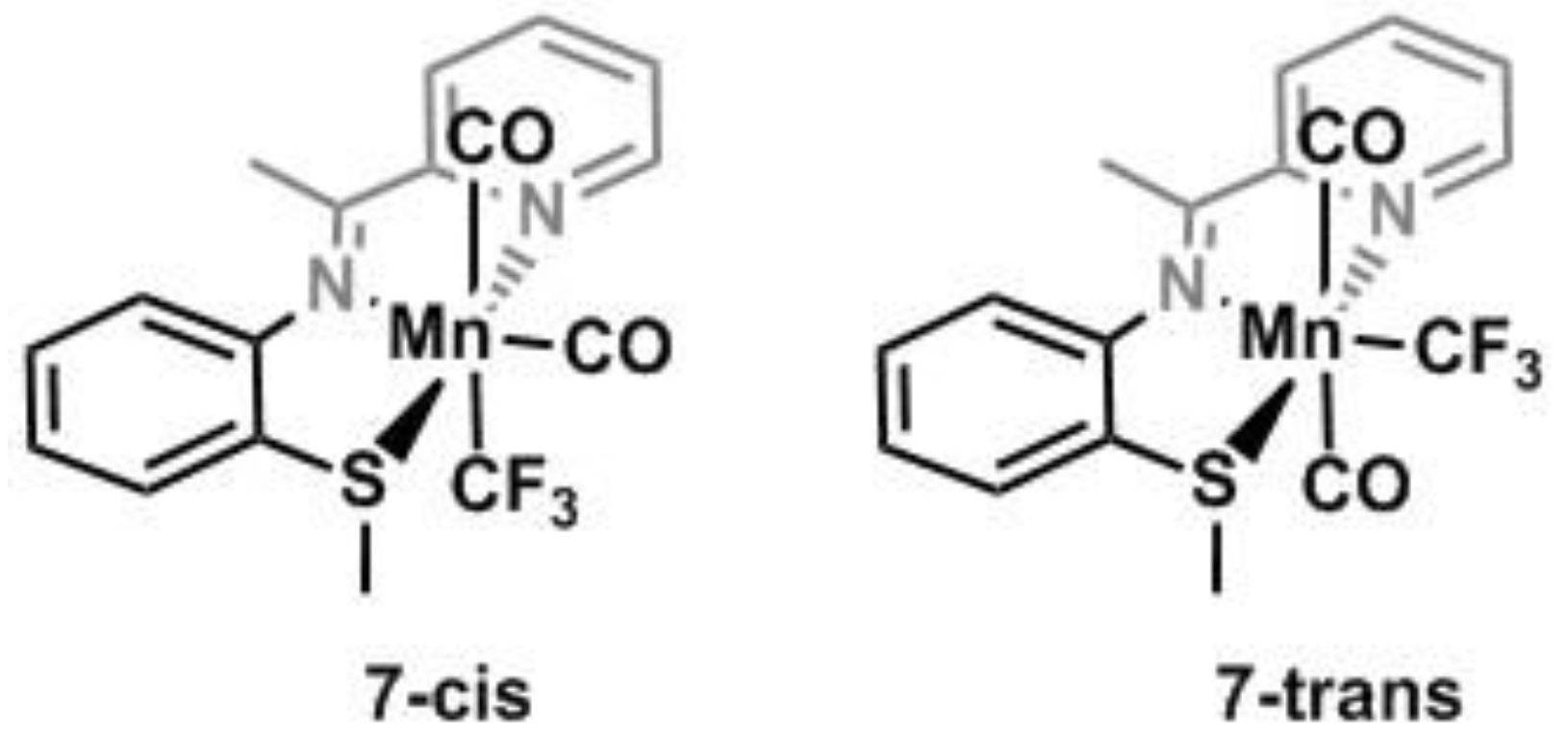
Synthesis of (7)-cis, (7)-trans. Me3NO (85.6 mg, 1.14 mmol; 3 equiv.) was added to a toluene solution (ca. 6 mL) of Mn(CO)5CF3/Mn(CO)5COCF3 (100 mg, 0.38 mmol) and the solution was stirred at RT for 2 h until the solution was light orange. NNS (97 mg, 0.38 mmol) was added to the solution after 3 h and the solution was refluxed at 110 °C overnight. After cooling, the solvent was removed under vacuum leaving an orange powder which was dissolved in a minimum of toluene and cooled to −32 °C overnight. The following day the solid was collected on a glass frit, washed with hexanes (3 × 1.5 mL) and dried under reduced pressure. Yield: 85 mg, 53% based on Mn(CO)5CF3/Mn(CO)5COCF3. IR (neat): 2960 (m), 2930 (m), 2850 (m), 2020 (s), 1920 (s), 1910 (s), 1470 (w), 1440 (w), 1380 (w), 1330 (w), 1260 (w), 1050 (s), 970 (m), 943 (m), 775 (w), 748 (w), 681 cm−1 (w). 1H NMR (300 MHz, CD3CN): 7-trans: δ 8.75 (broad singlet; 1H), 7.4–6.23 ppm (overlapping multiplets; aryl-H’s), 1.86 (broad singlet; 3H), 1.35 ppm (broad singlet; 3H); (7)-cis: 7.71 (broad multiplet; 1H), 7.4–6.23 (overlapping multiplets: aryl-H’s), 3.55 (broad singlet; 3H), 1.62 (broad singlet; 3H). 19F NMR (282 MHz, CD3CN): 7-trans: −20.43 ppm (br singlet, CF3); (7)-cis: −19.6 ppm (br s, CF3).
Synthesis of cis/trans-[(Phen)(CO)3Mn=CF2][OTf], (8). A glass vial was charged with 3 (25 mg, 0.064 mmol) and dissolved in DCM (3 mL). To this solution was added TMS-OTf (12 μL, 0.064 mmol) and the reaction was stirred at RT for 1.5 h (color changed from yellow to dark orange). The solvent was removed and the solid dried under reduced pressure for 2 h giving an orange solid. Yield: 28 mg, 84% yield. 1H NMR (300 MHz, CDCl3): δ 9.34 (broad singlet, 2H), 8.59 (broad singlet, 2H), 8.03 (broad singlet, 2H), 7.92 ppm (overlapping broad singlet, 2H). 19F NMR (282 MHz, CDCl3): major isomer: 155.6 (br s, Mn=CF2), −77.7 ppm (br s, OTf); minor isomer: 156.3 (br s, Mn=CF2), −78.2 ppm (br s, OTf).
Synthesis of [Mn=CF2][OTf] complexes. The above preparation of complex 8 can be applied as a general synthesis to obtain the Mn=CF2 adducts of several Mn–CF3 complexes as can be seen from 19F NMR spectra showing formation of new carbenes from complexes (4) and (7) (see Supplementary Materials).
4. Conclusions
In summary, we have described a convenient synthesis for various bi- and tri-dentate N-ligated Mn–CF3 carbonyl complexes in adequate to good yields. Our synthesis avoids the use of hazardous reagents such as Cd(CF3)2 used previously for the synthesis of Ln(CO)5−nMn–CF3 {L = MeCN} complexes [37]. This will allow for more in depth computational/experimental studies of Mn–CF3 electronic structure/reactivity. The structural data of these complexes show an unusual elongation of the Mn–CF3 bond when utilizing the terpyridine ligand and this may open the door towards CF3 insertion or transfer utilizing similar ancillary ligands. Additionally, this publication has shown that Mn–CF3 complexes undergo facile fluoride abstraction utilizing Lewis acids such as TMS-OTf to form hitherto unknown Mn=CF2 carbenes. These cationic, presumably electrophilic carbenes react with electron-rich olefins and further reactivity studies are in progress.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2304-6740/7/1/3/s1, Cif and Cif checked files. NMR and IR spectra.
Author Contributions
Conceptualization, A.L.D. and R.T.B.; Methodology, A.L.D., R.T.B.; Validation, A.L.D., J.G.D.G. and R.E.; Formal Analysis, A.L.D.; Investigation, A.L.D., J.G.D.G. and R.E.; Resources, R.T.B.; Data Curation, B.M.G.; Writing—Original Draft Preparation, A.L.D. and R.T.B.; Writing—Review and Editing, A.L.D. and R.T.B.; Visualization, A.L.D.; Supervision, A.L.D. and R.T.B.; Project Administration, A.L.D. and R.T.B.; Funding Acquisition, R.T.B.
Funding
This research was supported by NSERC grant RGPIN-2014-05841.
Acknowledgments
We thank the NSERC and the Canada Research Chairs program for generous financial support and the University of Ottawa, Canada Foundation for Innovation, and Ontario Ministry of Economic Development and Innovation for essential infrastructure. A.L.D. thanks the NSERC for a CGS-D scholarship. Special thanks to Wendy Pell for CV-data and Sharon Curtis and the John L. Holmes Mass Spectrometry Facility.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Steinborn, D. Fundamentals of Organometallic Catalysis; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Chiusoli, G.P.; Maitlis, P.M. (Eds.) Metal-Catalysis in Industrial Organic Processes; RSC Publishing: Cambridge, UK, 2006. [Google Scholar]
- Taw, F.L.; Clark, A.E.; Mueller, A.H.; Janicke, M.T.; Cantat, T.; Scott, B.L.; Hay, P.J.; Hughes, R.P.; Kiplinger, J.L. Titanium(IV) Trifluoromethyl Complexes: New Perspectives on Bonding from Organometallic Fluorocarbon Chemistry. Organometallics 2012, 31, 1484–1499. [Google Scholar] [CrossRef]
- Hughes, R.P. Adv. Organo-transition Metal Compounds Containing Perfluorinated Ligands. Organomet. Chem. 1990, 31, 183–267. [Google Scholar]
- Brothers, P.J.; Roper, W.R. Transition-metal Dihalocarbene Complexes. Chem. Rev. 1988, 88, 1293–1326. [Google Scholar] [CrossRef]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Blackwell: Chichester, UK, 2009. [Google Scholar]
- Chambers, R.D. Fluorine in Organic Chemistry; Blackwell: Oxford, UK, 2004. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Alonso, C.; de Marigorta, E.M.; Rubiales, G.; Palacios, F. Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev. 2015, 115, 1847–1935. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent Advances in Trifluoromethylation Methodology and new CF3-containing Drugs. J. Fluor. Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Jeschke, P. The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef]
- Im, J.; Walshe-Langford, G.E.; Moon, J.-W.; Löffler, F.E. Environmental Fate of the Next Generation Refrigerant 2,3,3,3-Tetrafluoropropene (HFO-1234yf). Environ. Sci. Technol. 2014, 48, 13181–13187. [Google Scholar] [CrossRef] [PubMed]
- Panferova, L.I.; Miloserdov, F.M.; Lishchynskyi, A.; Martinez Belmonte, M.; Benet-Buchholz, J.; Grushin, V.V. Well-Defined CuC2F5 Complexes and Pentafluoroethylation of Acid Chlorides. Angew. Chem. Int. Ed. 2015, 54, 5218–5222. [Google Scholar] [CrossRef]
- Konovalov, A.I.; Lishchynskyi, A.; Grushin, V.V. Mechanism of Trifluoromethylation of Aryl Halides with CuCF3 and the Ortho Effect. J. Am. Chem. Soc. 2014, 136, 13410–13425. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Tsubogo, T.; Litvinas, N.D.; Hartwig, J.F. A Broadly Applicable Copper Reagent for Trifluoromethylations and Perfluoroalkylations of Aryl Iodides and Bromides. Angew. Chem. Int. Ed. 2011, 50, 3793–3798. [Google Scholar] [CrossRef] [Green Version]
- Dubinina, G.G.; Furutachi, H.; Vicic, D.A. Active Trifluoromethylating Agents from Well-Defined Copper(I)–CF3 Complexes. J. Am. Chem. Soc. 2008, 130, 8600–8601. [Google Scholar] [CrossRef] [PubMed]
- Wiemers, D.M.; Burton, D.J. Pregeneration, Spectroscopic Detection and Chemical Reactivity of (Trifluoromethyl)copper, an Elusive and Complex Species. J. Am. Chem. Soc. 1986, 108, 832–834. [Google Scholar] [CrossRef]
- McLoughlin, V.C.R.; Thrower, J. Route to Fluoroalkyl-substituted Aromatic Compounds Involving Fluoroalkylcopper Intermediates. Tetrahedron 1969, 25, 5921–5940. [Google Scholar] [CrossRef]
- Choi, W.J.; Choi, S.; Ohkubo, K.; Fukuzumi, S.; Cho, E.J.; You, Y. Mechanisms and Applications of Cyclometalated Pt(II) Complexes in Photoredox Catalytic Trifluoromethylation. Chem. Sci. 2015, 6, 1454–1464. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of Arenes and Heteroarenes by Means of Photoredox Catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef]
- Natta, G. New Class of Polymers of α-olefin Having Exceptional Regularity of Structure. Atti. Acc. Naz. Lincei. 1955, 4, 61. [Google Scholar]
- Ziegler, K.; Holzkamp, E.; Breil, H. Polymerisation von Äthylen und anderen Olefinen. Angew. Chem. 1955, 67, 541. [Google Scholar]
- Natta, G.; Pino, P.; Corradini, P.; Danusso, F.; Mantica, E.; Mazzanti, G.; Moraglio, G. Crystalline High Polymers of α-Olefins. J. Am. Chem. Soc. 1955, 77, 1708–1710. [Google Scholar] [CrossRef]
- Boday, D.J. The State of Fluoropolymers. In Advances in Fluorine-Containing Polymers; Smith, D.W., Jr., Iacono, S.T., Boday, D.J., Kettwich, S.C., Eds.; American Chemical Society: Washington, DC, USA, 2012; pp. 1–5. [Google Scholar]
- Kim, C.U.; Lee, J.M.; Ihm, S.K. Emulsion polymerization of tetrafluoroethylene: Effects of reaction conditions on particle formation. J. Fluor. Chem. 1999, 96, 11–21. [Google Scholar] [CrossRef]
- Kim, C.U.; Lee, J.M.; Ihm, S.K. Emulsion polymerization of tetrafluoroethylene: Effects of reaction conditions on the polymerization rate and polymer molecular weight. J. Appl. Polym. Sci. 1999, 73, 777–793. [Google Scholar] [CrossRef]
- Sianesi, D.; Caporiccio, G. Stereospecific Polymerization of Perfluoroolefins. Makromol. Chem. 1963, 60, 213–222. [Google Scholar] [CrossRef]
- Huang, Y.; Li, J.; Zhou, J.; Zhu, Z.; Hou, G. π-Bis(Benzene)Chromium(0)-Catalyzed Oligomerization of Perfluoropropylene. J. Organomet. Chem. 1981, 205, 185–191. [Google Scholar] [CrossRef]
- Clot, E.; Megret, C.; Kraft, B.M.; Eisennstein, O.; Jones, W.D. Defluorination of Perfluoropropene Using Cp*2ZrH2 and Cp*2ZrHF: A Mechanism Investigation from a Joint Experimental-Theoretical Perspective. J. Am. Chem. Soc. 2004, 126, 5647–5653. [Google Scholar] [CrossRef] [PubMed]
- Smart, B.E. The Chemistry of Functional Groups, Supplement D; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons: New York, NY, USA, 1983; Chapter 14. [Google Scholar]
- Wilford, J.B.; Stone, F.G.A. Chemistry of the Metal Carbonyls. XXVIII. Addition of Rhenium Pentacarhonyl Hydride to Fluoro Olefins. Inorg. Chem. 1965, 4, 93–97. [Google Scholar] [CrossRef]
- Fujisawa, K.; Nabika, M. Development of New Polymerization Catalysts with Manganese(II) Complexes. Coord. Chem. Rev. 2013, 257, 119–129. [Google Scholar] [CrossRef]
- Algarra, A.G.; Grushin, V.V.; Macgregor, S.A. Natural Bond Orbital Analysis of the Electronic Structure of [LnM(CH3)] and [LnM(CF3)] Complexes. Organometallics 2012, 31, 1467–1476. [Google Scholar] [CrossRef]
- McClellan, W.R. Perfluoroalkyl and Perfluoroacyl Metal Carbonyls. J. Am. Chem. Soc. 1961, 83, 1598–1600. [Google Scholar] [CrossRef]
- Naumann, D.; Kaiser, M. A New Synthesis of Perfluoroorgano Manganese and Rhenium Compounds. Z. Anorg. Allg. Chem. 1995, 621, 812–816. [Google Scholar] [CrossRef]
- Banerjee, S.; Karunananda, M.K.; Bagherzadeh, S.; Jayarathne, U.; Parmelee, S.R.; Waldhart, G.W.; Mankad, N.P. Synthesis and Characterization of Heterobimetallic Complexes with Direct Cu-M Bonds (M = Cr, Mn, Co, Mo, Ru, W) Supported by N-Heterocyclic Carbene Ligands: A Toolkit for Catalytic Reaction Discovery. Inorg. Chem. 2014, 53, 11307–11315. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, S.E.A.; Durgaprasad, G.; Muthiah, K.A.T.; Rose, M.J. Tuning Coordination Modes of Pyridine/thioether Schiff Base (NNS) Ligands to Mononuclear Manganese Carbonyls. Dalton Trans. 2014, 43, 10725–10738. [Google Scholar] [CrossRef] [PubMed]
- Welch, K.D.; Dougherty, W.G.; Kassel, W.S.; Dubois, D.L.; Bullock, R.M. Synthesis, Structure and Reactions of Manganese Complexes Containing Diphosphine Ligands and Pendant Amines. Organometallics 2010, 29, 4532–4540. [Google Scholar] [CrossRef]
- Van Putten, R.; Uslamin, E.A.; Garbe, M.; Liu, C.; Gonzalez-de-Castro, A.; Lutz, M.; Junge, K.; Hensen, E.J.M.; Beller, M.; Lefort, L.; et al. Non-Pincer-Type Manganese Complexes as Efficient Catalysts for the Hydrogenation of Esters. Angew. Chem. Int. Ed. 2017, 56, 7531–7534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S. Tripodal Phosphine Ligands. Syntheses and Coordination Chemistry Toward Mn(I). J. Chin. Chem. Soc. 1992, 39, 611–616. [Google Scholar] [CrossRef]
- Luh, T.-Y. Trimethylamine N-oxide—A Versatile Reagent for Organometallic Chemistry. Coord. Chem. Rev. 1984, 60, 255–276. [Google Scholar] [CrossRef]
- Morrison, J.A. Trifluoromethyl-containing Transition Metal Complexes. Adv. Organomet. Chem. 1993, 35, 211–239. [Google Scholar]
- Leyssens, T.; Peeters, D.; Orpen, A.G.; Harvey, J.N. How Important is Metal-Ligand Back-bonding toward YX3 Ligands (Y = N, P, C, Si)? An NBO Analysis. Organometallics 2007, 26, 2637–2645. [Google Scholar] [CrossRef]
- Richmond, T.G.; Shriver, D.F. Electrophillic Halogen Exchange between Lewis Acids and Transition-Metal Perfluoroalkyl Complexes. Synthesis and Characterization of Transition Metal α-HaloAlkyl Complexes. Organometallics 1984, 3, 305–314. [Google Scholar] [CrossRef]
- Richmond, T.G.; Crespi, A.M.; Shriver, D.F. Nucleophillic, Electrophillic, and Homolytic Reaction Chemistry of Transition Metal Carbonyl Trihalomethyl (X = F, Cl, Br) Complexes. Organometallics 1984, 3, 314–319. [Google Scholar] [CrossRef]
- Harrison, D.J.; Daniels, A.L.; Guan, J.; Gabidullin, B.M.; Hall, M.B.; Baker, R.T. Nickel Fluorocarbene Metathesis with Fluoroalkenes. Angew. Chem. Int. Ed. 2018, 57, 5772–5776. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.J.; Daniels, A.L.; Korobkov, I.; Baker, R.T. d10 Nickel Difluorocarbenes and Their Cycloaddition Reactions with Tetrafluoroethylene. Organometallics 2015, 34, 5683–5686. [Google Scholar] [CrossRef]
- Harrison, D.J.; Lee, G.M.; Leclerc, M.C.; Korobkov, I.; Baker, R.T. Cobalt Fluorocarbenes: Cycloaddition Reactions with Tetrafluoroethylene and Reactivity of the Perfluorometallacyclic Products. J. Am. Chem. Soc. 2013, 135, 18296–18299. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Helquist, P. Cyclopropanation of Olefins with a Stable, Iron-containing Methylene Transfer Agent. J. Am. Chem. Soc. 1979, 101, 6473–6475. [Google Scholar] [CrossRef]
- O’Connor, E.J.; Brandt, S.; Helquist, P. (η5-C5H5)(CO)2FeCH2S+(CH3)2BF4−: A Methylene Transfer Reagent for the Direct Cyclopropanation of Alkenes. J. Am. Chem. Soc. 1987, 109, 3739–3947. [Google Scholar] [CrossRef]
Scheme 1.
General mechanism for 1,2-olefin insertion (Cossee-Arlman).

Scheme 2.
Unsuccessful formation of Mn–CF3 via Mn(II) (top) and Mn(I) (bottom).

Scheme 3.
Formation of easily substituted Mn–CF3 starting material.

Scheme 4.
Substitution of Mn–CF3 starting material with bidentate ligands.

Scheme 5.
Substitution of Mn–CF3 source starting material with tridentate ligands.

Figure 1.
ORTEP structures of 3 (left) and 6 (right) with 50% ellipsoids.

Scheme 6.
Structure of both isomers of Mn(NNS)–(CO)2CF3.

Scheme 7.
Formation of cis-/trans-complex (8).

Figure 2.
ORTEP structure of Mn(Phen)2(OTf)2, 9 (Mn–N1 = 2.2262 (3), Mn–N2 = 2.2397 (7) Å) with 50% ellipsoids.
Figure 2.
ORTEP structure of Mn(Phen)2(OTf)2, 9 (Mn–N1 = 2.2262 (3), Mn–N2 = 2.2397 (7) Å) with 50% ellipsoids.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
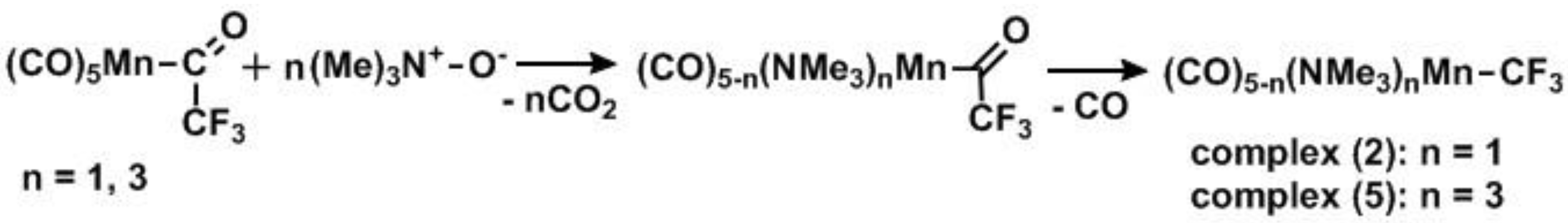{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected bond lengths (Å) for Mn–CF3 complexes.
| Complex | Mn–CCF3 | Mn–CCO | Mn–N |
|---|---|---|---|
| Mn(CO)5CF3 (1) | 2.056(5) | NA | NA |
| 1.789(5) (cis) | - | ||
| Mn(CO)3(Bpy)CF3 (3) | 2.039(7) | 1.780(7) (cis) | 2.041(2) |
| 1.823(1) (trans) | 2.034(8) | ||
| Mn(CO)2(Tpy)CF3 (6) | 2.096(1) | 1.772(3) (cis) | 2.021(1), 2.025(1) |
| 1.818(9) (trans) | 1.959(2) (N2) |
Table 2.
Selected CO stretching frequencies for Mn–CF3 complexes.
| Complex | CO Stretching Frequencies (cm−1) |
|---|---|
| Mn(CO)5CF3 (1) | 2140, 2040, 2010 |
| Mn(CO)3(Bpy)CF3 (3) | 2020, 1910 |
| Mn(CO)3(Phen)CF3 (4) | 2010, 1930 |
| Mn(CO)2(Tpy)CF3 (6) | 2020, 1900, 1850 |
| Mn(CO)2(NNS)CF3 (7) | 2020, 1910, 1900 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Daniels, A.L.; Da Gama, J.G.; Edjoc, R.; Gabidullin, B.M.; Baker, R.T. Synthesis and Reactivity of Mn–CF3 Complexes. Inorganics 2019, 7, 3. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7010003
AMA Style
Daniels AL, Da Gama JG, Edjoc R, Gabidullin BM, Baker RT. Synthesis and Reactivity of Mn–CF3 Complexes. Inorganics. 2019; 7(1):3. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7010003
Chicago/Turabian StyleDaniels, Alex L., Jason G. Da Gama, Racquel Edjoc, Bulat M. Gabidullin, and R. Tom Baker. 2019. "Synthesis and Reactivity of Mn–CF3 Complexes" Inorganics 7, no. 1: 3. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7010003
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.