
Non-Covalent Interactions of the Lewis Acids Cu–X, Ag–X, and Au–X (X = F and Cl) with Nine Simple Lewis Bases B: A Systematic Investigation of Coinage–Metal Bonds by Ab Initio Calculations



Abstract
:1. Introduction
- How do the distances r(Z⋯M), where Z is the acceptor atom of B, and the force constants kσ compare with those determined spectroscopically?
- Is De directly proportional to kσ?
- Can De be simply expressed as a product of a nucleophilicity NB assigned to B and an electrophilicity EMX assigned to M–X, and if so, do the NB agree with those determined earlier for the analogous hydrogen-bonded, halogen-bonded, and other non-covalently bound complexes?
- In view of the enhanced ionic character of a given M–X compared with that of the corresponding H–X, as alluded to earlier, are there any differences attributable to it?
- In addition, we shall consider the effects of normalising the De values with respect to the maximum positive values of the molecular electrostatic surface potentials (MESP) of the M–X molecules. Does this indicate whether the electrophilicity per unit positive potential along the molecular axis near to the atom M changes from F to Cl?
2. Computational Methods
3. Results
3.1. Comparison of Calculated and Experimental Properties of B⋯M–X
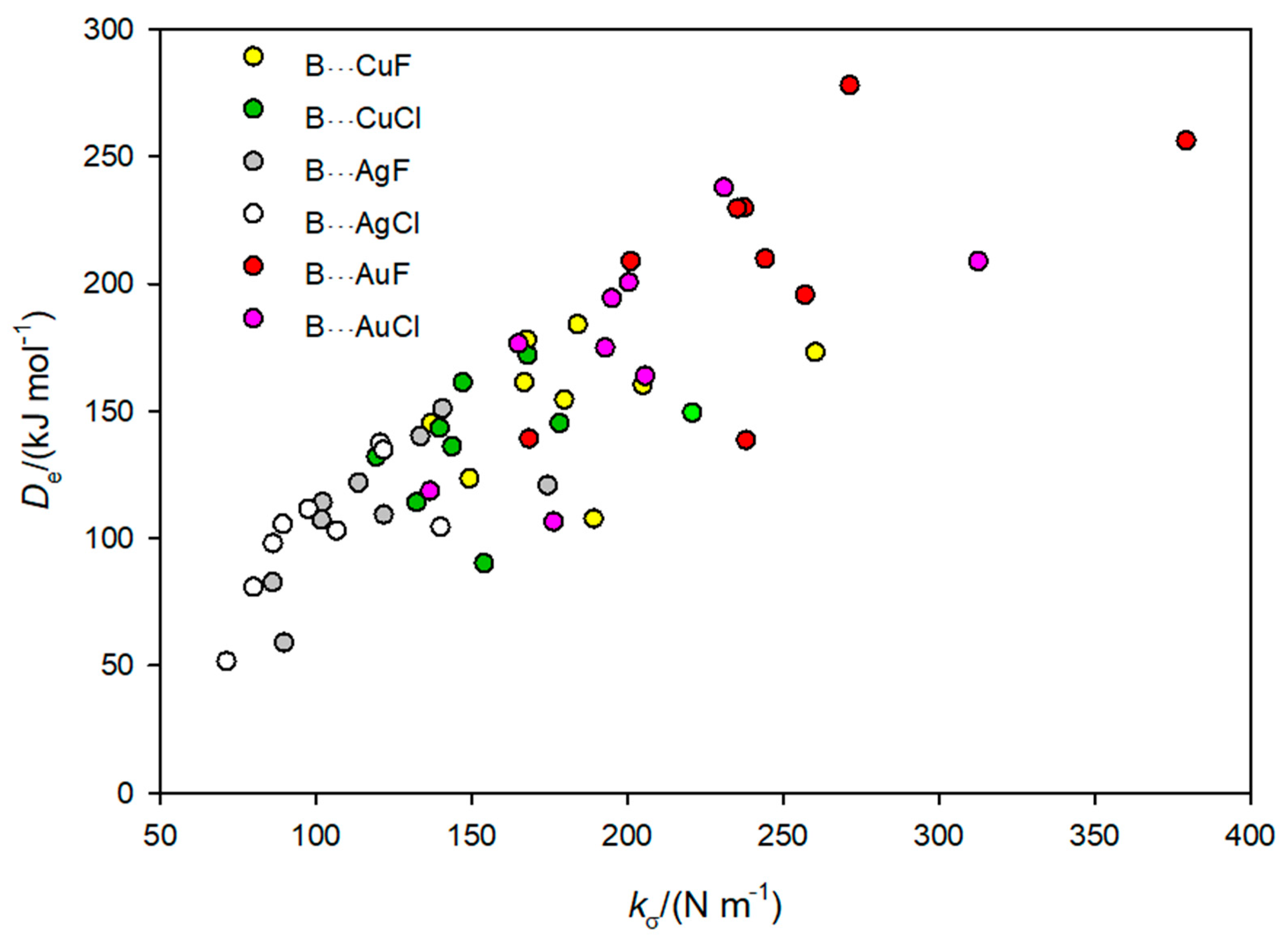
3.2. Is There a Linear Relationship between De and kσ for the B⋯M–X Complexes?
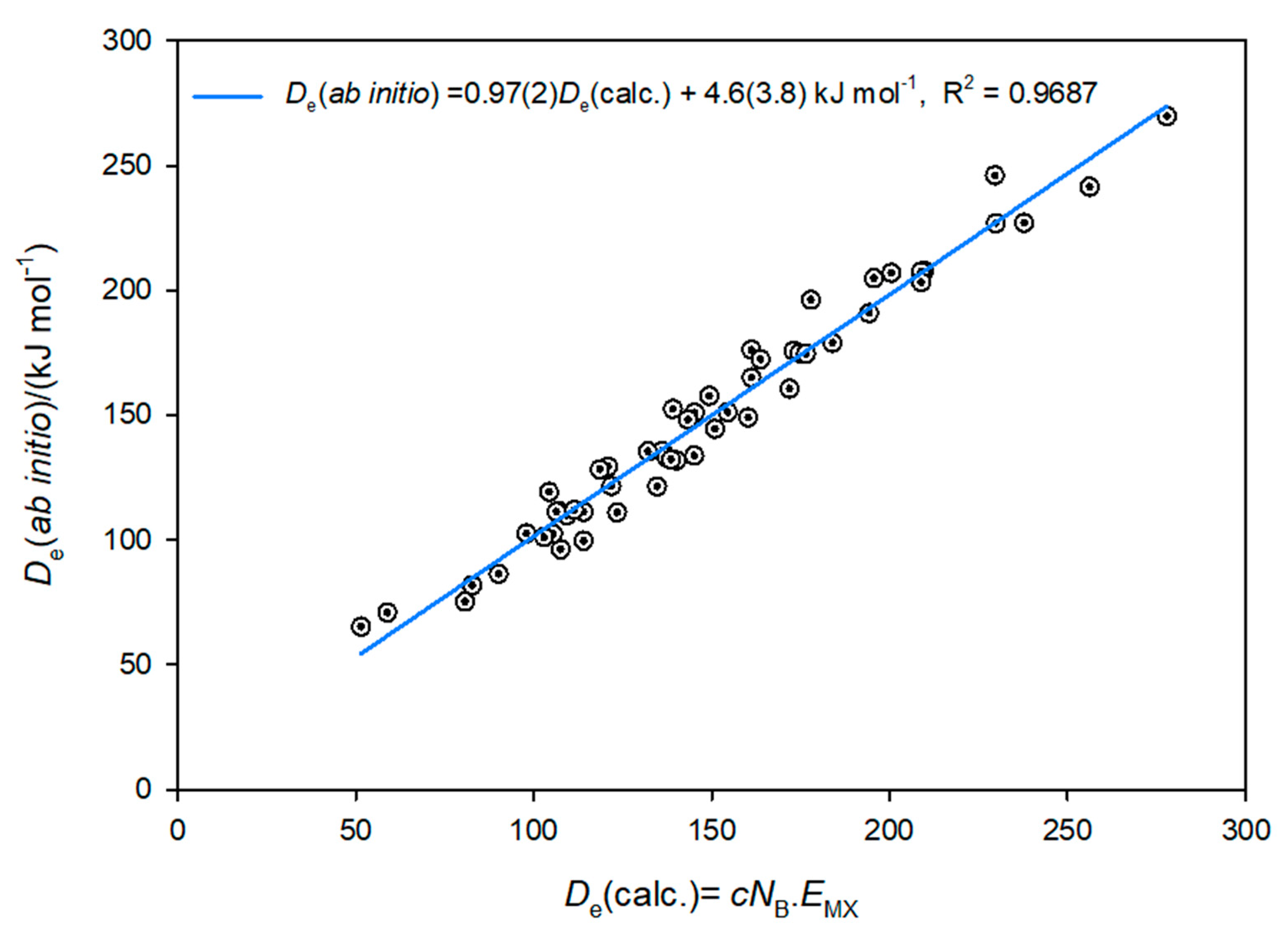
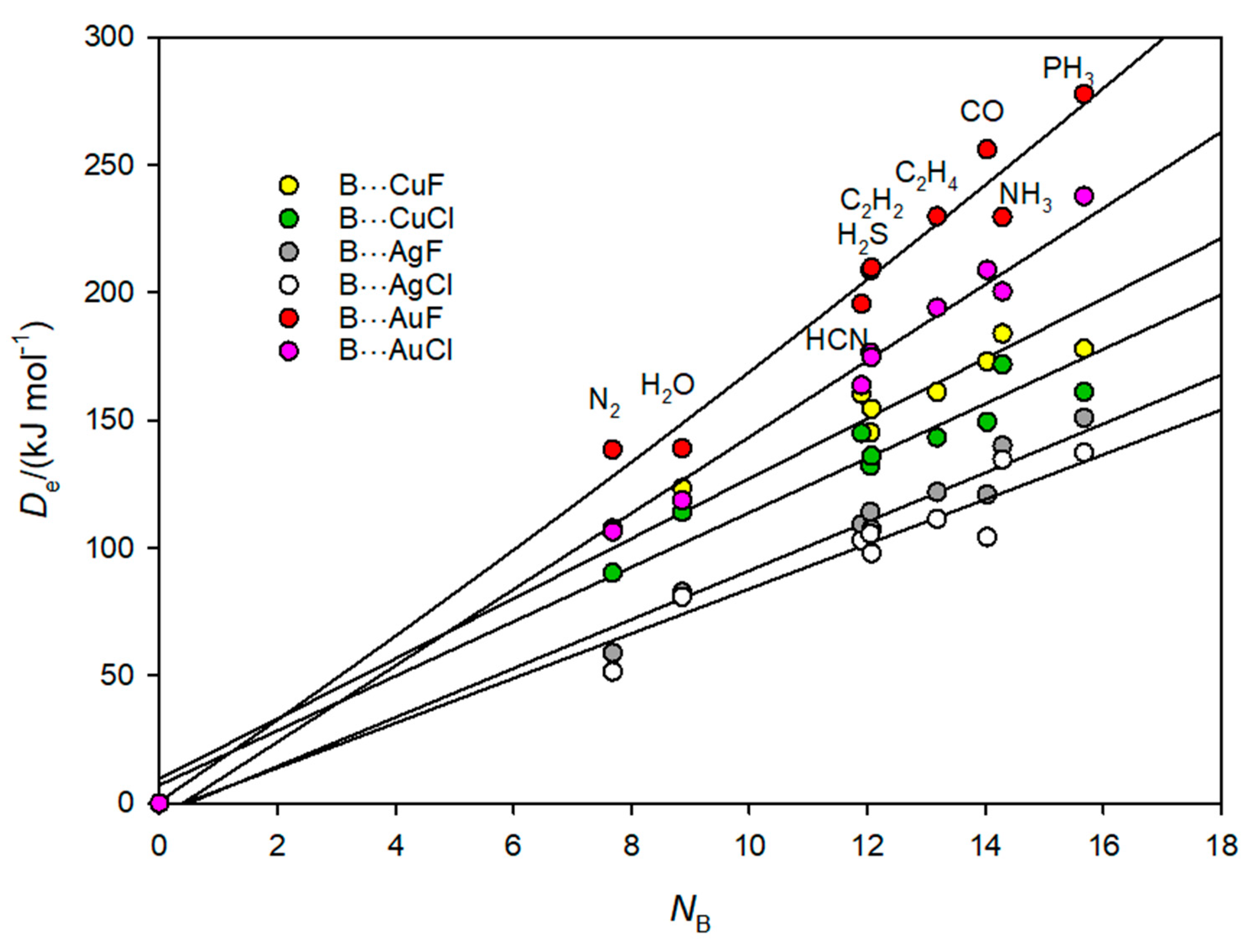
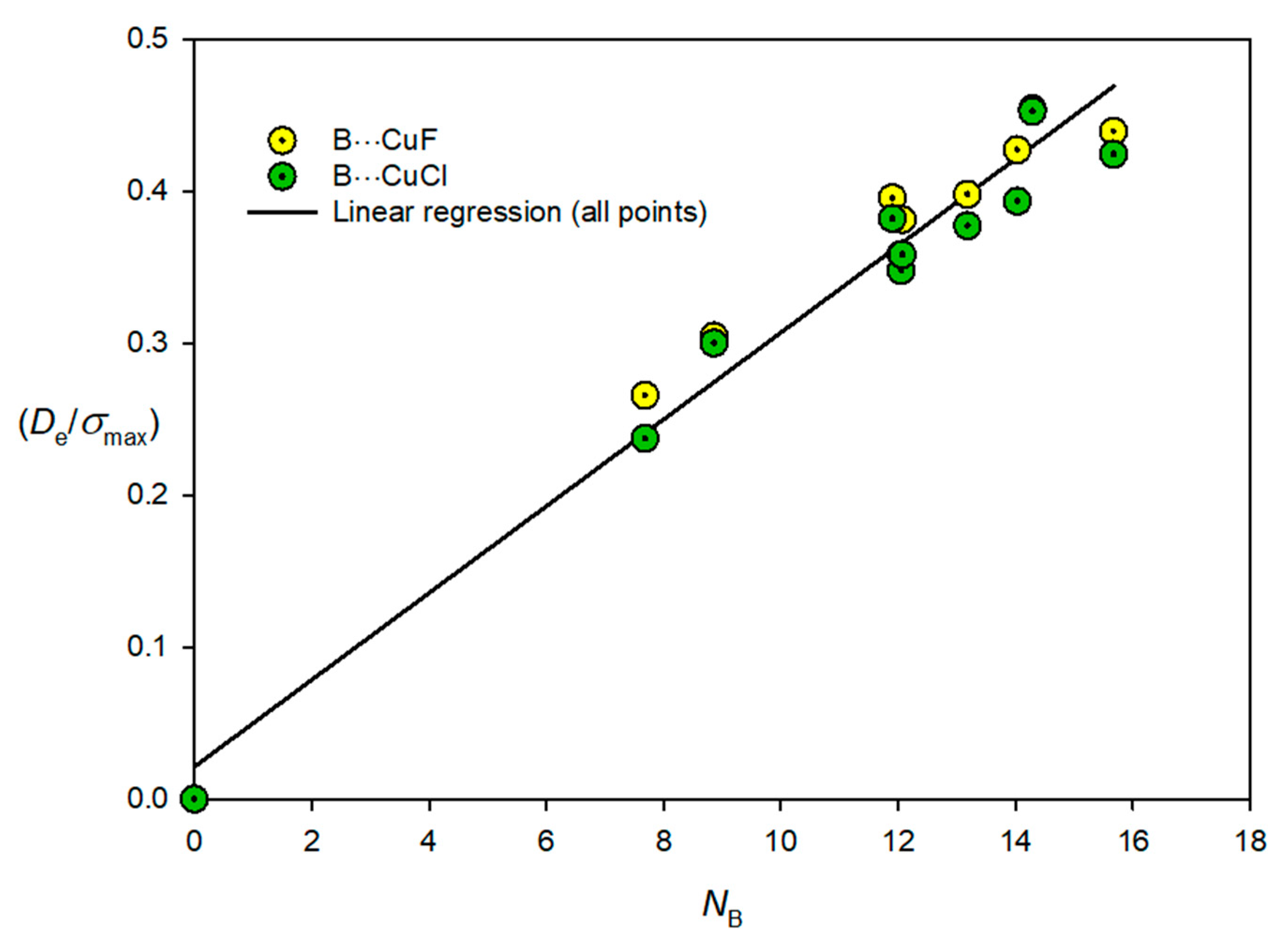
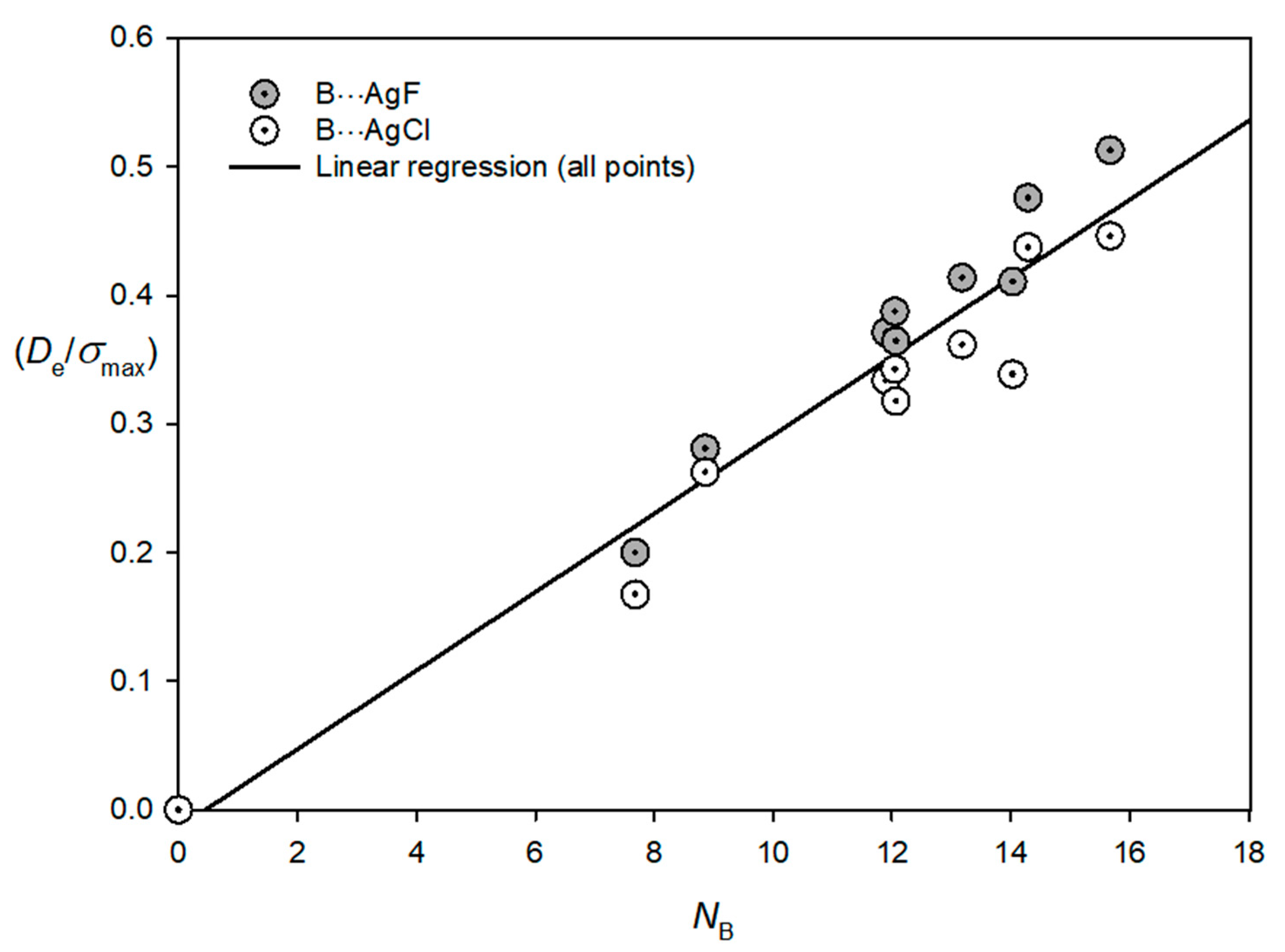
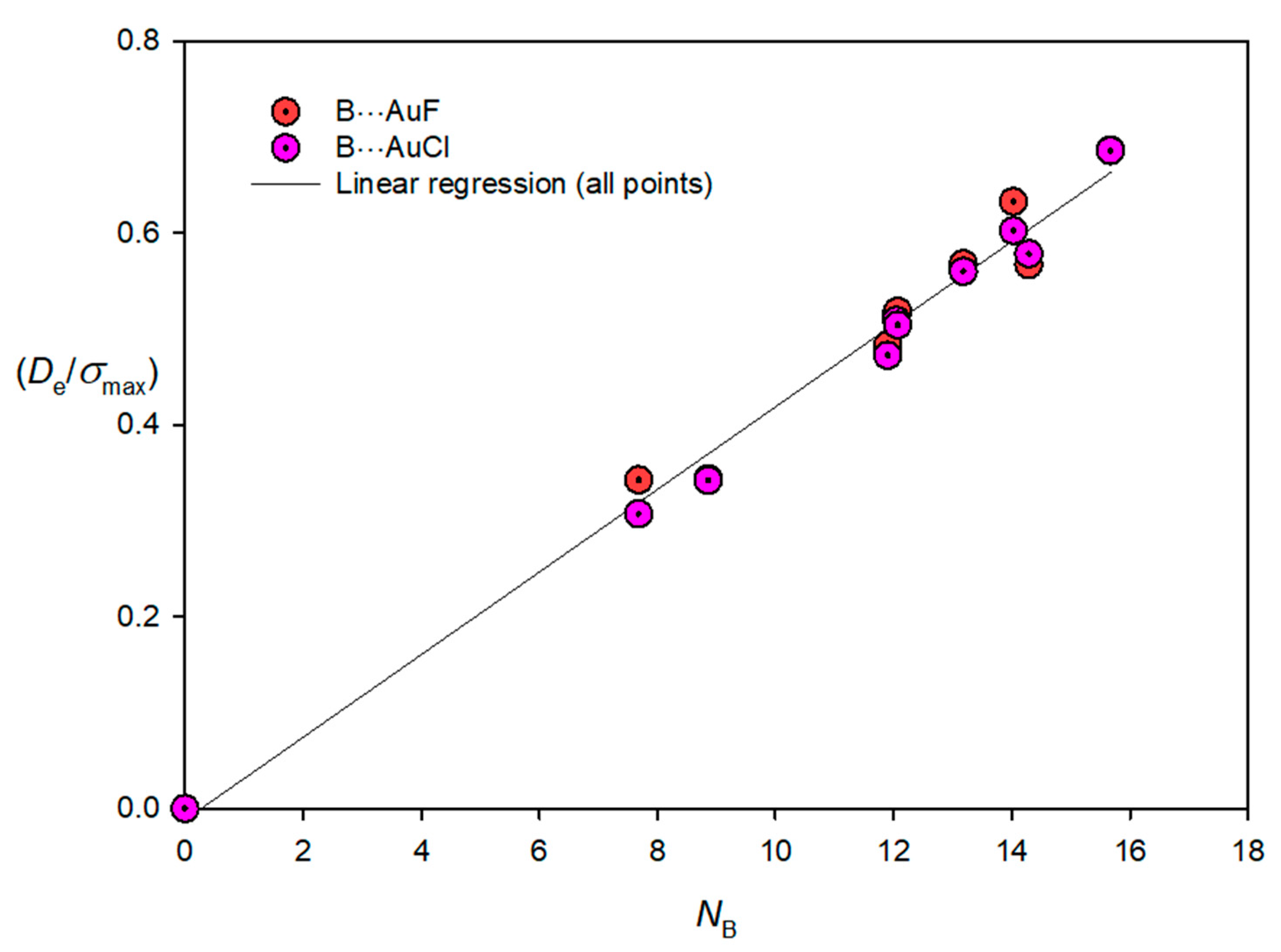
3.3. Electrophilicities of M–X and Nucleophilicities of B
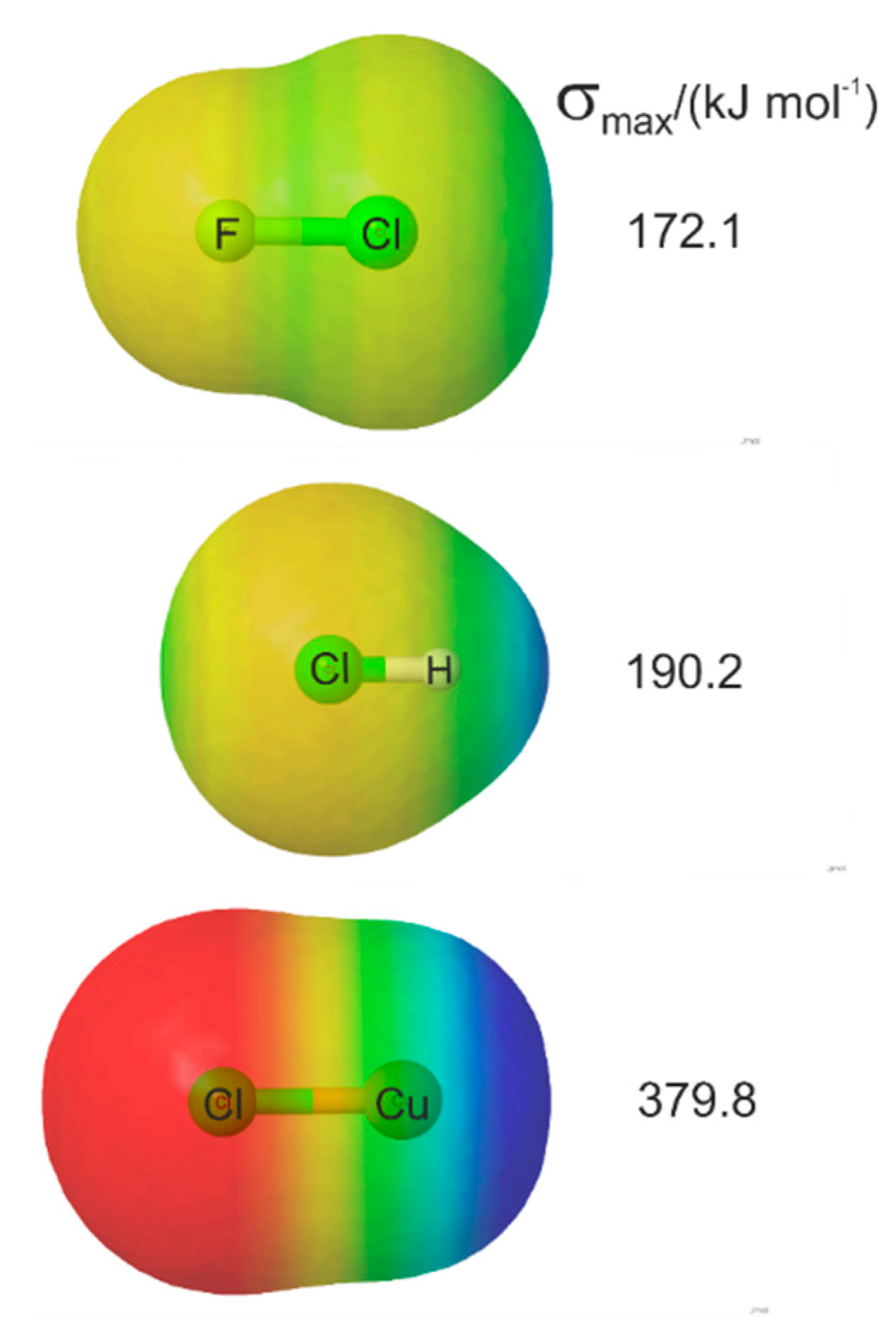
3.4. Molecular Electrostatic Surface Potentials and Reduced Electrophilicities of M–X Lewis Acids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, C.J.; Reynard, L.M.; Gerry, M.C.L. Pure Rotational Spectra, Structures, and Hyperfine Constants of OC-AuX (X = F, Cl, Br). Inorg. Chem. 2001, 40, 6123–6131. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.R.; Gerry, M.C.L. Microwave Spectra, Geometries, and Hyperfine Constants of OCCuX (X = F, Cl, Br). Inorg. Chem. 2001, 40, 6158–6166. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.R.; Gerry, M.C.L. Microwave Spectra, Geometries, and Hyperfine Constants of OCAgX (X = F, Cl, Br). Inorg. Chem. 2002, 41, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.G.; Matthews, S.L.; Poleshchuk, O.K.; Walker, N.R.; Legon, A.C. N2–Cu–F: A complex of dinitrogen and cuprous fluoride characterised by rotational spectroscopy. Angew. Chem. Int. Ed. 2006, 45, 6341–6343. [Google Scholar] [CrossRef]
- Harris, S.J.; Legon, A.C.; Walker, N.R.; Wheatley, D.E. Experimental detection and properties of H2O⋯Ag—Cl and H2S⋯Ag—Cl by rotational spectroscopy. Angew. Chem. Int. Ed. 2010, 49, 181–183. [Google Scholar] [CrossRef]
- Mikhailov, V.A.; Tew, D.P.; Walker, N.R.; Legon, A.C. H3N⋯AgCl: Synthesis in a supersonic jet and characterisation by rotational spectroscopy. Chem. Phys. Lett. 2010, 499, 16–20. [Google Scholar] [CrossRef]
- Mikhailov, V.A.; Roberts, F.J.; Stephens, S.L.; Harris, S.J.; Tew, D.P.; Harvey, J.N.; Walker, N.R.; Legon, A.C. Monohydrates of cuprous chloride and argentous chloride: H2O⋯CuCl and H2O⋯AgCl characterised by rotational spectroscopy and ab initio calculations. J. Chem. Phys. 2011, 134, 134305. [Google Scholar] [CrossRef]
- Stephens, S.L.; Tew, D.P.; Walker, N.R.; Legon, A.C. Monohydrate of argentous fluoride: H2O⋯AgF characterised by rotational spectroscopy and ab initio calculations. J. Mol. Spectrosc. 2011, 267, 163–168. [Google Scholar] [CrossRef]
- Walker, N.R.; Tew, D.P.; Harris, S.J.; Wheatley, D.E.; Legon, A.C. Characterisation of H2S⋯CuCl and H2S⋯AgCl isolated in the gas phase: A rigidly pyramidal geometry at sulphur revealed by rotational spectroscopy and ab initio calculations. J. Chem. Phys. 2011, 134, 014307. [Google Scholar] [CrossRef]
- Stephens, S.L.; Tew, D.P.; Mikhailov, V.A.; Walker, N.R.; Legon, A.C. A prototype transition metal-olefin complex C2H4⋯AgCl synthesised by laser ablation and characterised by rotational spectroscopy and ab initio methods. J. Chem. Phys. 2011, 135, 024315. [Google Scholar] [CrossRef]
- Stephens, S.L.; Mizukami, W.; Tew, D.P.; Walker, N.R.; Legon, A.C. Distortion of ethyne on formation of a π complex with silver chloride: C2H2⋯Ag−Cl characterised by rotational spectroscopy and ab initio calculations. J. Chem. Phys. 2012, 137, 174302. [Google Scholar] [CrossRef] [PubMed]
- Stephens, S.L.; Bittner, D.M.; Mikhailov, V.A.; Mizukami, W.; Tew, D.P.; Walker, N.R.; Legon, A.C. Changes in the geometries of C2H2 and C2H4 on coordination to CuCl revealed by broadband rotational spectroscopy and ab-initio calculations. Inorg. Chem. 2014, 53, 10722–10730. [Google Scholar] [CrossRef] [PubMed]
- Bittner, D.M.; Zaleski, D.P.; Stephens, S.L.; Tew, D.P.; Walker, N.R.; Legon, A.C. A monomeric complex of ammonia and cuprous chloride: H3N⋯CuCl isolated and characterised by rotational spectroscopy and ab initio calculations. J. Chem. Phys. 2015, 142, 144302. [Google Scholar] [CrossRef] [PubMed]
- Zaleski, D.P.; Stephens, S.L.; Tew, D.P.; Bittner, D.M.; Walker, N.R.; Legon, A.C. Distortion of ethyne when complexed with a cuprous and argentous halide: The rotational spectrum of C2H2⋯CuF. Phys. Chem. Chem. Phys. 2015, 17, 19230–19237. [Google Scholar] [CrossRef] [Green Version]
- Bittner, D.M.; Stephens, S.L.; Zaleski, D.P.; Tew, D.P.; Walker, N.R.; Legon, A.C. Gas Phase Complexes of H3N⋯CuF and H3N⋯CuI Studied by Rotational Spectroscopy and Ab Initio Calculations: The Effect of X (X = F, Cl, Br, I) in OC⋯CuX and H3N⋯CuX. Phys. Chem. Chem. Phys. 2016, 18, 13638–13645. [Google Scholar] [CrossRef] [Green Version]
- Legon, A.C.; Walker, N.R. What’s in a name? ‘Coinage-metal’ non-covalent bonds and their definition. Phys. Chem. Chem. Phys. 2018, 20, 19332–19338. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.A.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Regium bonds between Mn clusters (M = Cu, Ag, Au and n = 2–6) and nucleophiles NH3 and HCN. Phys. Chem. Chem. Phys. 2018, 20, 22498–22509. [Google Scholar] [CrossRef] [Green Version]
- Hoeft, J.; Lovas, F.J.; Tiemann, E.; Törring, T. Dipole moments of CuCl or AgCl. Z. Naturforsch. A 1970, 25, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Hensel, K.D.; Styger, C.; Jager, W.; Merer, A.J.; Gerry, M.C.L. Microwave spectra of metal chlorides produced using laser ablation. J. Chem. Phys. 1993, 99, 3321–3328. [Google Scholar] [CrossRef]
- Legon, A.C. A reduced radial potential energy function for the halogen bond and the hydrogen bond in complexes B⋯XY and B⋯HX, where X and Y are halogen atoms. Phys. Chem. Chem. Phys. 2014, 16, 12415–12421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkorta, I.; Legon, A.C. Strengths of non-covalent interactions in hydrogen-bonded complexes B⋯HX and halogen-bonded complexes B⋯XY (X, Y = F, Cl): An ab initio investigation. New J. Chem. 2018, 42, 10548–10554. [Google Scholar] [CrossRef]
- Alkorta, I.; Legon, A.C. Nucleophilicities of Lewis bases B and electrophilicities of Lewis acids as determined from the dissociation energies of complexes B⋯A involving hydrogen bonds, tetrel bonds, pnictogen bonds, chalcogen bonds and halogen bonds. Molecules 2017, 22, 1786. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Legon, A.C. Non-covalent interactions involving alkaline-earth atoms and Lewis bases B: An ab initio investigation of beryllium and magnesium bonds, B⋯MR2 (M = Be or Mg, and R = H, F or CH3). Inorganics 2019, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Hill, J.G.; Legon, A.C. An ab initio investigation of alkali–metal noncovalent bonds B⋯LiR and B⋯NaR (R = F, H or CH3) formed with simple Lewis bases B: The relative inductive effects of F, H and CH3. Phys. Chem. Chem. Phys. 2020, 22, 16421–16430. [Google Scholar] [CrossRef]
- Purvis, G.D., III; Bartlett, R.J. A full coupled-cluster singles and doubles model—The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef] [Green Version]
- Peterson, A.K.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Feller, D. The use of systematic sequences of wave functions for estimating the complete basis set, full configuration interaction limit in water. J. Chem. Phys. 1993, 98, 7059–7071. [Google Scholar] [CrossRef]
- Halkier, A.; Helgaker, T.; Jorgensen, P.; Klopper, W.; Olsen, J. Basis-set convergence of the energy in molecular Hartree–Fock calculations. Chem. Phys. Lett. 1999, 302, 437–446. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; et al. MOLPRO, Version 2012.1, A Package of Ab Initio Programs. 2012. Available online: http://www.molpro.net (accessed on 5 January 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision, A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 1 January 2021).
- Millen, D.J. Determination of stretching force constants of weakly bound dimers from centrifugal distortion constants. Can. J. Chem. 1985, 63, 1477–1479. [Google Scholar] [CrossRef]
- Bittner, D.M.; Walker, N.R.; Legon, A.C. A two force-constant model for complexes B⋯M−X (B is a Lewis base and MX is any diatomic molecule): Intermolecular stretching force constants from centrifugal distortion constants DJ or ΔJ. J. Chem. Phys. 2016, 144, 074308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costain, C.C. Determination of Molecular Structures from Ground State Rotational Constants. J. Chem. Phys. 1958, 29, 864–874. [Google Scholar] [CrossRef]
- Watson, J.K.G.; Roytburg, A.; Ulrich, W. Least-Squares Mass-Dependence Molecular Structures. J. Mol. Spectrosc. 1999, 196, 102–119. [Google Scholar] [CrossRef] [Green Version]
- Townes, C.H.; Schawlow, A.L. Microwave Spectroscopy; McGraw-Hill Book Company Inc.: New York, NY, USA, 1955; Chapter 9. [Google Scholar]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Understanding Regium Bonds and their Competition with Hydrogen Bonds in Au2:HX Complexes. ChemPhysChem 2019, 20, 1572–1580. [Google Scholar] [CrossRef]
- Zheng, B.; Liu, Y.; Wang, Z.; Zhou, F.; Liu, Y.; Ding, X.; Lu, T. Regium bonds formed by MX (M = Cu, Ag, Au; X = F, Cl, Br) with phosphine-oxide/phosphinous acid: Comparisons between oxygen-shared and phosphine-shared complexes. Mol. Phys. 2019, 117, 2443–2455. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Rivalry between Regium and Hydrogen Bonds Established within Diatomic Coinage Molecules and Lewis Acids/Bases. ChemPhysChem 2020, 21, 2557–2563. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lewis Base B | Lewis Acid, M–X | |||||
|---|---|---|---|---|---|---|
| Cu–F | Cu–Cl | Ag–F | Ag–Cl | Au–F | Au–Cl | |
| N2 | 107.6 | 90.2 | 58.9 | 51.5 | 138.6 | 106.4 |
| CO | 173.1 | 149.4 | 120.8 | 104.3 | 256.2 | 208.9 |
| C2H2 | 154.5 | 136.1 | 107.3 | 97.9 | 209.8 | 174.8 |
| CH2CH2 | 161.2 | 143.3 | 121.8 | 111.4 | 229.9 | 194.2 |
| PH3 | 177.9 | 161.2 | 151.0 | 137.4 | 277.9 | 237.8 |
| H2S | 145.2 | 132.1 | 114.0 | 105.5 | 208.8 | 176.5 |
| HCN | 160.3 | 145.1 | 109.3 | 102.9 | 195.6 | 163.8 |
| H2O | 123.4 | 114.0 | 82.7 | 80.8 | 139.1 | 118.6 |
| NH3 | 184.0 | 171.9 | 140.1 | 134.7 | 229.6 | 200.5 |
| Lewis Base B | Lewis Acid, M–X | |||||
|---|---|---|---|---|---|---|
| Cu–F | Cu–Cl | Ag–F | Ag–Cl | Au–F | Au–Cl | |
| N2 | 189.1 | 154.0 | 89.7 | 71.3 | 238.1 | 176.3 |
| CO | 260.3 | 220.8 | 174.4 | 140.0 | 379.3 | 312.6 |
| C2H2 | 179.7 | 143.7 | 101.8 | 86.2 | 244.2 | 192.8 |
| CH2CH2 | 166.9 | 139.7 | 113.7 | 97.6 | 237.5 | 195.0 |
| PH3 | 167.8 | 147.1 | 140.6 | 120.7 | 271.4 | 231.0 |
| H2S | 137.0 | 119.4 | 102.1 | 89.4 | 201.0 | 165.0 |
| HCN | 204.9 | 178.2 | 121.8 | 106.7 | 257.0 | 205.6 |
| H2O | 149.3 | 132.3 | 86.1 | 80.0 | 168.4 | 136.6 |
| NH3 | 184.0 | 168.0 | 133.5 | 121.7 | 235.3 | 200.4 |
| Lewis Base B | B⋯Cu–F | B⋯Cu–Cl | B⋯Ag–F | B⋯Ag–Cl | B⋯Au–F | B⋯Au–Cl | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Calc. | Exp. | Calc. | Exp. | Calc. | Exp. | Calc. | Exp. | Calc. | Exp. | Calc. | Exp. | |
| N2 | 1.815 | 1.790(2) a | 1.854 | - | 2.086 | - | 2.144 | - | 1.934 | - | 1.991 | - |
| CO | 1.785 | 1.76385(4) b | 1.817 | 1.79447(1) b | 1.970 | 1.96486(1) c | 2.019 | 2.01281(9) c | 1.859 | 1.847 d | 1.897 | 1.88593(6) d |
| C2H2 | 1.874 | 1.8474(7) e | 1.915 | 1.887(2) f | 2.139 | - | 2.192 | 2.1795(4) g | 2.002 | - | 2.043 | - |
| CH2CH2 | 1.895 | - | 1.934 | 1.908(12) g | 2.126 | - | 2.172 | 2.1697(4) h | 2.004 | - | 2.045 | - |
| PH3 | 2.134 | - | 2.168 | - | 2.287 | - | 2.331 | - | 2.188 | - | 2.224 | - |
| H2S | 2.154 | - | 2.190 | 2.1531(3) i | 2.355 | - | 2.400 | 2.3838(1) i | 2.239 | - | 2.285 | - |
| HCN | 1.819 | - | 1.851 | - | 2.061 | - | 2.101 | - | 1.940 | 1.987 | ||
| H2O | 1.918 | - | 1.943 | 1.925(5) j | 2.183 | 2.168(11) | 2.210 | 2.204(7) j | 2.085 | - | 2.132 | - |
| NH3 | 1.911 | 1.8928(6) k | 1.934 | 1.9182(13) l | 2.123 | - | 2.154 | 2.1545(8) m | 2.039 | - | 2.077 | - |
| Lewis Base B | NB | Lewis Acid M–X | EMX |
|---|---|---|---|
| N2 | 7.68 | Cu–F | 12.52 |
| CO | 14.03 | Cu–Cl | 11.24 |
| C2H2 | 12.07 | Ag–F | 9.22 |
| CH2CH2 | 13.18 | Ag–Cl | 8.49 |
| PH3 | 15.67 | Au–F | 17.21 |
| H2S | 12.05 | Au–Cl | 14.48 |
| HCN | 11.90 | ||
| H2O | 8.86 | ||
| NH3 | 14.29 |
| Molecule | σmax/(kJ mol−1) |
|---|---|
| Cu–F | 405.1 |
| Cu–Cl | 379.8 |
| Ag–F | 294.5 |
| Ag–Cl | 308.0 |
| Au–F | 404.8 |
| Au–Cl | 346.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkorta, I.; Walker, N.R.; Legon, A.C. Non-Covalent Interactions of the Lewis Acids Cu–X, Ag–X, and Au–X (X = F and Cl) with Nine Simple Lewis Bases B: A Systematic Investigation of Coinage–Metal Bonds by Ab Initio Calculations. Inorganics 2021, 9, 13. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9020013
Alkorta I, Walker NR, Legon AC. Non-Covalent Interactions of the Lewis Acids Cu–X, Ag–X, and Au–X (X = F and Cl) with Nine Simple Lewis Bases B: A Systematic Investigation of Coinage–Metal Bonds by Ab Initio Calculations. Inorganics. 2021; 9(2):13. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9020013
Chicago/Turabian StyleAlkorta, Ibon, Nicholas R. Walker, and Anthony C. Legon. 2021. "Non-Covalent Interactions of the Lewis Acids Cu–X, Ag–X, and Au–X (X = F and Cl) with Nine Simple Lewis Bases B: A Systematic Investigation of Coinage–Metal Bonds by Ab Initio Calculations" Inorganics 9, no. 2: 13. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9020013