Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments


LCM, CNRS, Ecole Polytechnique, IP Paris, Route de Saclay, 91128 Palaiseau, France
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(5), 58; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7050058
Submission received: 8 March 2019
/
Revised: 29 March 2019
/
Accepted: 17 April 2019
/
Published: 26 April 2019
(This article belongs to the Special Issue Binuclear Complexes)
Abstract
:This article presented the synthesis and characterization of original heterobimetallic species combining a divalent lanthanide fragment and a divalent nickel center bridged by the bipyrimidine ligand, a redox-active ligand. X-ray crystal structures were obtained for the Ni monomer (bipym)NiMe2, 1, as well as the heterobimetallic dimer compounds, Cp*2Yb(bipym)NiMe2, 2, along with 1H solution NMR, solid-state magnetic data, and DFT calculations only for 1. The reactivity with CO was investigated on both compounds and the stoichiometric acetone formation is discussed based on kinetic and mechanistic studies. The key role of the lanthanide fragment was demonstrated by the relatively slow CO migratory insertion step, which indicated the stability of the intermediate.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
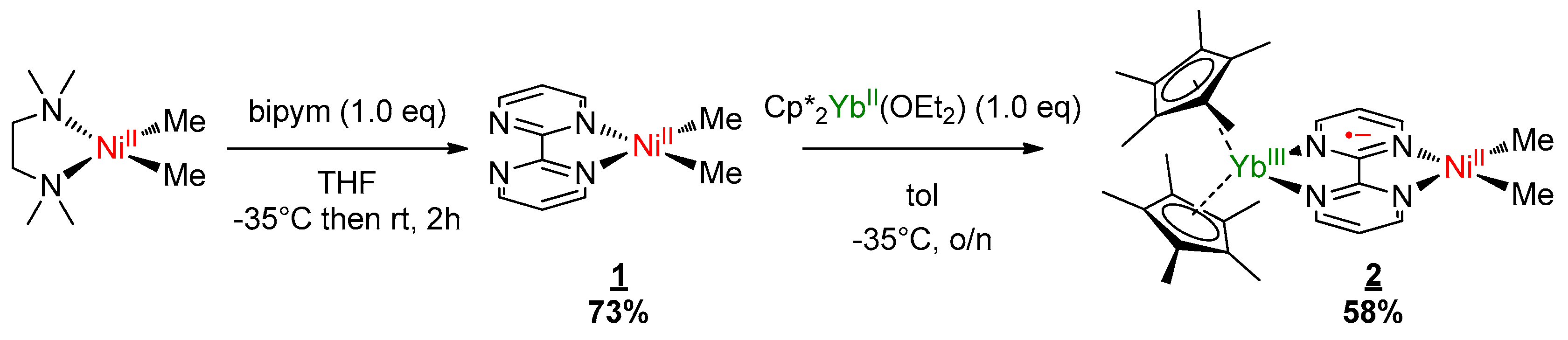{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Heterometallic complexes are important objects of study because both metallic fragments have a role to play in the chemical reaction and/or properties of interest [1,2,3]. In nature, the active sites of many enzymes are bimetallic, and the understanding of the role of each metallic part is crucial to the design of appropriate models [4,5,6,7]. In some cases, the role of one fragment is purely structural and only facilitates the reaction at the other metal center, while in most cases, both fragments have a role and either participate in the fate of the chemical reaction or complete it [8]. This particular case was extensively studied in terms of the tandem-reaction catalysts, where, for example, one metal is the source of one reaction, the other of a second one, and the substrate undergoes two chemical transformations in one pot [9,10,11,12]. Another elegant use of the bimetallic complexes is found in the chemical cooperation between both metal fragments, allowing a reactivity that would not occur efficiently with only one of these taken separately [13]. The recent interest in photochemically active bimetallic complexes is witness to these developments [14,15].
In our group, we designed bimetallic complexes with a slightly different approach. We combined a reductive divalent lanthanide fragment with a transition metal fragment, which possessed a ligand that could be eventually reduced or oxidized upon coordination. These studies have recently led us to develop a system with Pd and the bipyrimidine ligand (bipym) in which the palladium can be stabilized at the PdIV for several hours at room temperature [16]. This result was rendered possible using divalent ytterbium, which can reduce the palladium/bipym complex and have an impact on the overall electronic structure of the bimetallic assembly. Thus, the two metallic fragments cooperate by means of their electronic correlation. The use of divalent lanthanide is purposeful, since as they are strong single electron reductants [17,18,19,20], the divalent lanthanides adapt their electronic structures depending on the ligand that is used and form multiconfigurational electronic states [21,22,23], which allow the tuning of their properties with the redox-active ligand [24,25,26,27,28,29].
High oxidation states such as PdIV or NiIV are actively sought after because of their scarcity, but also because they allow important reactions [30,31,32,33,34]. Notably, the oxidative addition from PdII or NiII centers is an important step because it may avoid the difficult use of low-valence Pd and Ni species as catalysts, and their electrophilic character could also prevent the use of other electrophile metal fragments which have less-abundant resources, such as Rh, Ir, or Pt [35,36]. For example, methanol is transformed to acetic acid using carbon monoxide (CO) in a reaction that uses Ir (Cativa) [37,38] or Pt (Monsanto) [39] catalysts. In these reactions, the crucial step of CO insertion is performed after the oxidative addition at the high oxidation state of the metal center [40].
In this work, we developed a new complex that possesses a Ni metal center and bipym as the ligand and combined it with the Cp*2Yb divalent lanthanide fragment. The synthesis and characterization of the bimetallic complex is presented, as well as reactivity studies implying CO insertion.
2. Results and Discussions
2.1. Synthesis and X-ray Diffraction
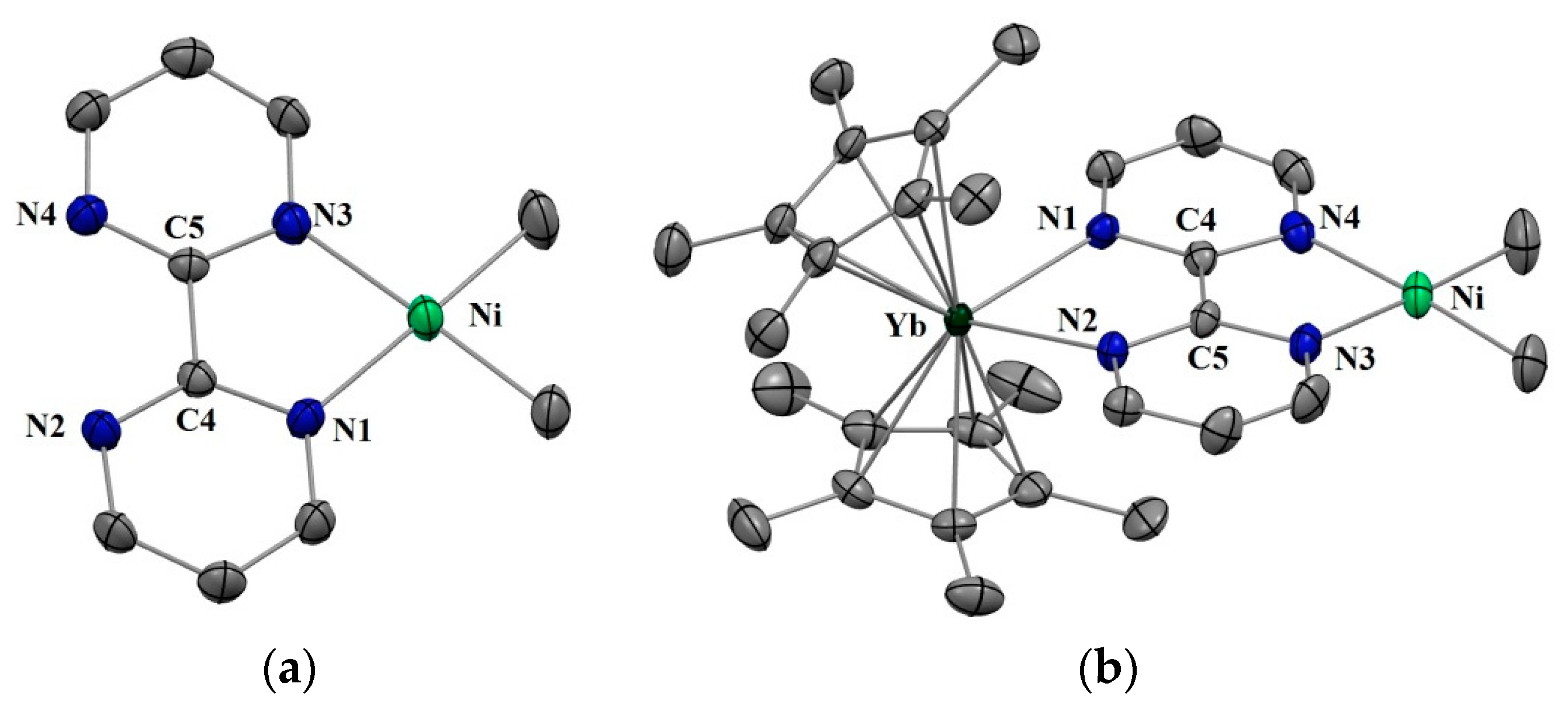
Complexes 1 and 2 were synthesized according to similar published procedures for similar Pd complexes (Scheme 1) [16]. The (tmeda)NiMe2 [41] was suitable for the ligand exchange in THF with bipyrimidine (bipym) and the reaction yielded (bipym)NiMe2 (1) as dark X-ray-suitable crystals in good yield after the solution was cooled to −35 °C. Note that this synthesis method avoided the formation of the (bipym) (NiMe2)2 dimer as well as free bipym, which are both very problematic for the next step. An Oak Ridge Thermal Ellipsoid Plot (ORTEP) of 1 is shown in Figure 1, and the main metric parameters are available in Supplementary Materials. Complex 1 was dissolved in toluene and cooled down to −35 °C and the addition of a room temperature toluene solution of the Cp*2Yb(OEt2) complex led to a dark-brown solution, which yielded X-ray-suitable dark-brown crystals of the heterobimetallic dimer Cp*2Yb(bipym)NiMe2, 2, when cooled down to −35 °C. An ORTEP is shown in Figure 1, and the main metric parameters are available in the Supplementary Materials. Comparing the solid-state structures of 1 and 2, the first noteworthy feature is the C4–C5 distance, which was strongly reduced in 2 (1.403(4) Å) compared to that in 1 (1.482(5) Å). This is similar to the observation made with the Pd complex [16] and is due to an electron transfer from the ytterbium center that reduced the bipyrimidine ligand. Accordingly, the average Cp centroid–Yb distance was 2.31(1) Å, which is lower than that when the fragment is divalent and similar to that when it is trivalent [25]. The Ni–N (1.959(2) and 1.956(3) Å) and Ni–C (1.930(3) and 1.925(1) Å) average distances were similar in both 1 and 2, respectively, which is indicative of a similar oxidation state in both complexes.
2.2. Solution NMR and Solid-State Magnetism
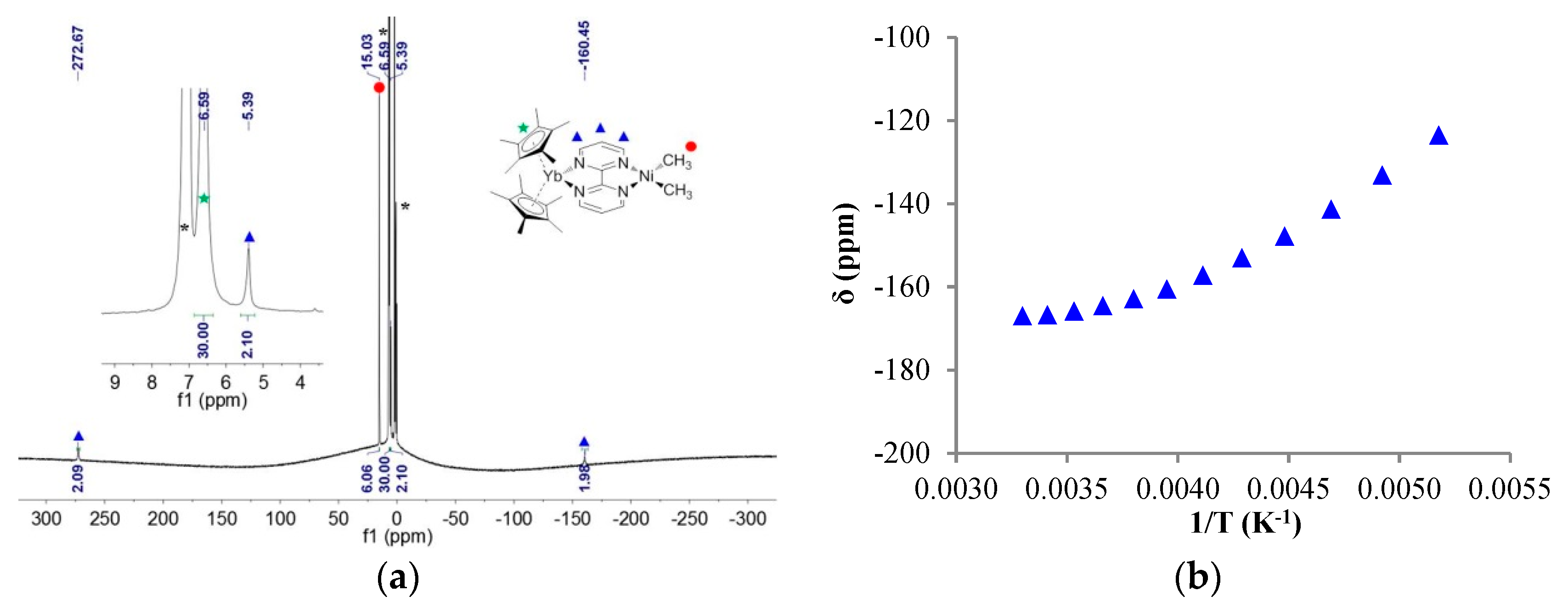
The 1H NMR of 1 showed only four signals: three for the bipym ligand, integrating each for two protons, and one for the methyl fragments, integrating for six protons, in good agreement with a C2v symmetry in solution. In 1, all signals were found in the typical diamagnetic range. The 1H NMR of 2 at 253 K in tol-d8 was very different: three signals integrated for two protons at 272.67, 5.39, and −160.45 ppm, one signal integrated for six protons at 15.03 ppm, and one for 30 protons at 6.59 ppm (Figure 2). The latter signal was easily attributed to the protons of the Cp* fragments, while the former were attributed to the bipym ligand and the methyl fragments, respectively. The spectrum is in agreement with a C2v symmetry in solution, and the clear paramagnetism of the signals agrees with an electron transfer from the divalent ytterbium to the bipym ligand. In such a situation, the ytterbium metal center becomes trivalent and is f13 (one hole on the f-shell) while the bipym is a radical anion.
Considering the paramagnetism measured by NMR, multiple possibilities arise for the electronic ground state of 2: a triplet state or a singlet state with a low-lying triplet state. The chemical shifts of each resonance were plotted versus 1/T (Figure S5), and an example is given in Figure 2. It is clear from these plots that the behavior did not strictly follow Curie’s law, since the plots are curved. Two reasons may explain this behavior: (i) the bipym ligand in 2 was in exchange with free bipym and the chemical shift depended on the equilibrium thermodynamics at a given temperature, or (ii) the magnetic behavior of 2 did not follow Curie’s law because of a magnetic exchange coupling between the two single electrons. To test these two propositions, the solid-state magnetism was measured.
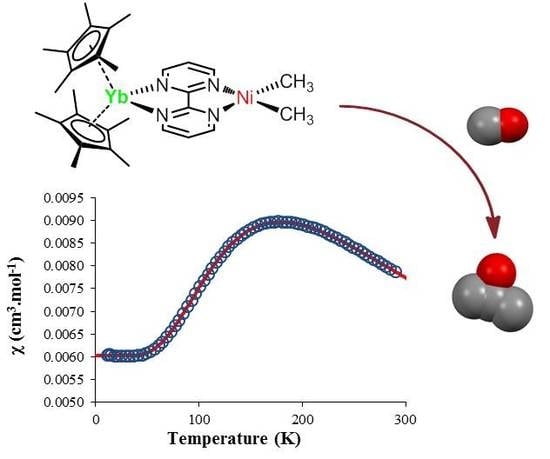
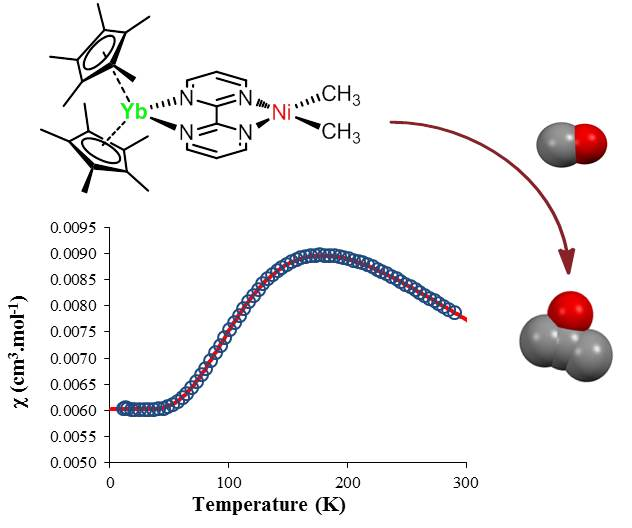
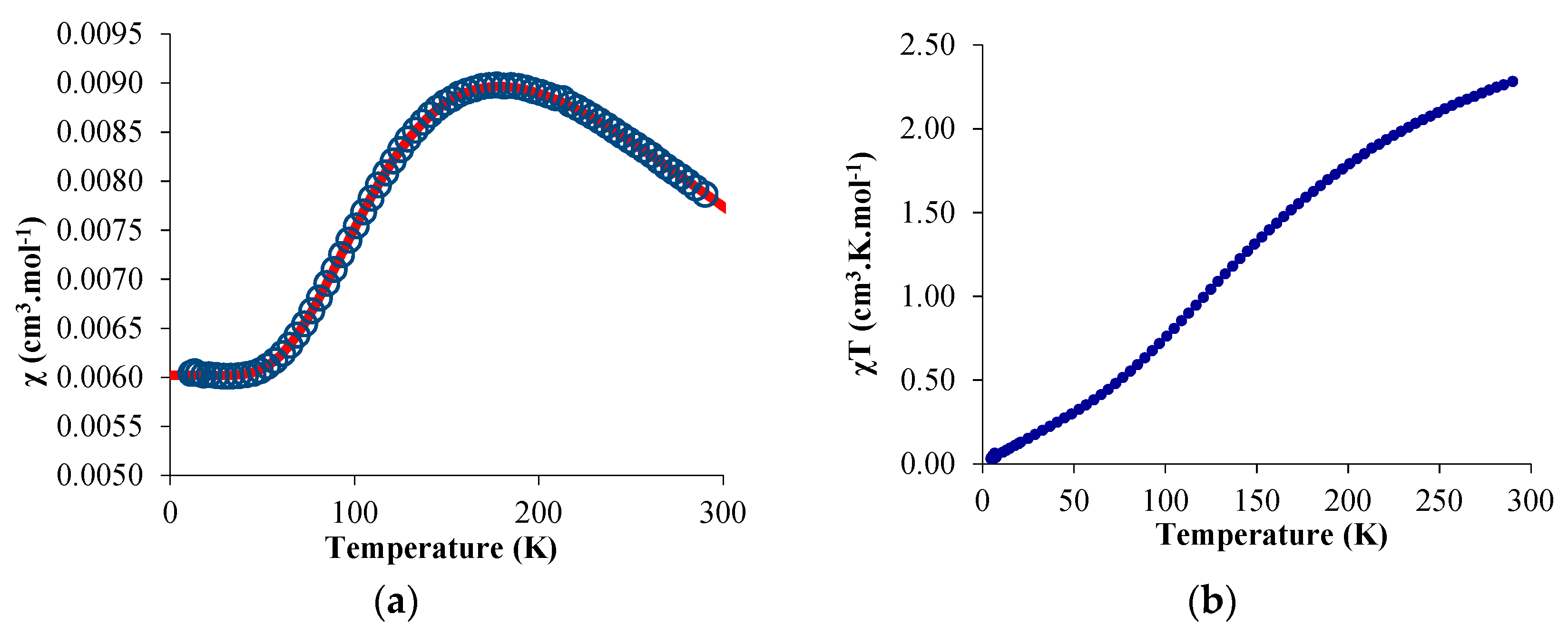
The temperature-dependent magnetic data were recorded and χ and χT were plotted versus T and are represented in Figure 3. The χT value decreased gradually with decreasing temperature until the value approached zero, with two inflection points around 65 and 170 K. The near-zero value at low temperature indicates a singlet ground state while the value of 2.3 emu·K·mol−1 is close to the theoretical value obtained by the sum of a 2F state (f13) and a 2S state (bipym·−). The χ value versus the temperature is more indicative of the magnetic behavior of 2: a maximum was obtained at 170 K while the low-temperature data was low and independent of the temperature (temperature-independent paramagnetism (TIP) or Van Vleck paramagnetism) [42,43]. Such a behavior can be explained by an anti-ferromagnetic coupling between the single electron located on the ytterbium center and that on the bipym ligand. The TIP was indicative of a low-lying triplet state above the singlet ground state. The χT value at room temperature agrees with a substantial population of the triplet at this temperature and therefore a low-lying triplet excited state. Using a modified version of the Bleaney–Bowers equation (Equation (1)) [44], the data were fitted using an average g-value of 3.78, a TIP of 0.00602 emu·mol−1, and a 2J value of −275.2 cm−1. The average g-value that was used was relatively close to the one measured for the similar trivalent ytterbium complexes, that is, 3.315 for Cp*2Yb(bipy)+ [45]. The red curve is the fit of the data with the Equation (1), and is very satisfactory. However, the value of both TIP and g-average are in very high agreement with a high anisotropy in this system. In order to account for the anisotropy, we used another model from Lukens et al. that is more adapted to highly anisotropic compounds such as the lanthanide complexes [46]. Assuming the c12 is close to 1 in 2 (which means that the oxidation state is close to purely trivalent) and using the g-value of the similar Cp*2Yb(bipy)+ complex [45], the 2J value was estimated to be −245.65 cm−1—a similar value to that found with the simple Bleaney–Bowers model (Equation (1)). It indicates that the exchange coupling was rather strong, compares well with the value found in the literature for the Cp*2Yb fragment and other N-aromatic heterocycles [46], and is typical for this type of species [47].
The magnetism as well as the 1H solution NMR spectroscopy agreed with a singlet ground state and a triplet state that is substantially populated. Strong spin density was located on the bipym ligand as indicated by the X-ray crystallography, and the exchange coupling was monitored by the solid-state magnetism. However, in order to probe the reactivity at the nickel center, it is important to quantify how the electronic density is organized on the transition metal. For this, we turned to theoretical computations on 1.
2.3. Theoretical Computations on 1
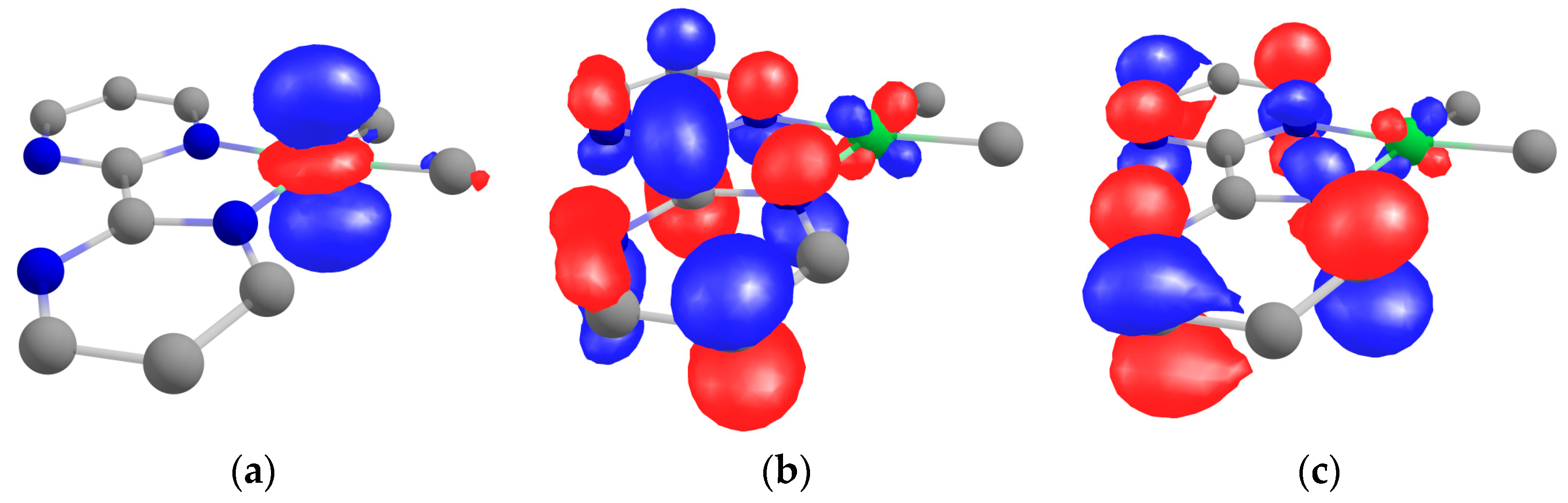
Theoretical computations were performed on 1 at the DFT level with three different functionals: PBE, PBE0, and TPSSh (see the Materials and Methods section for more information). The difference in geometry is given in the Supplementary Materials, and the differences were minor compared to the X-ray data (see Table S7). The HOMO was clearly the dz2 of the nickel center, while the LUMO and LUMO +1 were orbitals located in the bipym ligand with the b1 symmetry (in C2v) (Figure 4). The LUMO possessed much density on the C–C bond that linked the two pyrimidine rings, the LUMO +1 had a node at these positions. The density on the nickel center was only residual, and the estimated atomic orbital Mulliken contribution from the nickel to these two MOs were 8.3% for the LUMO and 4.6% for the LUMO +1.
According to the magnetism, the ground state of 2 was a singlet state while the triplet state was low in energy. We did not perform DFT computations on 2 because the electronic state was likely to be multiconfigurational considering the previous data published on similar compounds [21,22,23,24,25,27] and the multi-referential calculations are more difficult considering the number of atoms. However, the magnetism and the X-ray crystal structure were indicative of spin density localized on the C–C bond that linked the two pyrimidine rings, which was in agreement with an electron transfer from the Cp*2Yb fragment to the LUMO of 1. Another interesting feature was the high-energy dz2 located on the Ni center that is ready for reactivity. However, the addition of MeI or MeOTf on 1 and 2 only led to the very fast formation of ethane, even at low temperature. If it is indicative of some reactivity, the lack of intermediates observed in the course of the reaction does not allow the drawing of any conclusions on the possible mechanism. Therefore, we turned to the reaction of 1 and 2 with CO.
2.4. Reactivity with Carbon Monoxide
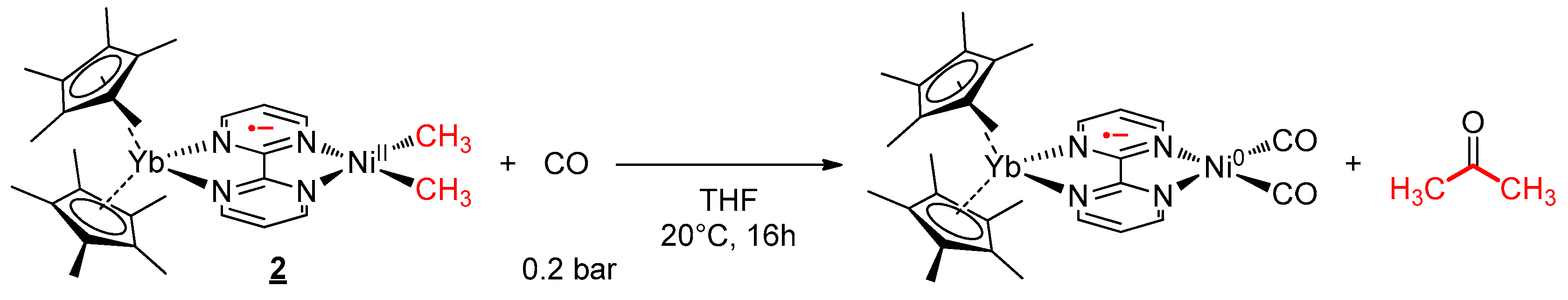
The reactivity with CO and group 10 alkyl complexes has been well investigated over the years. We were interested in knowing whether the presence of the lanthanide fragment in 2 influenced the mechanism of such a reaction. Thus, the reactivity at a low concentration of CO (0.2 bar) was investigated in both complexes 1 and 2 in THF. The reaction occurred rapidly and was followed by 1H NMR spectroscopy. After a few minutes of reaction, in the case of complex 2, new signals appeared at 259.9, 12.4, and −0.1 ppm, while the starting material concentration decreased slowly (see Figure S12). As the reaction time evolved, the intensity of these new signals increased then dropped until the end of the reaction. Similar to 2, in the complex 1 system, new signals appeared at 9.2 and 2.2 ppm with time, and the rapid formation of acetone was clearly observed (see Figure S19). These new signals were attributed to the acyl intermediate after the CO migratory insertion in the Ni–C bond. The prior coordination of the CO on the nickel center through the loaded dz2 orbital was not observed. The formation of acetone (Scheme 2) was monitored with time. The apparition of this ketone indicates a reductive elimination from the acyl-methyl divalent nickel center to form Ni0 species. Considering the presence of CO in excess, the formation of bis-CO Ni0 species is likely. At the end of the reaction, the signals of the intermediates disappeared, and unfortunately, at this point, the reduced complex could not be isolated. Instead, the formation of a small amount of free bipym ligand as well as the Cp*2Yb(bipym)YbCp*2 [48] dimer was observed, indicating the disassociation of the reduced Ni0 complex.
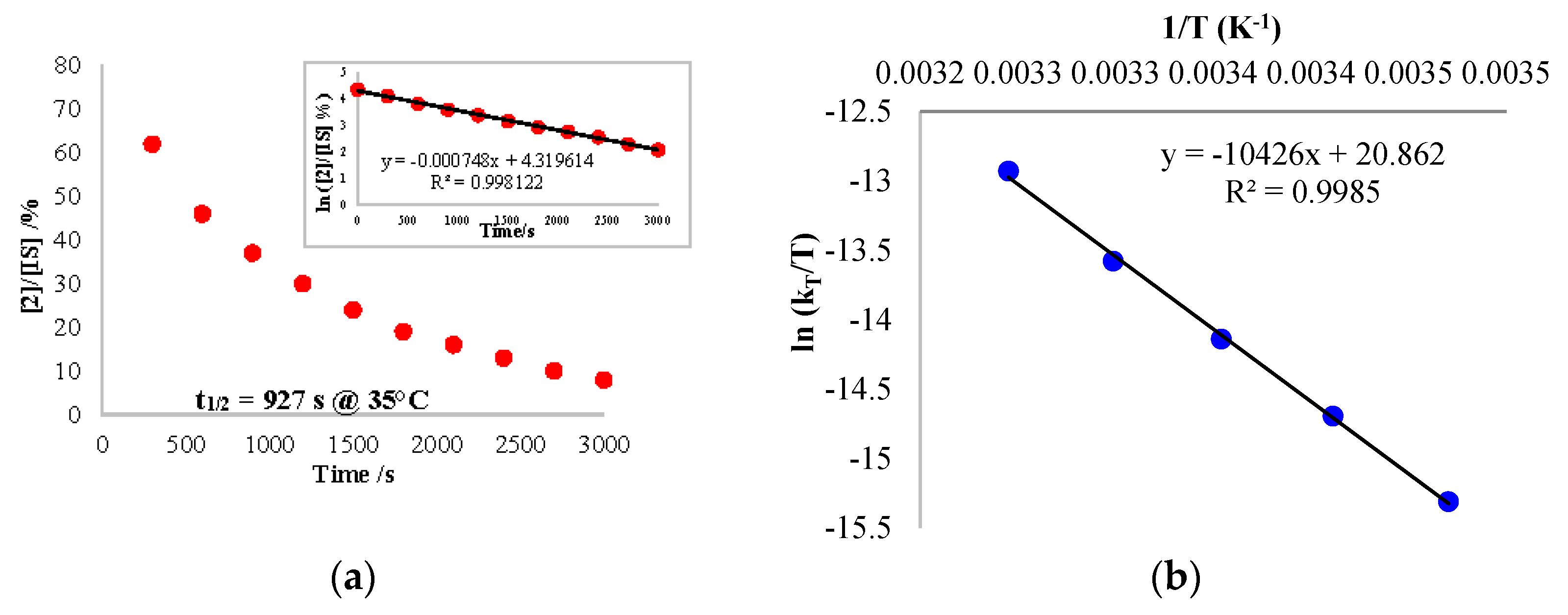
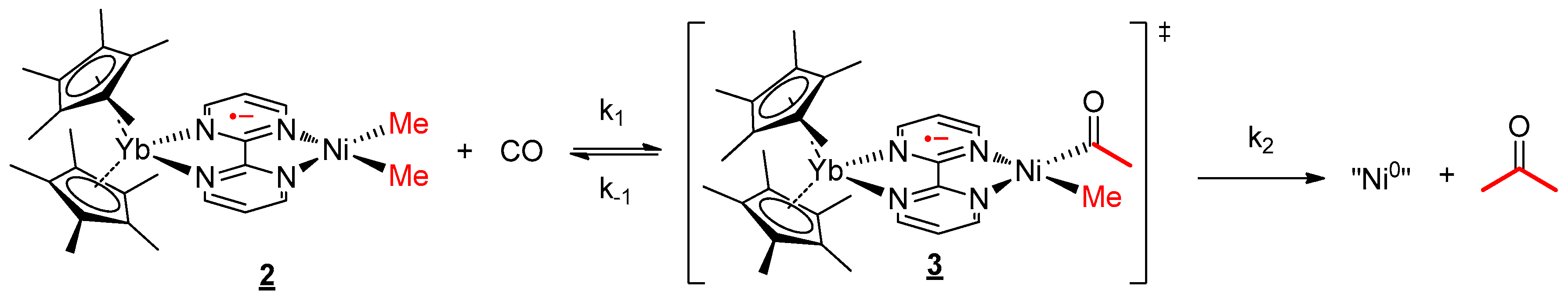
The reaction kinetics was followed by analysis with 1H NMR spectroscopy, using the decrease of the signal of 2. A mono-exponential variation decrease of the concentration of 2 was recorded at 35 °C (Figure 5a). The variation of the concentration of 2 did not modify the observed rate of the reaction at the same temperature at moderate concentrations of 2 (0.0073–0.029 mol/L). At higher concentrations, there was no longer a large excess of CO and the rate evolved. This information indicates that the rate overall order was pseudo first-order in 2 only when CO was in excess. The mechanism as well as the rate law can be written accordingly (Scheme 3 and Equation (2)):
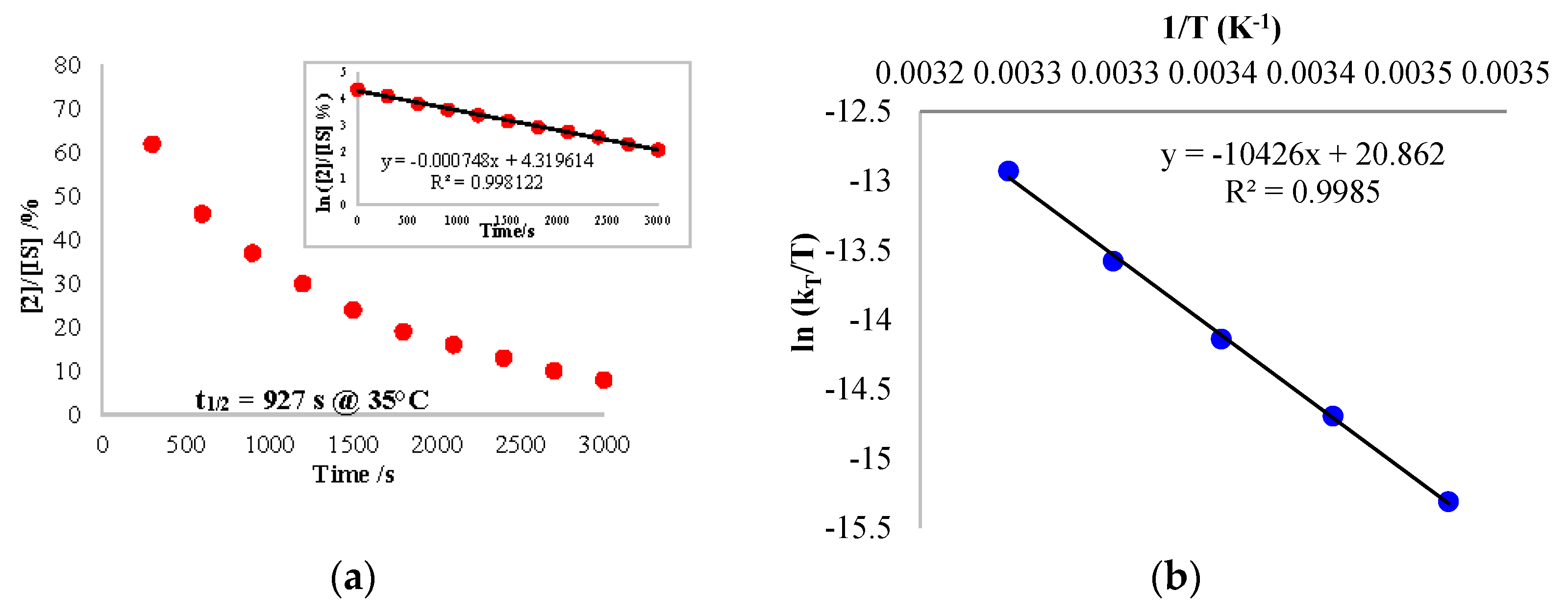
Using the rate law, the same analysis over a temperature range from 15 to 35 °C allowed us to perform an Eyring analysis (Figure 5b). The activation parameters obtained were 20.7 kcal·mol−1 and −5.75 cal·mol−1·K−1. The moderate ∆H≠ is in agreement with a rather slow reaction at room temperature while the small negative ∆S≠ indicates that the reductive elimination (entropically favorable) was not the rate-determining step (RDS) or it was balanced with a step with similar rate that was not favorable in entropy. On the other hand, the migratory insertion had a more modest effect on the entropy and could correspond to the RDS in this case. Without the information on the C coordination, it is not possible to conclude at this stage.
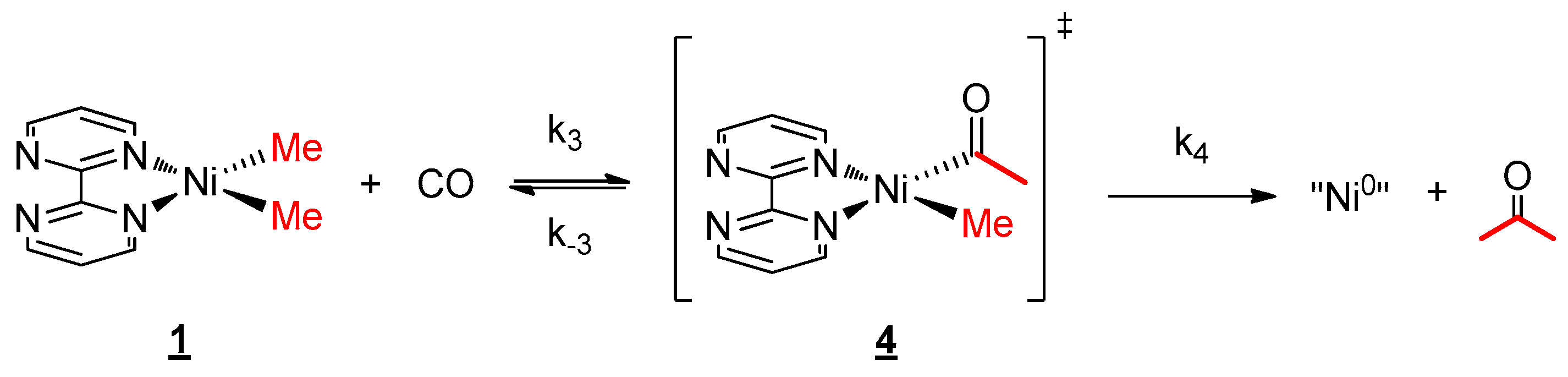
The kinetics of the reaction with CO and 1 were also monitored over time by 1H NMR spectroscopy in order to estimate the influence of the divalent lanthanide fragment. Having discovered that the overall reaction was also following a pseudo first-order kinetics (Scheme 4), an Eyring plot was constructed with the same temperature range as the one with 2 and CO, revealing the activation parameters ∆H≠ and ∆S≠ as 17.06 kcal mol−1 and −16.41 cal mol−1 K−1, respectively (see Table S2 and Figure S18). The decent negative ∆S≠ value explains that the RDS is likely to be the reductive elimination, or more precisely, the entropically favorable rate k4 was much larger than k3 (see Supplementary Materials). Besides, the lesser ∆H≠ than the one of 2 indicates that there was a decreased barrier in the reaction without the organolanthanide fragment. This conclusion can also be proved with the much higher half-life time of complex 2 at 20 °C (94.7 min) compared to the one of 1 (39.0 min, see Tables S1 and S2). The comparison of these data allows the deduction that the CO migratory insertion intermediate will be much more stable in the presence of lanthanide fragments.
3. Materials and Methods
All reactions were performed using standard Schlenk-line techniques or in argon- or nitrogen-filled gloveboxes (MBraun, Garching, Germany). All glassware was dried at 140 °C for at least 12 h prior to use. Tetrahydrofuran (THF), THF-d8, toluene, and toluene-d8 were dried over sodium, degassed, and transferred under reduced pressure in a cold flask.
1H NMR spectra were recorded in 5-mm tubes adapted with a J. Young valve on Bruker AVANCE II or III-300 MHz (Bruker, Billerica, MA, USA). 1H chemical shifts were expressed relative to TMS (Tetramethylsilane) in ppm. Magnetic susceptibility measurements were made for all samples on powder in sealed quartz tubes at 0.5 and 20 kOe in a 7 T Cryogenic SX600 SQUID magnetometer (Cryogenic, London, UK). Diamagnetic corrections were made using Pascal’s constants. Elemental analyses were obtained from Mikroanalytisches Labor Pascher (Remagen, Germany).
(Tmeda)NiMe2 [41] and Cp*2Yb(OEt2) [49] complexes were synthetized according to published procedures. The bipyrimidine from TCI (Tokyo, Japan) was sublimated prior to use.
3.1. Synthesis of (bipym)Ni(Me)2 (1)
(tmeda)NiMe2 (146 mg, 0.71 mmol, 1.0 equiv.) and bipyrimidine (113 mg, 0.71 mmol, 1.0 equiv.) were respectively dissolved in cold THF (−35 °C). Transferring the bipyrimidine solution dropwise into the greenish yellow nickel solution at ambient temperature gave a dark-colored mixture after stirring for several minutes. Then, the mixture was stirred for 2 h and was stored at −35 °C in order to crystallize. Black crystalline product was obtained after one night and isolated in 73% yield (105 mg, 0.42 mmol). Yield: 73%. 1H NMR (300 MHz, 293 K, thf-d8): δ (ppm) = 9.32 (m, 2H, bipym), 9.12 (m, 2H, bipym), 7.67 (m, 2H, bipym), 0.06 (s, 6H, Ni–Me). 13C NMR (75 MHz, 273 K, thf-d8): δ (ppm) = 162.0, 156.6, 154.6, 124.4, −5.2. Anal. calcd. for C10H12N4Ni: C, 48.64; H, 4.90; N, 22.69; found: C, 47.00; H, 4.59; N, 21.00.
3.2. Synthesis of (Cp*)2Yb(bipym)Ni(Me)2 (2)
Cp*2Yb(OEt2) (200 mg, 0.39 mmol, 1.0 equiv.) and (bipym)NiMe2 (98 mg, 0.40 mmol, 1.02 equiv.) were dissolved in toluene, respectively, and cooled down to −35 °C. Transferring the green Cp*2Yb solution dropwise into the Nickel solution at ambient temperature gave a dark-brown mixture once the addition was finished. Then, the mixture was stored at −35 °C in order to crystallize. Dark-brown crystals were obtained after several hours, and were isolated after washing three times with n-pentane, in 58% yield (156 mg, 0.22 mmol). Yield: 58%. 1H NMR (300 MHz, 293 K, thf-d8): δ (ppm) = 246.83 (s, 2H, bipym), 15.91 (s, 6H, –Me), 9.02 (s, 2H, bipym), 6.09 (s, 30H, Cp*), −172.34 (s, 2H, bipym). Anal. calcd for C30H42N4NiYb: C, 52.19; H, 6.13; N, 8.11; found: C, 53.07; H, 6.06; N, 7.31.
3.3. Theoretical Calculations
All calculations were performed using the ORCA 4.0.0.2 software [50]. The geometry optimizations were done at three different levels of theory (PBE [51], PBE0 [52], and TPSSh [53,54]) using scalar relativistic ZORA Hamiltonian with ZORA-def2-TZVP basis set [55] and SARC/J auxiliary basis set for Coulomb fitting [56,57,58]. Each time, dispersion corrections were added to the functional used in the D3 framework proposed by Grimme [59] with the addition of the Becke–Johnson damping (D3BJ) [41]. Frequencies were calculated (analytically for PBE and PBE0 and numerically for TPSSh) to ensure these structures corresponded to energy minima. Single-point energy calculations starting from PBE optimized geometry were then performed in gas phase and in a toluene continuum with the CPCM method [60].
3.4. Crystal Structure Determinations
The structure resolution was accomplished using the SHELXS-97 and SHELXT [61] programs, and the refinement was done with the SHELXL program [62,63]. The structure solution and the refinement were achieved with the PLATON software [64]. Pictures of the compound structure were obtained using the MERCURY software. During the refinement steps, all atoms except hydrogen atoms were refined anisotropically. The position of the hydrogen atoms was determined using residual electronic densities. Finally, in order to obtain a complete refinement, a weighting step followed by multiple loops of refinement was done. The structures have been deposited in the CCDC with #1901938 (1) and 1901939 (2).
3.5. CO Migratory Insertion Studies
Reactivity tests were conducted in 5 mm NMR tubes adapted with a J. Young valve by adding CO gas directly to a degassed frozen solution of 1 or 2 and letting it react at room temperature. Kinetic analysis was performed following the 1H NMR resonances. The concentration of 1 was normalized by benzene residue (used as internal standard) in the deuterated solvent and complex 2 was referred to the toluene (used as internal standard), which crystalized in the cell. Integration of the NMR signals required care.
4. Conclusions
We successfully synthesized an original molecule with full characterizations containing reductive divalent lanthanide and reactive NiMe2 fragments with a redox-active bridging ligand. The strong electron correlation occurred due to the lanthanide fragment and therefore largely influenced the reactivity behavior in carbon monoxide migratory insertion. Complex 2 had a singlet ground state and a substantially populated triplet state. It was also shown that due to the electron transfer, strong spin density was on the N-heteroatom ligand to the reactive metal center. These considerations led to the increased stability of the CO migratory insertion step, forming an acyl-methyl intermediate. This work provides us with a new strategy for the further mechanistic study of carbonylation reactions.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2304-6740/7/5/58/s1. 1H NMR characterization (Figures S1–S5), Kinetic analysis (Figures S6–S19 and Tables S1 and S2), Magnetism (Figure S20), Crystallographic data (Figures S21–S23 and Tables S3–S6), DFT Calculation data (Figures S24 and S25; Tables S7–S12).
Author Contributions
D.W. synthesized and characterized both compounds and studied the reactivity. M.T. realized the theoretical computations. M.C. realized the X-ray diffraction analysis. J.M. conducted the magnetic measurements and analyzed all the data with D.W. and G.N. G.N. had the original idea, managed all aspects of the project, and wrote the structure of the manuscript. All authors contributed to the editing of the manuscript.
Funding
This work is part of a project that has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program under grant agreement No. 716314.
Acknowledgments
CNRS and École polytechnique are thanked for funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Buchwalter, P.; Rosé, J.; Braunstein, P. Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef] [PubMed]
- Sil, A.; Ghosh, U.; Mishra, V.K.; Mishra, S.; Patra, S.K. Synthesis, structure, electrochemical, and spectroscopic properties of hetero-bimetallic Ru(II)/Fe(II)-alkynyl organometallic complexes. Inorg. Chem. 2019, 58, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Saito, K.; Kikuchi, S.; Akita, M.; Inagaki, A. Visible-light-controlled homo-and copolymerization of styrenes by a bichromophoric Ir–Pd catalyst. Chem. Commun. 2015, 51, 5717–5720. [Google Scholar] [CrossRef]
- Dobbek, H.; Gremer, L.; Kiefersauer, R.; Huber, R.; Meyer, O. Catalysis at a dinuclear [CuSMo(O)OH] cluster in a CO dehydrogenase resolved at 1.1-Å resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 15971–15976. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, T.S.; Hollingsworth, R.L.; Lord, R.L.; Groysman, S. Cooperative bimetallic reactivity of a heterodinuclear molybdenum–copper model of Mo–Cu CODH. Dalton Trans. 2018, 47, 10017–10024. [Google Scholar] [CrossRef] [PubMed]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase enzymes and their synthetic models: The role of metal hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, S.; Bruschi, M.; De Gioia, L.; Le Roy, C.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. FeMo heterobimetallic dithiolate complexes: Investigation of their electron transfer chemistry and reactivity toward acids, a density functional theory rationalization. Inorg. Chem. 2019, 58, 679–694. [Google Scholar] [CrossRef]
- Ogo, S.; Mori, Y.; Ando, T.; Matsumoto, T.; Yatabe, T.; Yoon, K.-S.; Hayashi, H.; Asano, M. One model, two enzymes: Activation of hydrogen and carbon monoxide. Angew. Chem. Int. Ed. 2017, 56, 9723–9726. [Google Scholar] [CrossRef]
- Mankad, N.P. Selectivity effects in bimetallic catalysis. Chem. Eur. J. 2016, 22, 5822–5829. [Google Scholar] [CrossRef]
- Pye, D.R.; Mankad, N.P. Bimetallic catalysis for C–C and C–X coupling reactions. Chem. Sci. 2017, 8, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Ajamian, A.; Gleason, J.L. Two birds with one metallic stone: Single-pot catalysis of fundamentally different transformations. Angew. Chem. Int. Ed. 2004, 43, 3754–3760. [Google Scholar] [CrossRef]
- Mata, J.A.; Hahn, F.E.; Peris, E. Heterometallic complexes, tandem catalysis and catalytic cooperativity. Chem. Sci. 2014, 5, 1723–1732. [Google Scholar] [CrossRef]
- Lorber, C.; Vendier, L. Imido–titanium/molybdenum heterobimetallic systems. Switching from η6-arene to fischer-type aminocarbene complexes by tuning reactivity conditions. Organometallics 2010, 29, 1127–1136. [Google Scholar] [CrossRef]
- Stark, H.S.; Altmann, P.J.; Sproules, S.; Hess, C.R. Structural characterization and photochemical properties of mono- and bimetallic Cu–Mabiq complexes. Inorg. Chem. 2018, 57, 6401–6409. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Kawashima, M.; Yamashita, H. Visible-light-enhanced suzuki–miyaura coupling reaction by cooperative photocatalysis with an Ru–Pd bimetallic complex. Chem. Commun. 2014, 50, 14501–14503. [Google Scholar] [CrossRef]
- Goudy, V.; Jaoul, A.; Cordier, M.; Clavaguéra, C.; Nocton, G. Tuning the Stability of Pd(IV) Intermediates using a redox non-innocent ligand combined with an organolanthanide fragment. J. Am. Chem. Soc. 2017, 139, 10633–10636. [Google Scholar] [CrossRef]
- Goudy, V.; Xémard, M.; Karleskind, S.; Cordier, M.; Alvarez Lamsfus, C.; Maron, L.; Nocton, G. Phenylacetylene and carbon dioxide activation by an organometallic samarium complex. Inorganics 2018, 6, 82. [Google Scholar] [CrossRef]
- Xémard, M.; Cordier, M.; Louyriac, E.; Maron, L.; Clavaguéra, C.; Nocton, G. Small molecule activation with divalent samarium triflate: A synergistic effort to cleave O2. Dalton Trans. 2018, 47, 9226–9230. [Google Scholar] [CrossRef]
- Evans, W.J.; Ulibarri, T.A.; Ziller, J.W. Isolation and x-ray crystal structure of the first dinitrogen complex of an f-element metal, [(C5Me5)2Sm]2N2. J. Am. Chem. Soc. 1988, 110, 6877–6879. [Google Scholar] [CrossRef]
- Jaroschik, F.; Momin, A.; Nief, F.; Le Goff, X.F.; Deacon, G.B.; Junk, P.C. Dinitrogen reduction and C–H activation by the divalent organoneodymium complex [(C5H2tBu3)2Nd(µ-I)K([18]crown-6)]. Angew. Chem. Int. Ed. 2009, 48, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.H.; Walter, M.D.; Kazhdan, D.; Hu, Y.-J.; Lukens, W.W.; Bauer, E.D.; Maron, L.; Eisenstein, O.; Andersen, R.A. Decamethylytterbocene complexes of bipyridines and diazabutadienes: Multiconfigurational ground states and open-shell singlet formation. J. Am. Chem. Soc. 2009, 131, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.H.; Kazhdan, D.; Werkema, E.L.; Walter, M.D.; Lukens, W.W.; Bauer, E.D.; Hu, Y.-J.; Maron, L.; Eisenstein, O.; Head-Gordon, M.; et al. Intermediate-valence tautomerism in decamethylytterbocene complexes of methyl-substituted bipyridines. J. Am. Chem. Soc. 2010, 132, 17537–17549. [Google Scholar] [CrossRef] [PubMed]
- Nocton, G.; Lukens, W.L.; Booth, C.H.; Rozenel, S.S.; Melding, S.A.; Maron, L.; Andersen, R.A. Reversible sigma C–C bond formation between phenanthroline ligands activated by (C5Me5)2Yb. J. Am. Chem. Soc. 2014, 136, 8626–8641. [Google Scholar] [CrossRef]
- Nocton, G.; Booth, C.H.; Maron, L.; Andersen, R.A. Thermal dihydrogen elimination from Cp*2Yb(4,5-diazafluorene). Organometallics 2013, 32, 1150–1158. [Google Scholar] [CrossRef]
- Nocton, G.; Booth, C.H.; Maron, L.; Andersen, R.A. Influence of the torsion angle in 3,3′-dimethyl-2,2′-bipyridine on the intermediate valence of Yb in (C5Me5)2Yb(3,3′-Me2-bipy). Organometallics 2013, 32, 5305–5312. [Google Scholar] [CrossRef]
- Jacquot, L.; Xémard, M.; Clavaguéra, C.; Nocton, G. Multiple one-electron transfers in bipyridine complexes of Bis(phospholyl) thulium. Organometallics 2014, 33, 4100–4106. [Google Scholar] [CrossRef]
- Nocton, G.; Booth, C.H.; Maron, L.; Ricard, L.; Andersen, R.A. Carbon–hydrogen bond breaking and making in the open-shell singlet molecule, Cp*2Yb(4,7-Me2phen). Organometallics 2014, 44, 6819–6829. [Google Scholar] [CrossRef]
- Nocton, G.; Ricard, L. Reversible C–C coupling in phenanthroline complexes of divalent samarium and thulium. Chem. Commun. 2015, 51, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Jaoul, A.; Clavaguera, C.; Nocton, G. Electron transfer in tetramethylbiphosphinine complexes of Cp*2Yb and Cp*2Sm. New J. Chem. 2016, 40, 6643–6649. [Google Scholar] [CrossRef]
- Dick, A.R.; Kampf, J.W.; Sanford, M.S. Unusually stable palladium(IV) Complexes: Detailed mechanistic investigation of C–O bond-forming reductive elimination. J. Am. Chem. Soc. 2005, 127, 12790–12791. [Google Scholar] [CrossRef] [PubMed]
- Shabashov, D.; Daugulis, O. Auxiliary-assisted palladium-catalyzed arylation and Alkylation of sp2 and sp3 carbon–hydrogen bonds. J. Am. Chem. Soc. 2010, 132, 3965–3972. [Google Scholar] [CrossRef] [PubMed]
- Rovira, M.; Roldán-Gómez, S.; Martin-Diaconescu, V.; Whiteoak, C.J.; Company, A.; Luis, J.M.; Ribas, X. Trifluoromethylation of a well-defined square-planar aryl–NiII complex involving NiIII/CF3· and NiIV–CF3 intermediate species. Chem. Eur. J. 2017, 23, 11662–11668. [Google Scholar] [CrossRef] [PubMed]
- Camasso, N.M.; Sanford, M.S. Design, synthesis, and carbon–heteroatom coupling reactions of organometallic nickel(IV) complexes. Science 2015, 347, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- D’Accriscio, F.; Borja, P.; Saffon-Merceron, N.; Fustier-Boutignon, M.; Mézailles, N.; Nebra, N. C–H bond trifluoromethylation of arenes enabled by a robust, high-valent nickel(IV) complex. Angew. Chem. Int. Ed. 2017, 56, 12898–12902. [Google Scholar] [CrossRef] [PubMed]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d transition metals for C–H activation. Chem. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Han, F.-S. Transition-metal-catalyzed Suzuki–Miyaura cross-coupling reactions: A remarkable advance from palladium to nickel catalysts. Chem. Soc. Rev. 2013, 42, 5270–5298. [Google Scholar] [CrossRef]
- Peng, J.-B.; Wu, F.-P.; Wu, X.-F. First-row transition-metal-catalyzed carbonylative transformations of carbon electrophiles. Chem. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Maitlis, P.M.; Haynes, A.; Sunley, G.J.; Howard, M.J. Methanol carbonylation revisited: Thirty years on. J. Chem. Soc. Dalton Trans. 1996, 2187–2196. [Google Scholar] [CrossRef]
- Paulik, F.E.; Roth, J.F. Novel catalysts for the low-pressure carbonylation of methanol to acetic acid. Chem. Commun. 1968, 1578a. [Google Scholar] [CrossRef]
- Haynes, A.; Maitlis, P.M.; Morris, G.E.; Sunley, G.J.; Adams, H.; Badger, P.W.; Bowers, C.M.; Cook, D.B.; Elliott, P.I.P.; Ghaffar, T.; et al. Promotion of iridium-catalyzed methanol carbonylation: Mechanistic studies of the cativa process. J. Am. Chem. Soc. 2004, 126, 2847–2861. [Google Scholar] [CrossRef]
- Kaschube, W.; Pörschke, K.R.; Wilke, G. tmeda-Nickel-Komplexe: III. (N,N,N′,N′-Tetramethylethylendiamin)-(dimethyl)nickel(II). J. Organomet. Chem. 1988, 355, 525–532. [Google Scholar] [CrossRef]
- Nocton, G.; Ricard, L. N-aromatic heterocycle adducts of bulky [1,2,4-(Me3C)3C5H2]2Sm: Synthesis, structure and solution analysis. Dalton Trans. 2014, 43, 4380–4387. [Google Scholar] [CrossRef] [PubMed]
- Van Vleck, J.H. The Theory of Electric and Magnetic Susceptibilities; Oxford University Press: London, UK, 1932. [Google Scholar]
- O’Connor, C.J. Magnetochemistry—advances in theory and experimentation. In Progress in Inorganic Chemistry; Lippard, S.J., Ed.; Wiley: Hoboken, NJ, USA, 1982; Chapter 4. [Google Scholar]
- Lukens, W.W.; Walter, M.D. Quantifying exchange coupling in f-ion pairs using the diamagnetic substitution method. Inorg. Chem. 2010, 49, 4458–4465. [Google Scholar] [CrossRef]
- Lukens, W.W.; Magnani, N.; Booth, C.H. Application of the hubbard model to Cp*2Yb(bipy), a model system for strong exchange coupling in lanthanide systems. Inorg. Chem. 2012, 51, 10105–10110. [Google Scholar] [CrossRef]
- Scarborough, C.C.; Wieghardt, K. Electronic structure of 2,2′-bipyridine organotransition-metal complexes. Establishing the ligand oxidation level by density functional theoretical calculations. Inorg. Chem. 2011, 50, 9773–9793. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.J.; Boncella, J.M.; Andersen, R.A. Preparation of coordination compounds of Cp*2Yb with heterocyclic nitrogen bases: Examples of antiferromagnetic exchange coupling across bridging ligands. Organometallics 2002, 21, 4622–4631. [Google Scholar] [CrossRef]
- Tilley, T.D.; Boncella, J.M.; Berg, D.J.; Burns, C.J.; Andersen, R.A.; Lawless, G.A.; Edelman, M.A.; Lappert, M.F. Bis[Bis(Trimethylsilyl)Amido]Bis(Diethyl Ether)Ytterbium and (Diethyl Ether)Bis(η5-Pentamethylcyclopentadienyl)Ytterbium. In Inorganic Syntheses; Ginsberg, A.P., Ed.; Wiley: Hoboken, NJ, USA, 2007; Chapter 27; pp. 146–150. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-electron scalar relativistic basis sets for third-row transition metal atoms. J. Chem. Theo. Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-electron scalar relativistic basis sets for the lanthanides. J. Chem. Theo. Comput. 2009, 5, 2229–2238. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallgr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Spek, A. Single-crystal structure validation with the program PLATON. J. App. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of the bimetallic complex. bipym: bipyrimidine ligand.
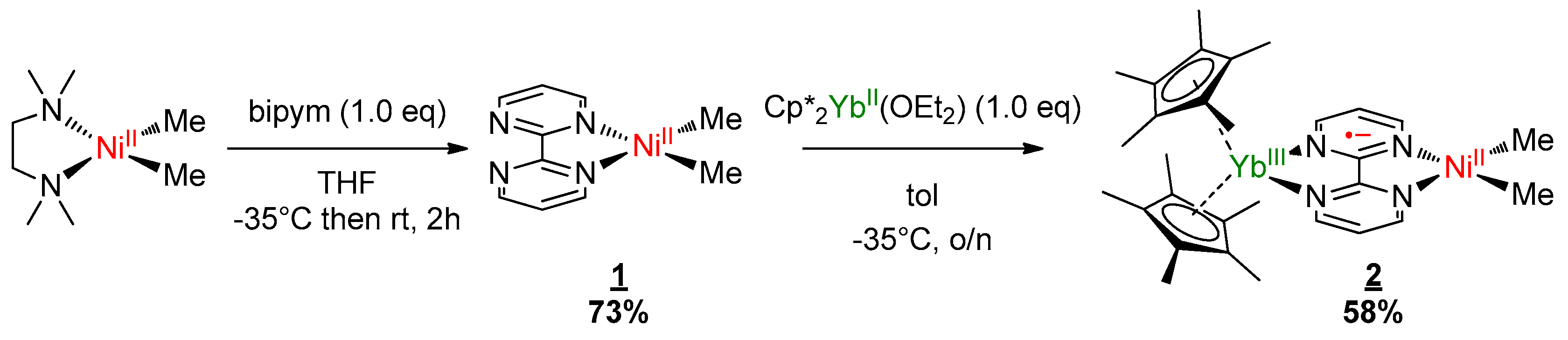
Figure 1.
(a) Oak Ridge Thermal Ellipsoid Plot (ORTEP) of (bipym)NiMe2 (1) (only one of the molecules of the cell is shown) (b) ORTEP of Cp*2Yb(bipym)NiMe2 (2). Hydrogen atoms have been removed for clarity and thermal ellipsoids are at the 50% level.
Figure 1.
(a) Oak Ridge Thermal Ellipsoid Plot (ORTEP) of (bipym)NiMe2 (1) (only one of the molecules of the cell is shown) (b) ORTEP of Cp*2Yb(bipym)NiMe2 (2). Hydrogen atoms have been removed for clarity and thermal ellipsoids are at the 50% level.
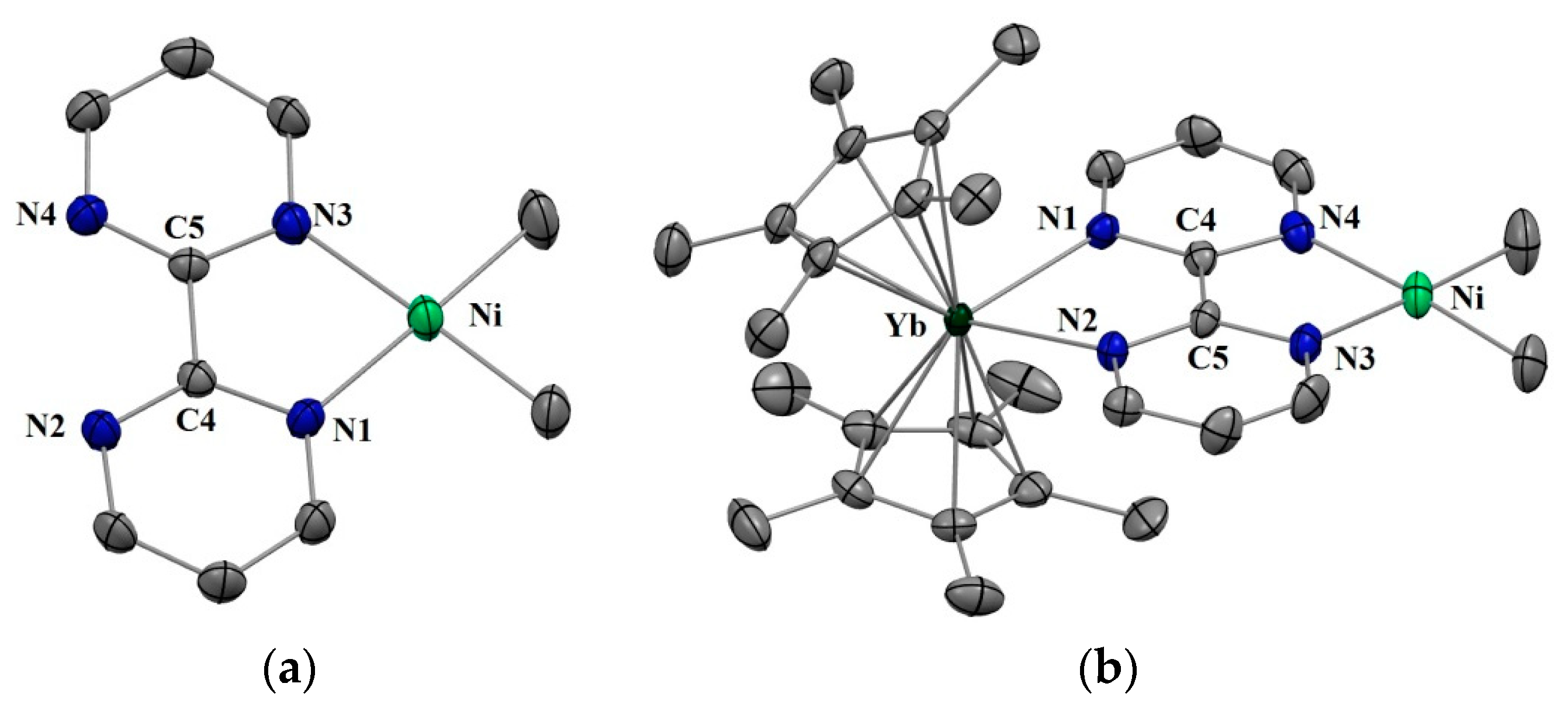
Figure 2.
(a) 1H NMR of 2 at 253 K, solvent and grease impurities are indicated by an asterisk (*); (b) Variable temperature 1H NMR chemical shifts (δ) versus 1/T (K−1) of the proton at −160.5 ppm on the previous spectrum.
Figure 2.
(a) 1H NMR of 2 at 253 K, solvent and grease impurities are indicated by an asterisk (*); (b) Variable temperature 1H NMR chemical shifts (δ) versus 1/T (K−1) of the proton at −160.5 ppm on the previous spectrum.
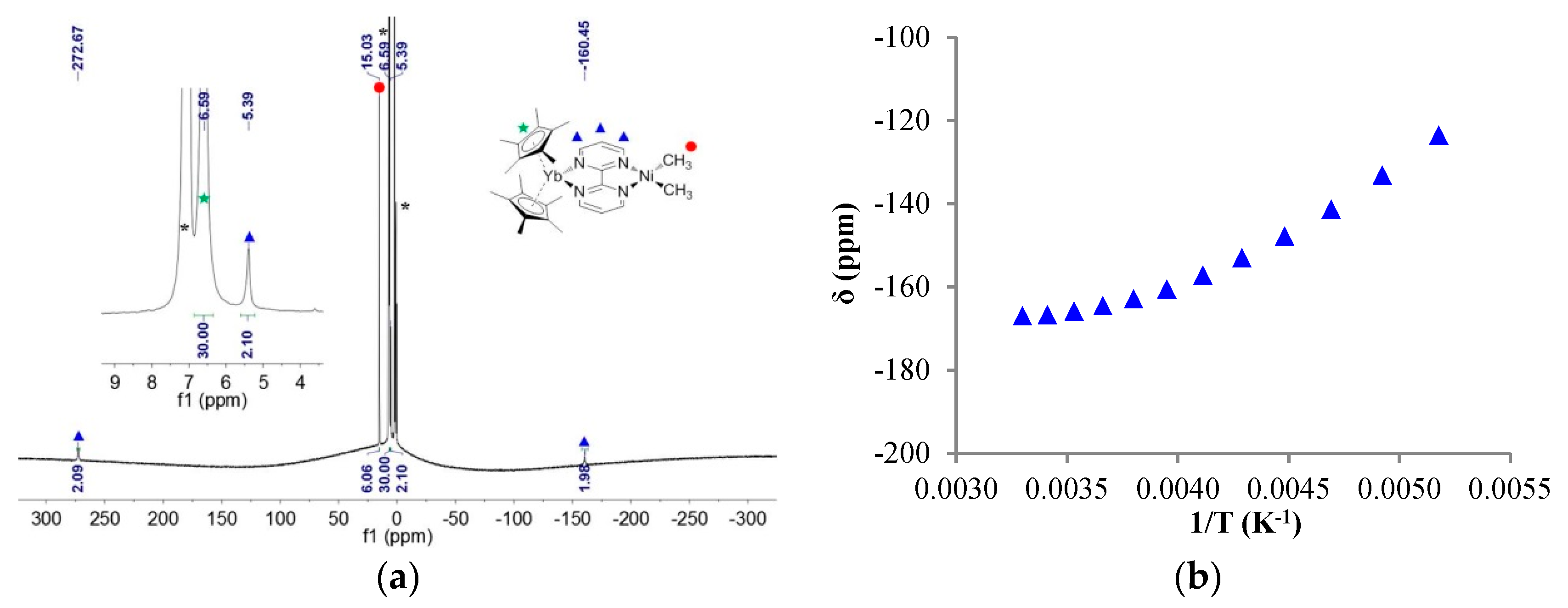
Figure 3.
Solid-state temperature-dependent magnetic data at 0.5 T: (a) plot of χ versus T and of (b) χT versus T. The solid line represents the fit of the magnetic data (see text).
Figure 3.
Solid-state temperature-dependent magnetic data at 0.5 T: (a) plot of χ versus T and of (b) χT versus T. The solid line represents the fit of the magnetic data (see text).
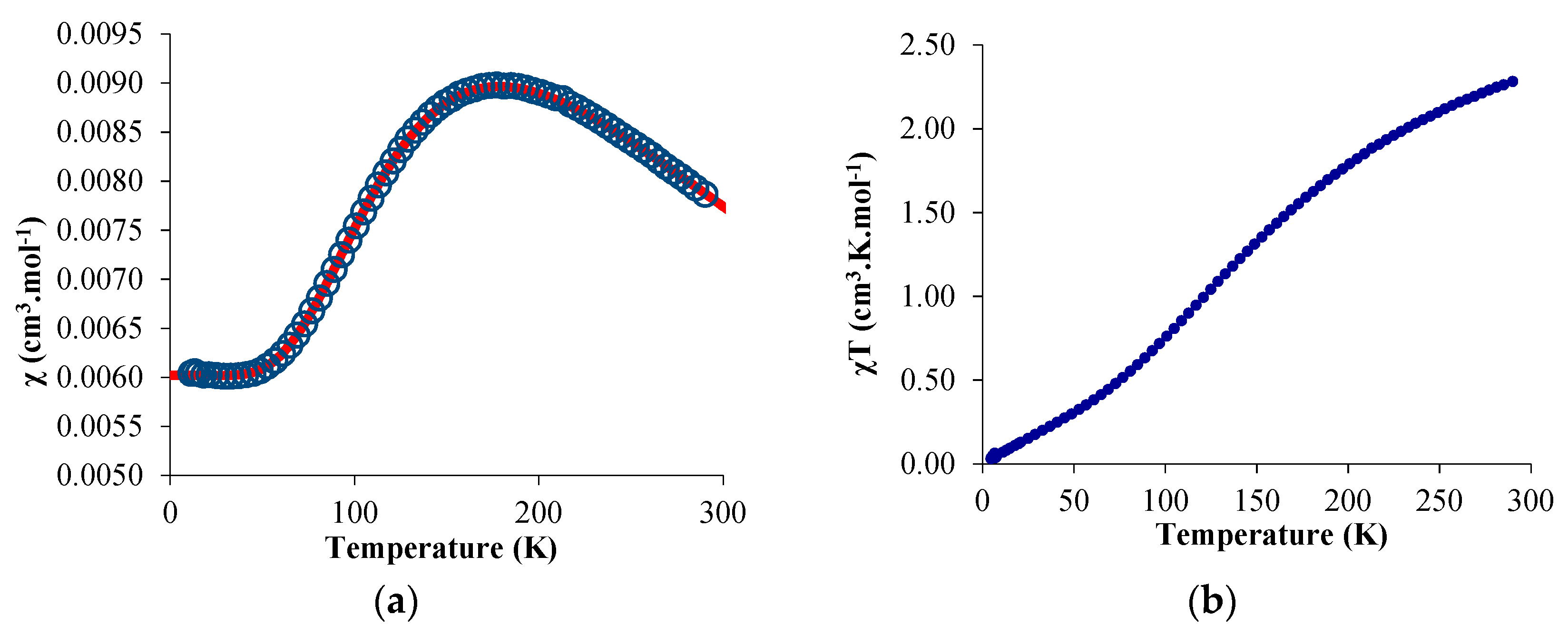
Figure 4.
Kohn−Sham orbitals of 1 at the TPSSh level of theory calculated taking PBE optimized geometry: (a) HOMO, (b) LUMO, and (c) LUMO +1.
Figure 4.
Kohn−Sham orbitals of 1 at the TPSSh level of theory calculated taking PBE optimized geometry: (a) HOMO, (b) LUMO, and (c) LUMO +1.

Scheme 2.
Reactivity of 2 with carbon monoxide.

Scheme 3.
Mechanism for the reaction of 2 with CO.
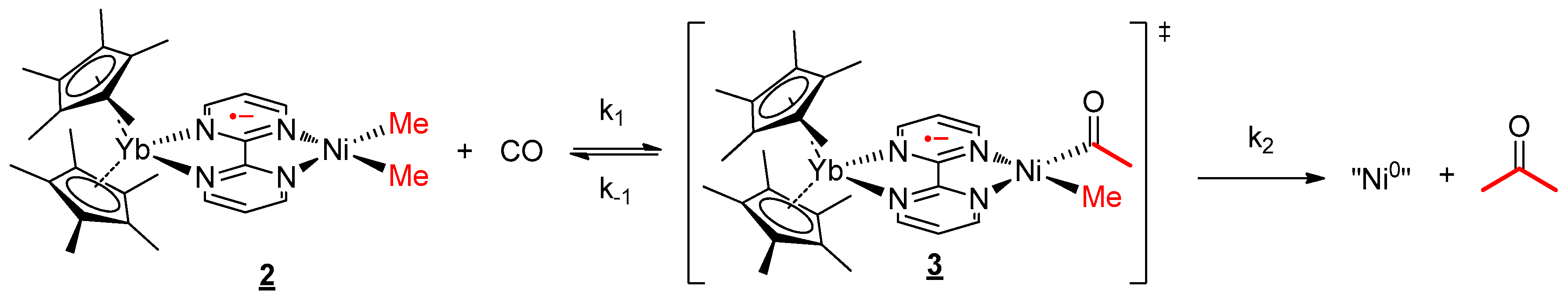
Figure 5.
(a) Plot of the concentration of 2/IS (normalized) over the reaction time at 35 °C. The insert shows the kinetics order. (b) Eyring plot analysis for the reaction of 2 with carbon monoxide. The internal standard (IS) used was the toluene molecules crystallized in the cell.
Figure 5.
(a) Plot of the concentration of 2/IS (normalized) over the reaction time at 35 °C. The insert shows the kinetics order. (b) Eyring plot analysis for the reaction of 2 with carbon monoxide. The internal standard (IS) used was the toluene molecules crystallized in the cell.
Scheme 4.
Mechanism for the reaction of 1 with CO.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, D.; Moutet, J.; Tricoire, M.; Cordier, M.; Nocton, G. Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments. Inorganics 2019, 7, 58. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7050058
AMA Style
Wang D, Moutet J, Tricoire M, Cordier M, Nocton G. Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments. Inorganics. 2019; 7(5):58. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7050058
Chicago/Turabian StyleWang, Ding, Jules Moutet, Maxime Tricoire, Marie Cordier, and Grégory Nocton. 2019. "Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments" Inorganics 7, no. 5: 58. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7050058
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.