
Arene-Osmium(II) Complexes in Homogeneous Catalysis



Departamento de Química Orgánica e Inorgánica, Facultad de Química, Universidad de Oviedo, Julián Clavería 8, E-33006 Oviedo, Spain
*
Authors to whom correspondence should be addressed.
Inorganics 2021, 9(7), 55; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9070055
Submission received: 23 June 2021
/
Revised: 9 July 2021
/
Accepted: 11 July 2021
/
Published: 12 July 2021
(This article belongs to the Special Issue Metal Arene Complexes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Although the application of arene-osmium(II) complexes in homogeneous catalysis has been much less studied than that of their ruthenium analogues, different works have shown that, in some instances, a comparable or even superior effectiveness can be achieved with this particular class of compounds. This review article focuses on the catalytic applications of arene-osmium(II) complexes. Among others, transfer hydrogenation, hydrogenation, oxidation, and nitrile hydration reactions, as well as different C-C bond forming processes, are comprehensively discussed.
1. Introduction
The isolation of bis(benzene)chromium by Fischer and Hafner in the 1950s marked the starting point of a fruitful research field in organometallic chemistry devoted to study the synthesis and properties of transition-metal complexes with π-coordinated arene ligands [1], compounds that have found many applications in modern chemistry [2,3,4,5,6,7,8,9]. Among them, half-sandwich (η6-arene)-ruthenium(II) complexes, introduced for the first time by Winkhaus and Singer in 1967 [10], have been particularly studied given (i) the ease of access to the dimeric precursors [{RuCl(µ-Cl)(η6-arene)}2] by dehydrogenation of cyclohexadiene derivatives with RuCl3·nH2O and (ii) the rich reactivity of these dimers which allows the facile exchange and functionalization of the coordinated arene, as well as the introduction a wide variety of mono-, bi- and tridentate P-, N-, O-, S-, or C-donor ligands in the coordination sphere of the metal by cleavage of the chloride bridges and chloride ligands substitution reactions [11,12,13,14,15,16,17]. Indeed, (η6-arene)-ruthenium(II) complexes currently represent one of the most versatile families of organometallic ruthenium compounds since the high structural diversity that can be achieved within this family of complexes enables their application in different fields, such as medicinal and supramolecular chemistry [18,19,20,21,22,23,24,25,26,27,28] and, obviously, homogeneous catalysis [29,30,31,32,33,34,35].
Although the behavior of (η6-arene)-osmium(II) complexes mimics to some extent that of their ruthenium counterparts, the chemistry of the former has been comparatively much less studied [11,36,37,38,39]. However, their catalytic potential is well documented and different works have shown that, in some instances, a comparable or even superior effectiveness can be achieved with this particular class of compounds when compared to their ruthenium analogues, or to related half-sandwich complexes of other metals more commonly employed in homogeneous catalysis, such as rhodium and iridium. The aim of the present review article is to provide a comprehensive overview of the developments achieved in this particular research area, thus complementing other reviews and accounts previously published covering the application of osmium-based catalysts in selected organic transformations (mainly hydrogenation, dehydrogenation and isomerization processes) [35,40,41,42,43].
2. Transfer Hydrogenation and Hydrogenation Reactions
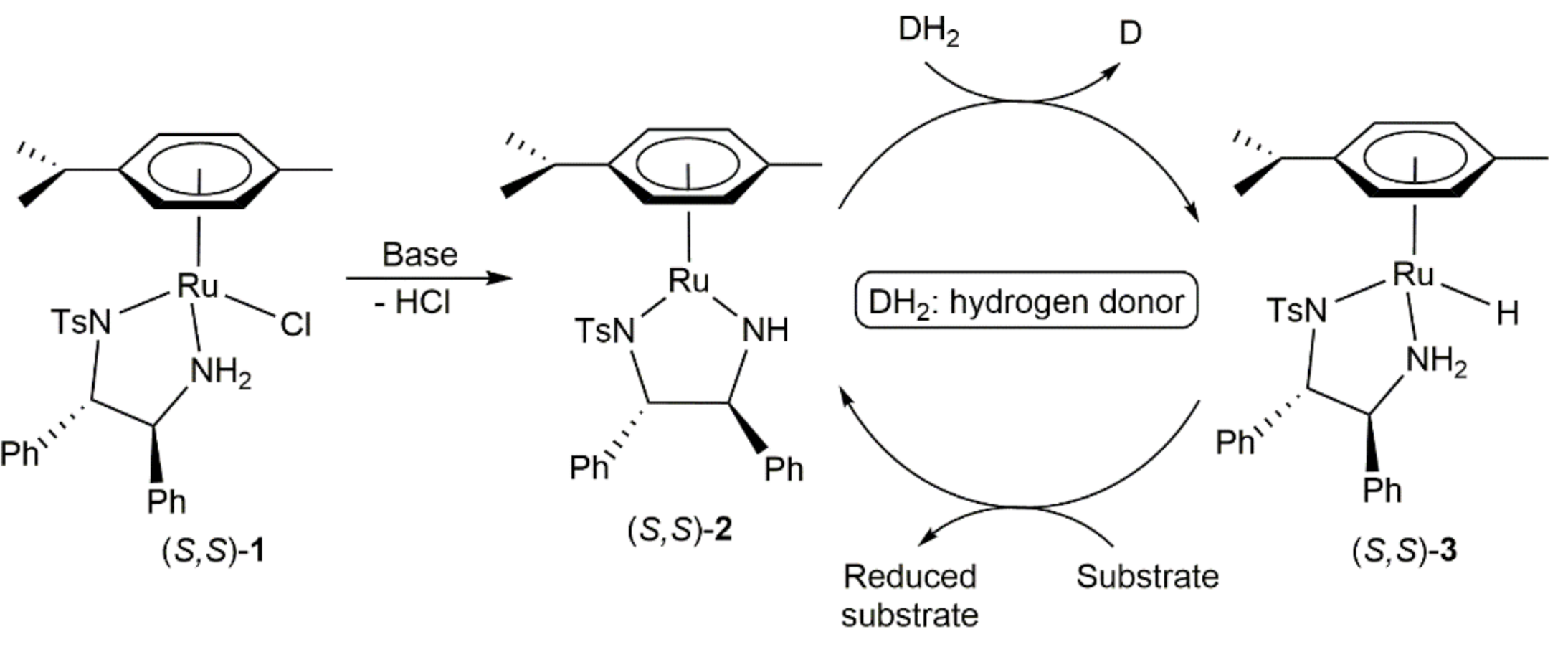
During the last three decades, metal-catalyzed transfer hydrogenation processes have attracted a great academic interest [44,45,46], representing nowadays a reliable synthetic tool for the reduction of carbonyl compounds and other unsaturated substrates, which in addition has found applications in industry [47,48]. A wide range of transition- and main group-metals have been successfully involved in such transformations, with ruthenium complexes playing a prominent role [44]. In this context, we must highlight the arene-ruthenium(II) derivatives (R,R)- and (S,S)-[RuCl(η6-p-cymene){TsNCH(Ph)CH(Ph)NH2}] (1; Ts = p-toluenesulfonate), reported by Noyori and co-workers at the end of 1990s [49,50], capable to reduce a broad range of substrates in high yields and with an outstanding enantioselectivity (up to > 99% ee). The key point for their high activity and stereoselection lies in the presence of a NH2 group coordinated to the metal center and the formation of the 16-electron complexes [Ru(η6-p-cymene){TsNCH(Ph)CH(Ph)NH}] (2) and the hydride derivatives [RuH(η6-p-cymene){TsNCH(Ph)CH(Ph)NH2}] (3), that have been identified as the active species of the catalytic cycle (Scheme 1).
Transfer hydrogenation reactions with these catalysts proceed through a ligand-assisted outer-sphere mechanism in which the substrate to be reduced has no direct interaction with the metal center. Instead, a six-membered ring transition state, generated by H-bonding interaction between the hydride and amine ligands of 3 with the substrate, takes place (Figure 1). This interaction allows for the simultaneous transfer of the hydride and NH proton, thus leading to the reduced product and the regeneration 2 [51]. The catalyst is configurationally defined at the Ru center because a favored diastereoisomer of the hydride intermediate 3 is generated during each cycle and its chirality is relayed to the product, which explains the high enantioselectivities usually observed.
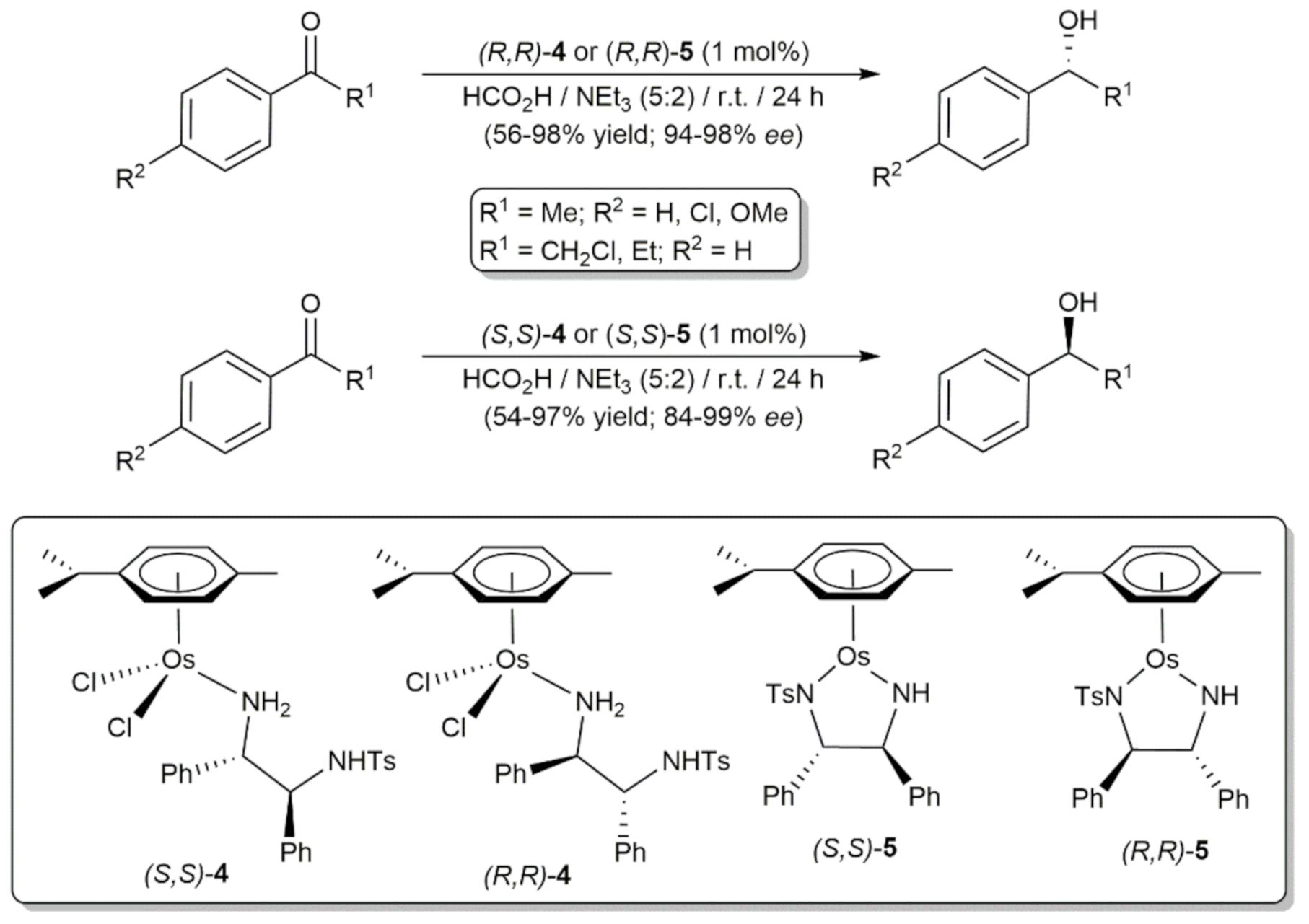
Stimulated by the great success achieved by this type of Ru(II) catalysts, Wills, Sadler and co-workers synthetized in 2015 the related air-stable 18- and 16-electron Os(II) derivatives (R,R)/(S,S)-[OsCl2(η6-p-cymene){TsNHCH(Ph)CH(Ph)NH2}] (4) and (R,R)/(S,S)-[Os(η6-p-cymene){TsNCH(Ph)CH(Ph)NH}] (5), respectively, and studied their behavior in the asymmetric transfer hydrogenation of aromatic ketones with formic acid as the hydrogen donor (Scheme 2) [52]. The catalytic reactions, performed at r.t. with a metal loading of 1 mol% in the formic acid/triethylamine azeotrope, led to the corresponding alcohols in moderate to high yields after 24 h, with enantioselectivities comparable to those reached with the 16-electron Ru(II) complexes (R,R)- and (S,S)-2 under identical conditions. However, we must note that conversions were in general lower as a consequence of the slower kinetics associated with the ligands substitution processes at osmium.
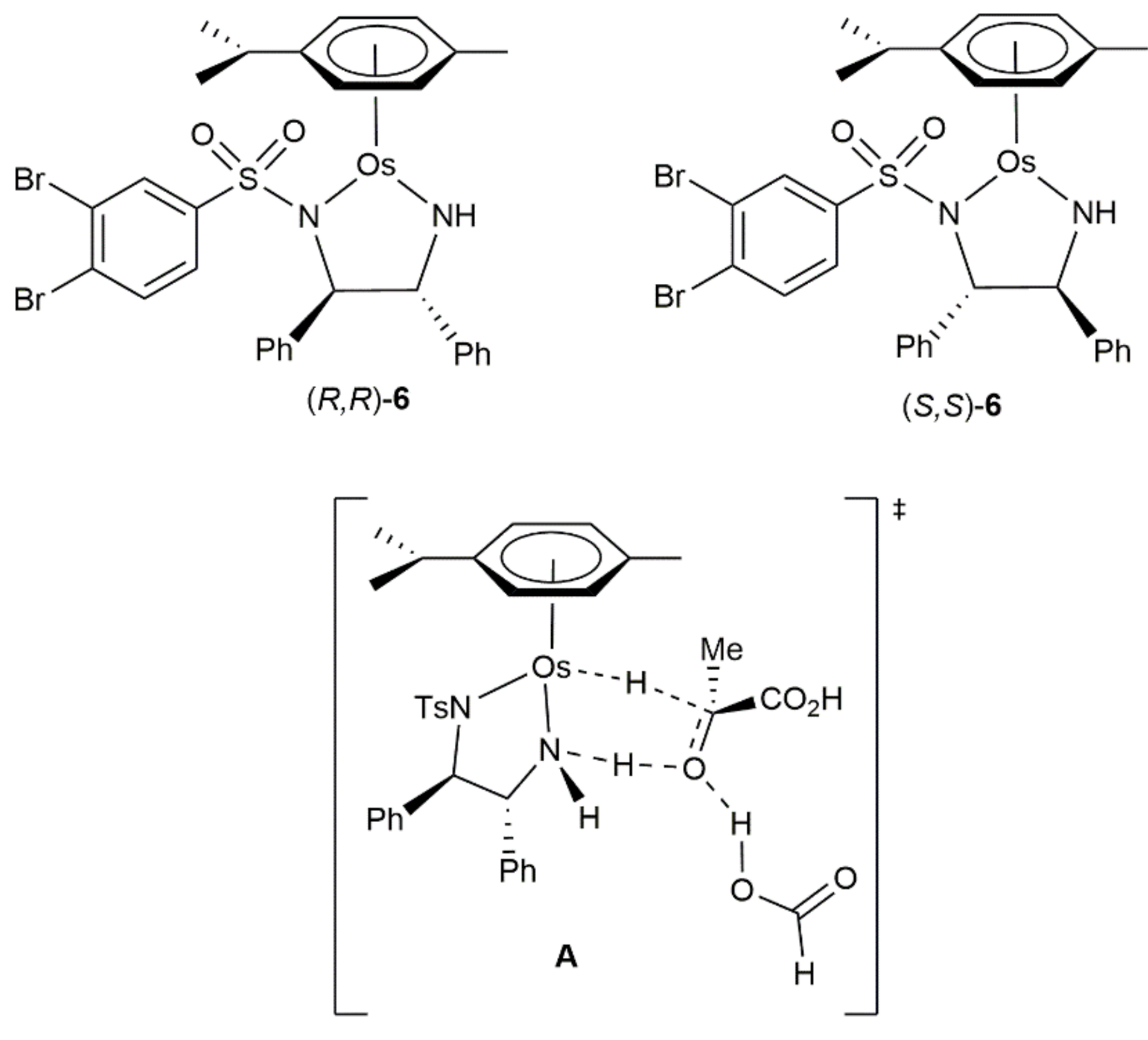
In a subsequent study, the same group achieved the enantioselective reduction of pyruvate to l-lactate and D-lactate (up to 84% ee) catalyzed by complexes (R,R)-5 and (S,S)-5, respectively, in human cancer cells employing non-toxic concentrations of sodium formate as the hydrogen source [53]. Inductively coupled plasma-mass spectrometry (ICP-MS) experiments on fractionated cancer cells treated with compounds (R,R)/(S,S)-5 suggested that the catalytic reactions may take place in the cytosol. In addition, insights into the intracellular stability of these catalysts, was gained with the bromide-labelled derivatives (R,R)/(S,S)-6 (Figure 2) employing a combination of X-ray fluorescence (XRF) elemental mapping and ICP-MS techniques [54]. The results obtained pointed to a rapid degradation of the complexes, with the brominated N,N-donor ligands being easily displaced by reaction with endogeneous thiols and translocated into the nucleus, and the resulting osmium-containing fragments excreted from the cells via the P-glycoprotein membrane pumps (please note that osmium catalysis in cells is an emerging and promising area of research since catalytic anticancer metallodrugs could minimize side-effects, lead to novel mechanisms of action and widen their therapeutic applications [55,56]). It should also be mentioned at this point that Density Functional Theory (DFT) calculations on the asymmetric transfer hydrogenation of pyruvic acid into lactic acid catalyzed by (R,R)-[Os(η6-p-cymene){TsNCH(Ph)CH(Ph)NH}] (5) in formic acid where performed by Wang and Yang, revealing that the enantio-determining hydride and proton transfer step is assisted by one formic acid molecule (A in Figure 2) [57].
Asymmetric transfer hydrogenation of ketones using 2-propanol as solvent and hydrogen donor was successfully accomplished by Faller and Lavoie with an arene-osmium(II) catalyst generated in situ from dimer [{OsCl(μ-Cl)(η6-p-cymene)}2] and the chiral amino-alcohol ligand (1R,2S)-(+)-cis-1-amino-2-indanol (Scheme 3) [58]. The process, which required of the assistance of a base (KOtBu), was highly enantioselective, yielding the corresponding alcohol products with ee´s ≥ 89%. It is worth mentioning that both the conversions and enantioselectivities observed in these reactions were comparable to those previously described with the [{RuCl(μ-Cl)(η6-p-cymene)}2]/(1R,2S)-(+)-cis-1-amino-2-indanol combination [59].
The same catalytic system was further employed for the asymmetric transfer hydrogenation of different para- and ortho-substituted benzaldehyde-α-d derivatives [60]. As shown in Scheme 4, the corresponding alcohols were in all the cases generated in almost quantitative yield, albeit with only a modest to moderate enantioselectivity (up to 60% ee). As in the precedent case, similar conversions and enantiomeric excesses (26–68% ee), were reached when [{RuCl(μ-Cl)(η6-p-cymene)}2], or the related chloride-bridged dimers [{RhCl(μ-Cl)Cp*}2] and [{IrCl(μ-Cl)Cp*}2] (Cp* = pentamethylcyclopentadienyl), were used as the metal sources.
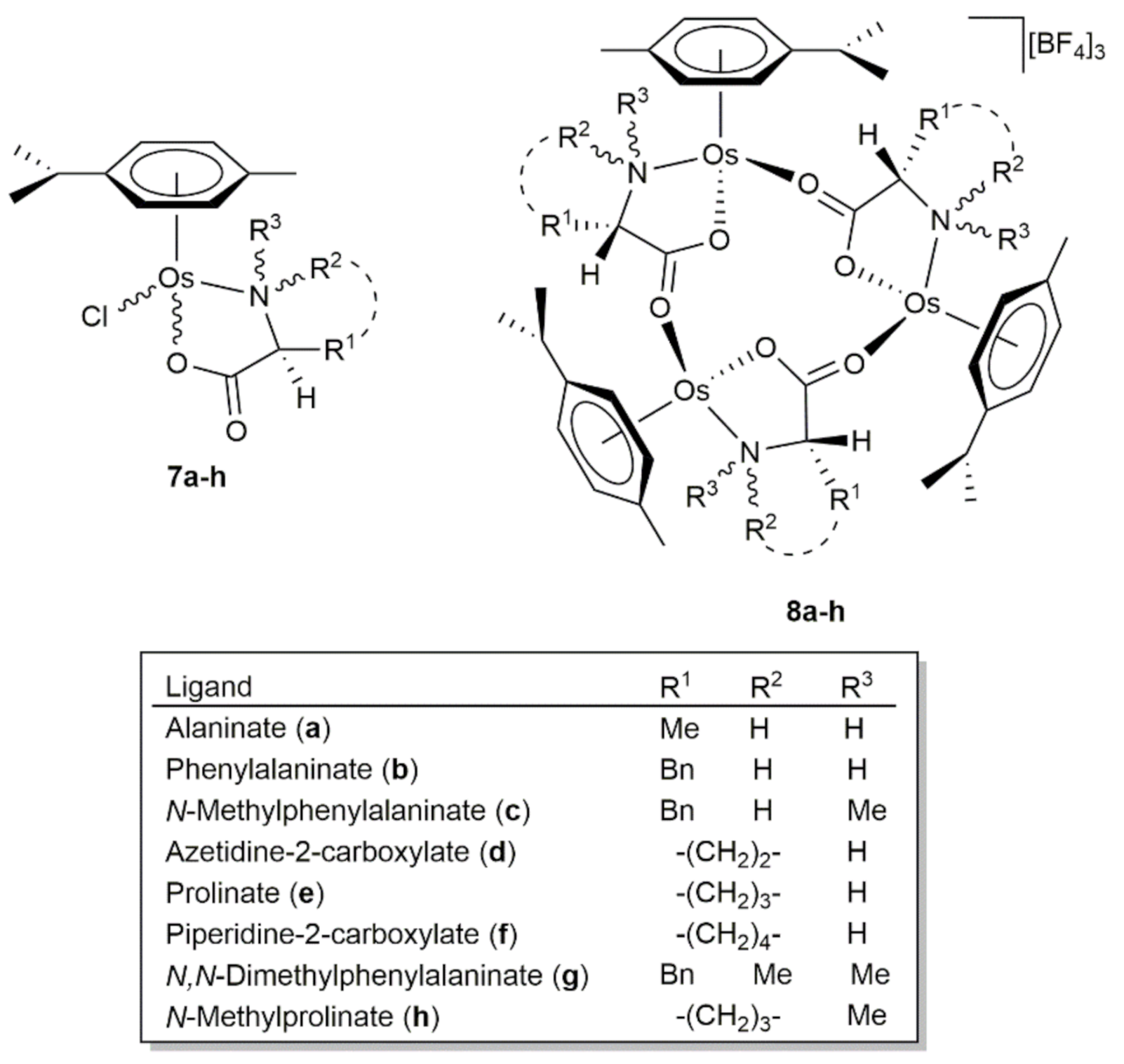
The asymmetric transfer hydrogenation of ketones was also studied by Carmona and co-workers employing as catalysts different mono- and trinuclear arene-osmium(II) complexes featuring chelated l-α-amino carboxylate ligands (Figure 3) [61,62]. The mononuclear derivatives 7a-h were generated by reacting [{OsCl(μ-Cl)(η6-p-cymene)}2] with two equivalents of the corresponding l-α-amino acid in the presence of KOH and, during their formation, the metal (7a,b,g) or the metal and nitrogen atoms (7c-f,h) became stereogenic centers. Regardless of the ligand, all of them were isolated as non-separable mixtures of the two possible epimers at the metal (in the case of 7c-f,h, only one epimer at nitrogen was formed) and subjected to catalysis as the diastereomeric mixture. Concerning the trinuclear species 8a-h, they were synthesized from 7a-h by chloride abstraction with AgBF4 and the trimerization process proceeded with chiral self-recognition, i.e., only the diastereoisomers with equal configurations at the metal (ROsROsROs and SOsSOsSOs) were formed.
In the presence of a base (NaCO2H), complexes 7–8a-h were active catalysts for the transfer hydrogenation of acetophenone using 2-propanol as solvent and hydrogen donor. The reactions, performed at 83 °C with a catalyst loading of 1.5 mol%, led to 1-phenylethanol in variable yields (18–97%) after 1–24 h with up to 72% ee (reached with trimer 8e). The reaction rate was strongly influenced by the substitution pattern of the nitrogen atom in the aminocarboxylate ligand, with the rate order observed NH2 > NHR > NR2 suggesting that a mechanism similar to that operating with the Noyori´s catalyst also applies to these complexes. The catalytic study was extended to other substrates (4´-methylacetophenone, 4´-chloroacetophenone, 1-indanone, and 3-methyl-2-cyclohexen-1-one) using the prolinate-derived trimer 8e as catalyst, which was able to generate the corresponding alcohols with up to 82% ee, albeit with disparate conversions (from 8 to 86% after 1–2 h) [61,62].
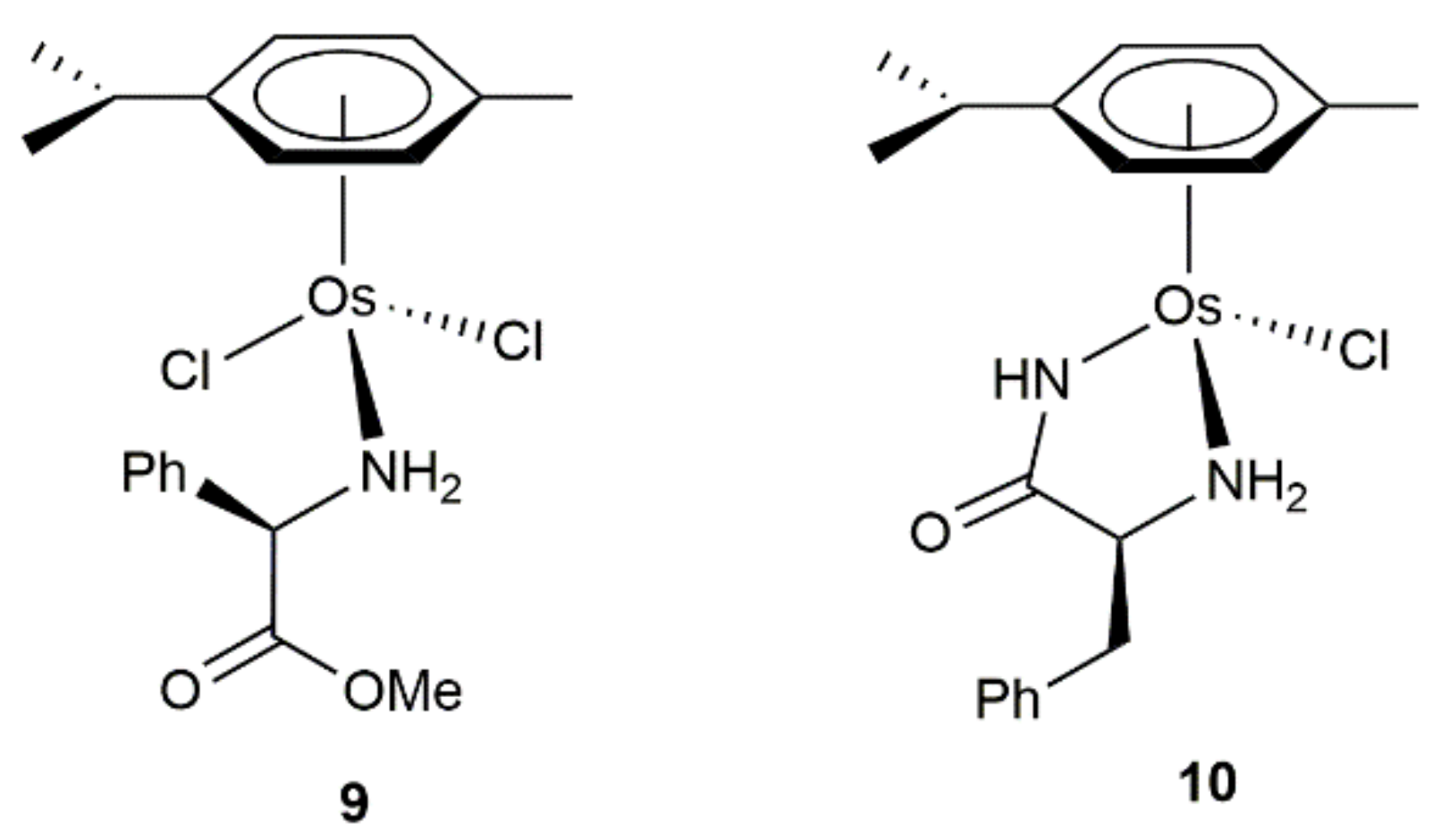
The base-assisted transfer hydrogenation of different saturated and α,β-unsaturated ketones by 2-propanol with complexes 9 and 10, containing as ligands enantiomerically pure (R)-phenylglycine methylester and (S)-phenylalanine amide, respectively, was additionally reported by Sarfraz and co-workers (see Figure 4) [63]. The conversions (from 11 to 100%) and selectivities (reduction of the C=C or C=O bond) observed were strongly dependent on the nature of the substrates and the temperature employed (r.t. or 70 °C), without indicating whether the reduction process occurs with any enantioselection.
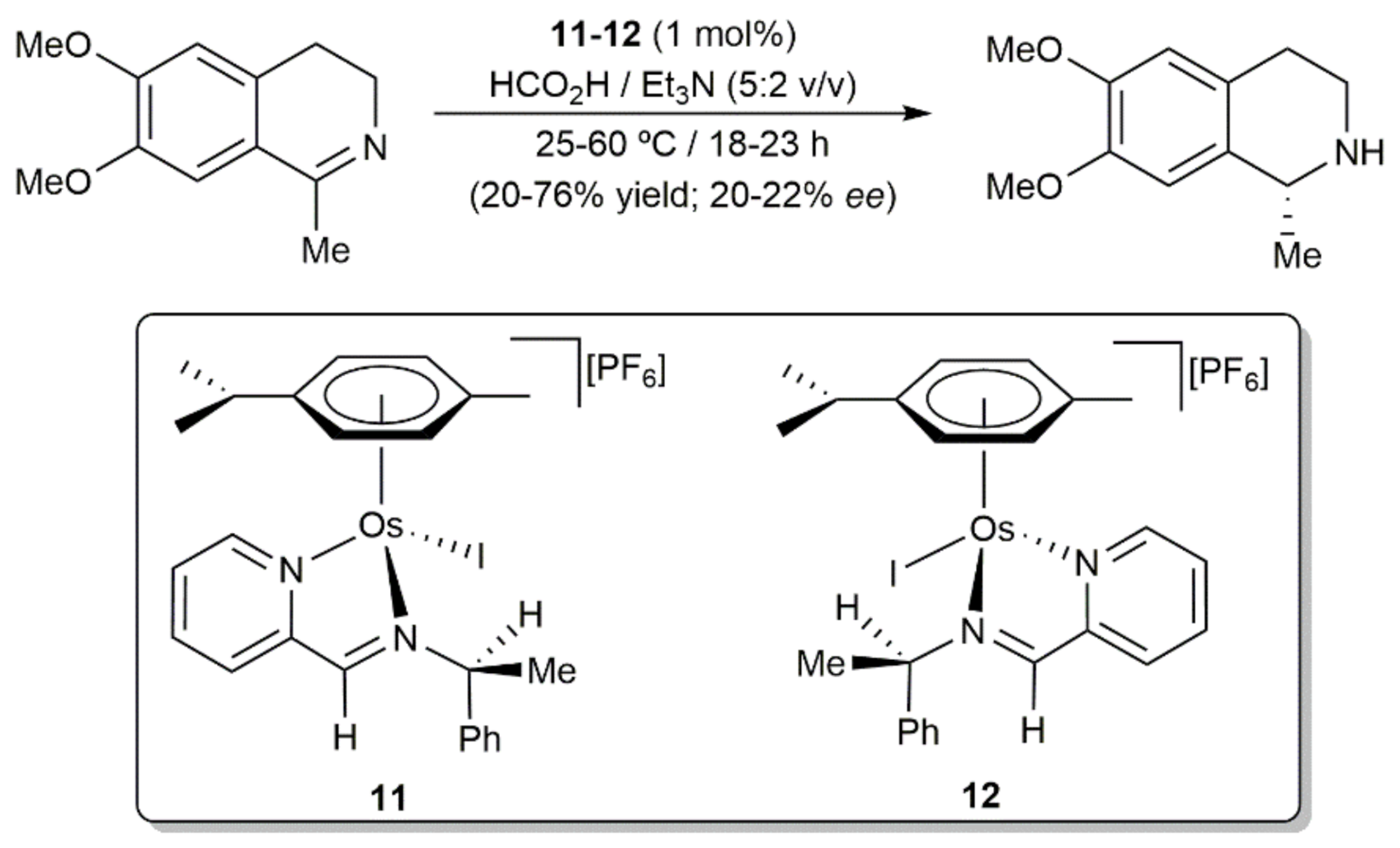
Within the field of asymmetric transformations, we must finally mention that Sadler and co-workers reported on the transfer hydrogenation of the cyclic imine 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline with the iodide osmium(II) complexes 11 and 12, employing formic acid as the hydrogen source (Scheme 5) [64]. Compounds 11 and 12 were generated by reacting methanolic solutions of dimer [{OsI(μ-I)(η6-p-cymene)}2] with N-(2-pyridylmethylene)-(S)-phenylethylamine and N-(2-pyridylmethylene)-(R)-phenylethylamine, respectively, in the presence of NH4PF6, and isolated in a diastereomerically pure manner by fractional crystallization. As shown in the scheme, their catalytic activities were only moderate and their chiral induction very low.
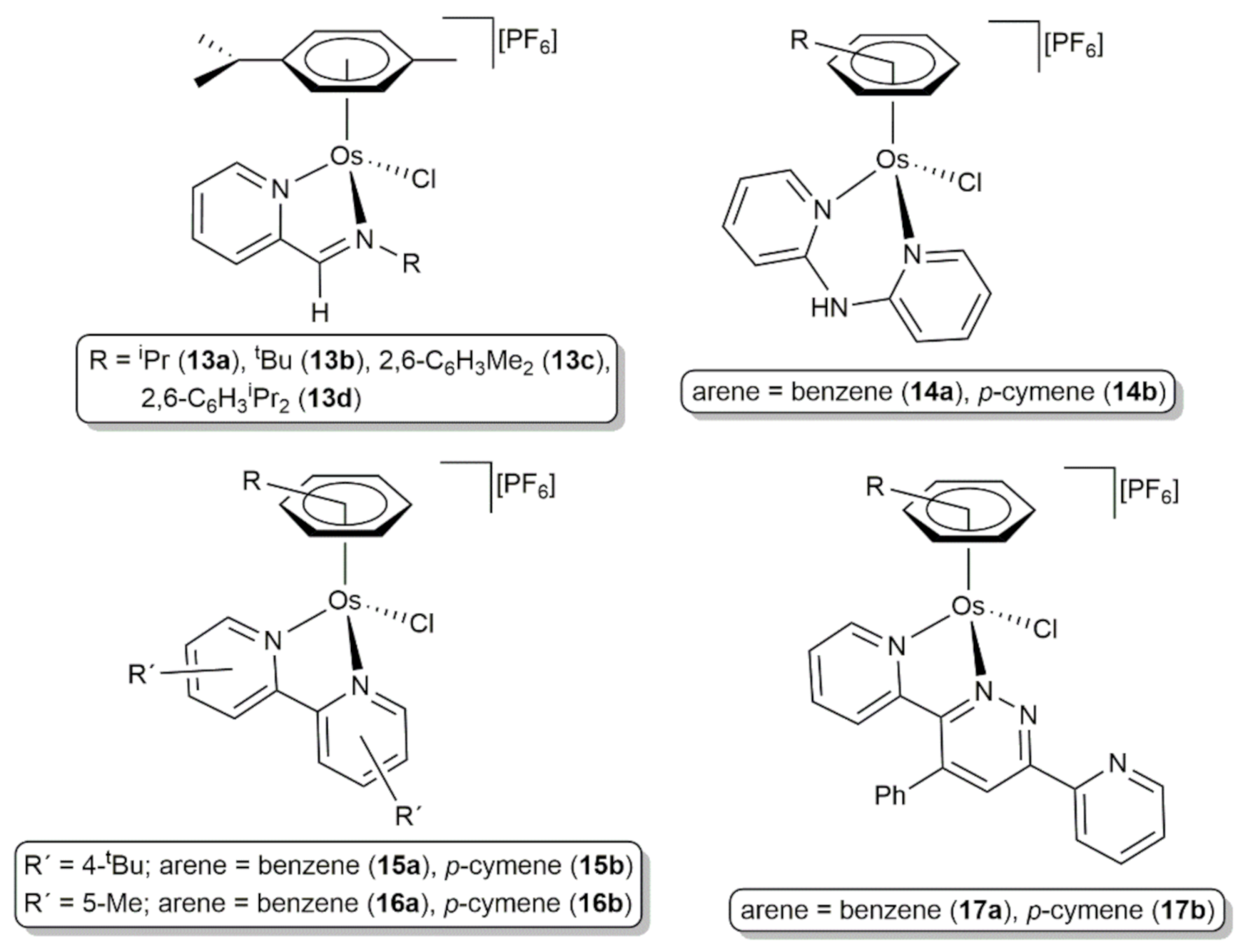
The related cationic Os(II) complexes 13a-d with chelated achiral imino-pyridine ligands, as well as compounds 14–17a-b featuring different bidentate N,N-donor ligands (see Figure 5), proved to be also capable to promote the transfer hydrogenation of ketones by 2-propanol when combined with NaOH [65]. In particular, performing the catalytic reactions with an osmium loading of 0.05 mol%, all of them were able to selectively convert cyclohexanone into cyclohexanol in ≥90% yield after 3–4 h of heating at 82 °C (turnover number (TON) and frequency (TOF) values up to 1932 and 640 h−1, respectively). Additional experiments with a range of different cyclic and acyclic ketones and the pyridazine-based complexes 17a,b confirmed the generality of the process and pointed to a comparable reactivity of these species to that of analogous ruthenium(II) systems.
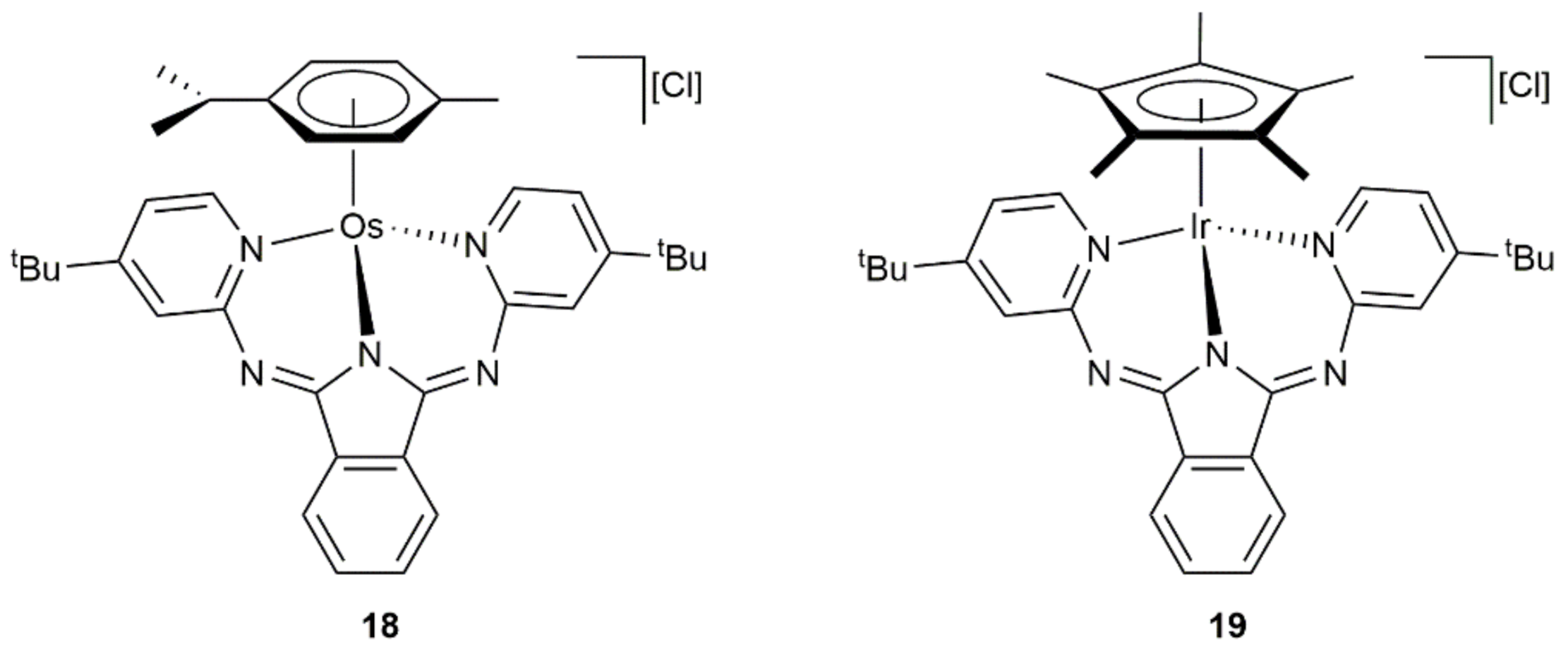
Transfer hydrogenation of acetophenone and cyclohexanone by 2-propanol was also successfully accomplished by Gade and co-workers with the cationic complex 18 containing a κ3-(N,N,N)-coordinated bis(pyridylimino)isoindolate ligand (Figure 6) [66]. Thus, performing the reactions at 80 °C with 5 mol% of 18, in conjunction with KOH, the corresponding alcohols were generated in quantitative yield after 2 h, results comparable to those achieved with the related Ir(III) derivative 19.
On the other hand, application of the arene-osmium(II) complexes 20–21b, containing a sterically hindered β-diketiminate ligand, as catalysts in olefin hydrogenation reactions was described by Phillips and co-workers in 2013 [67]. As exemplified with the selective hydrogenation of limonene into 2-p-menthene depicted in Scheme 6, both showed an effectiveness superior to that of their ruthenium analogues 20–21a, with the saturated 18-electron chloride complex 20b featuring the highest activity (similar observations were made when styrene and cyclohexene were employed as substrates). In the same work, the cationic 16-electron derivatives 21a-b were also screened in the catalytic dehydrocoupling of N,N-dimethylamine borane (DMAB). Performing the reactions in THF at 42 °C with a catalyst loading of 0.5 mol%, quantitative dehydrogenation of DMAB was observed with both complexes. However, for this particular transformation, the ruthenium derivative 21a turned out to be much more active, completing the reaction in only 0.5 h (versus the 30 h required by 21b).
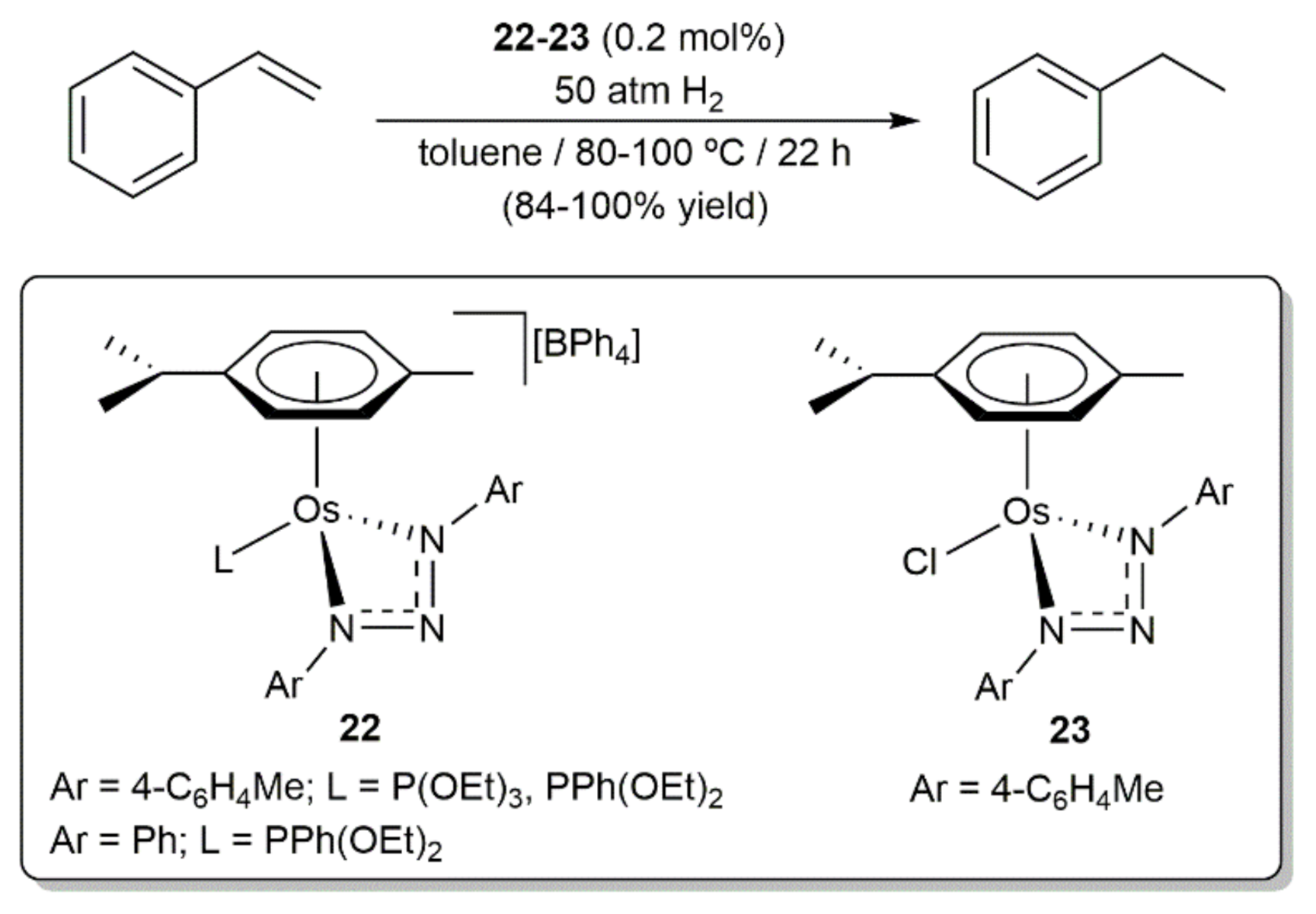
Hydrogenation of styrene was additionally described by Albertin and co-workers employing complexes 22 and 23, containing chelating 1,3-bis(aryl)triazenide ligands, as catalysts (Scheme 7) [68]. Performing the reactions with an osmium loading of 0.2 mol%, in toluene at 80–100 °C and under 50 atm of H2, all of them were able to generate ethylbenzene in high yield (≥84%), regardless of their cationic or neutral nature.
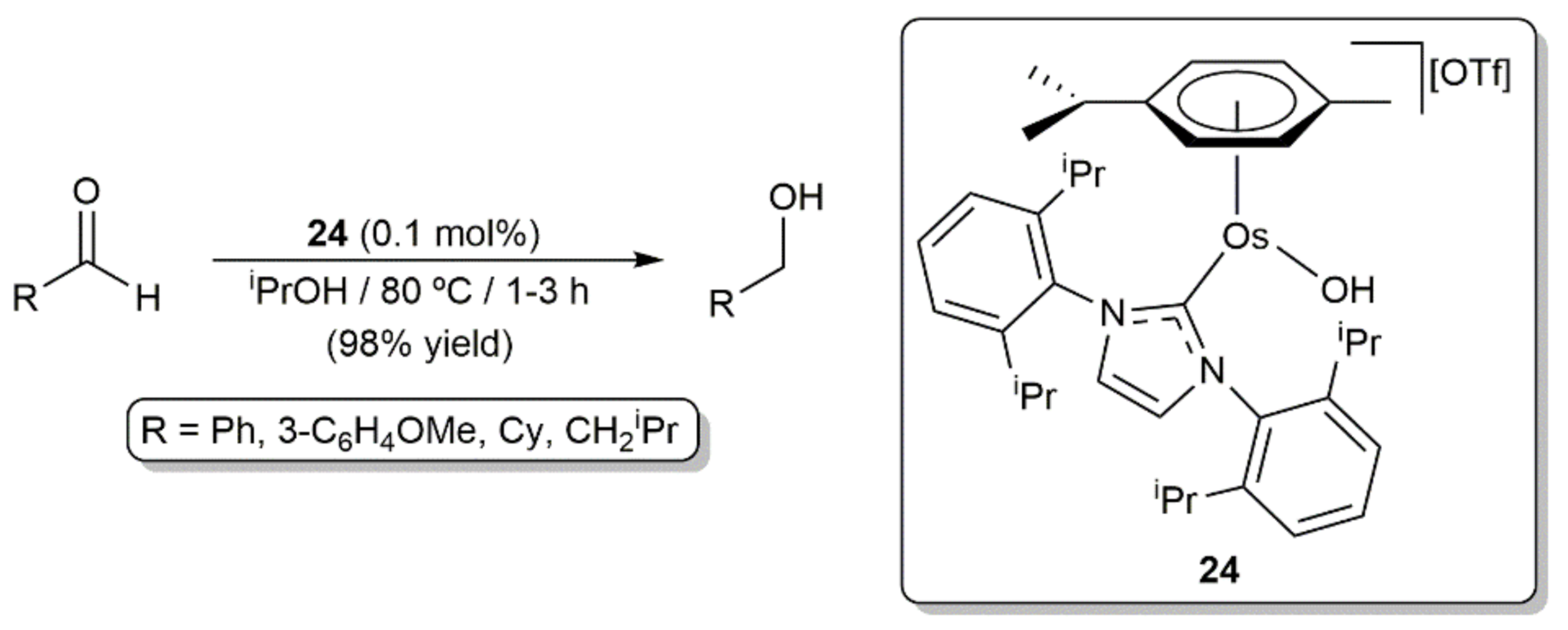
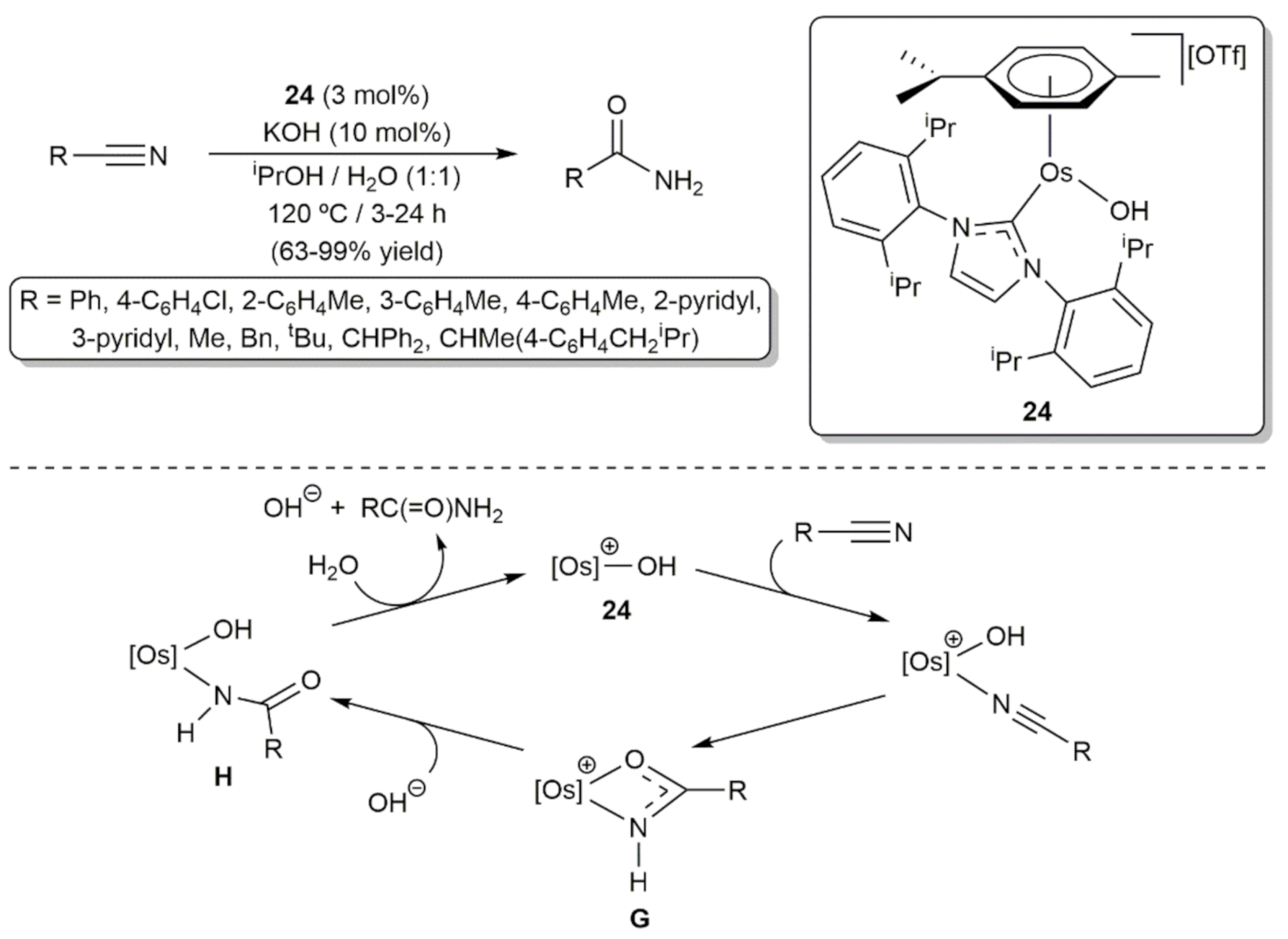
On the other hand, the involvement of arene-osmium(II) complexes with N-heterocyclic carbene (NHC) ligands as catalysts in hydrogenation and transfer hydrogenation processes has also been documented. In this context, the first example was described by Castarlenas, Esteruelas, and co-workers in 2008, who demonstrated the ability of the 16-electron hydroxo complex [Os(OH)(η6-p-cymene)(IPr)][OTf] (24; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene) to promote the transfer hydrogenation of aldehydes by 2-propanol under base-free conditions [69]. As shown in Scheme 8, the primary alcohol products were obtained in excellent yield (98% in all the cases) and short times by performing the catalytic reactions at 80 °C with 0.1 mol% of 24 (TOF up to 980 h−1). Complex 24 was also active in the reduction of acetophenone, but a longer reaction time (22 h) and a higher catalyst loading (1 mol%) was in this case needed to obtain the desired 1-phenylethanol in only 72% yield (TOF = 3.3 h−1). The behavior of 24 towards 2-propanol was explored by the authors, observing the clean and immediate formation at −30 °C of the hydride derivative [OsH{κ1-(O)-O=CMe2}(η6-p-cymene)(IPr)][OTf], which probably acts as the catalytically active species.
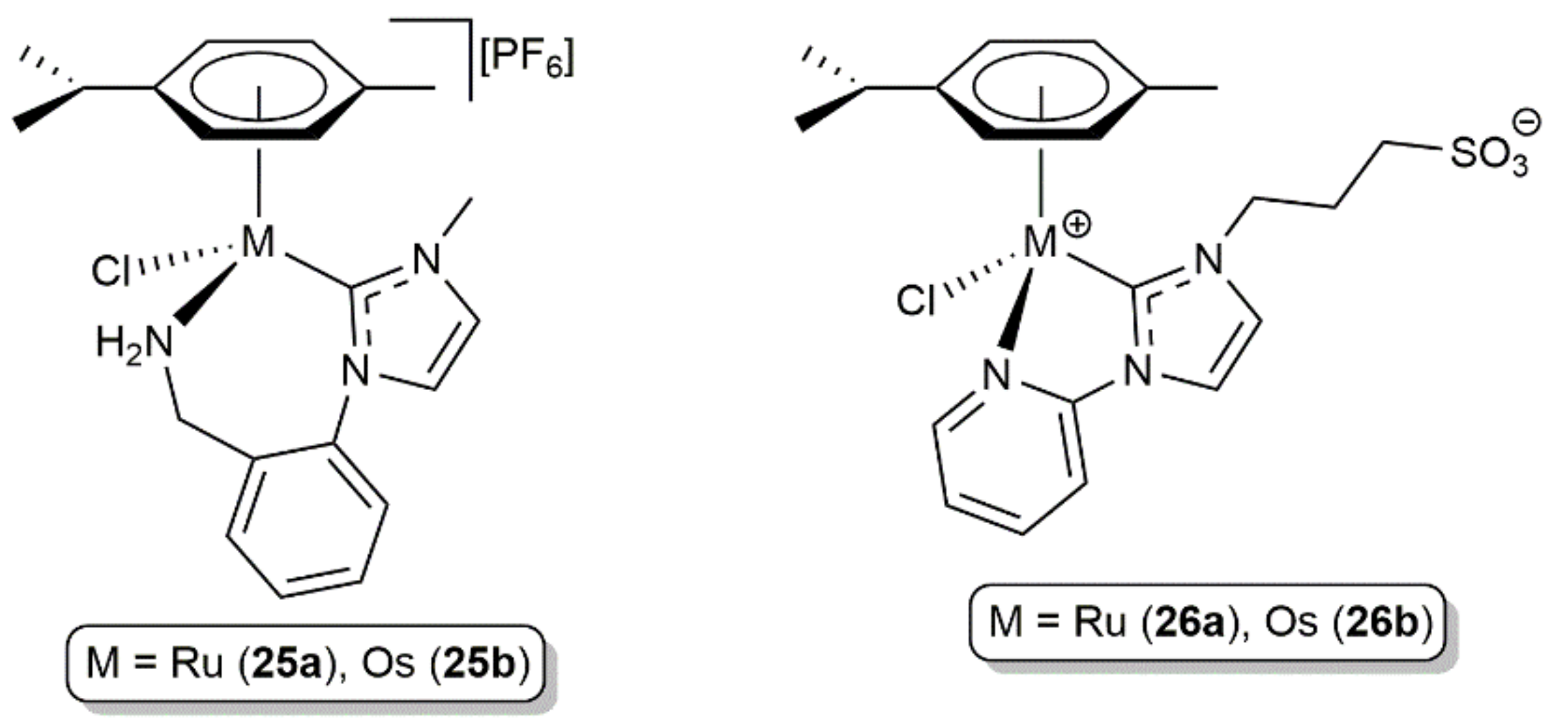
Some years later, Morris and co-workers synthesized the osmium complex 25b, featuring a chelating NHC ligand with a tethered primary amine donor (Figure 7), and compared its catalytic behavior in the hydrogenation of acetophenone with that of the analogue ruthenium(II) derivative 25a [70]. Both complexes required the presence of base (KOtBu) to operate, with the activity of 25b being remarkably lower than that of 25a (23% conversion after 3 h at 50 °C in iPrOH using 25 bar of H2 and a catalyst/substrate/base ratio of 1/8/200 vs quantitative conversion after 1.5 h under identical conditions). The higher oxophilic nature of osmium, capable to form more stable intermediate alkoxo complexes, was invoked by the authors to explain the difference in activity found. Similar observations were made by Kühn and co-workers when studying the hydrogenation and transfer hydrogenation of acetophenone in aqueous medium with the hydrophilic complexes 26a-b (see Figure 7) [71].
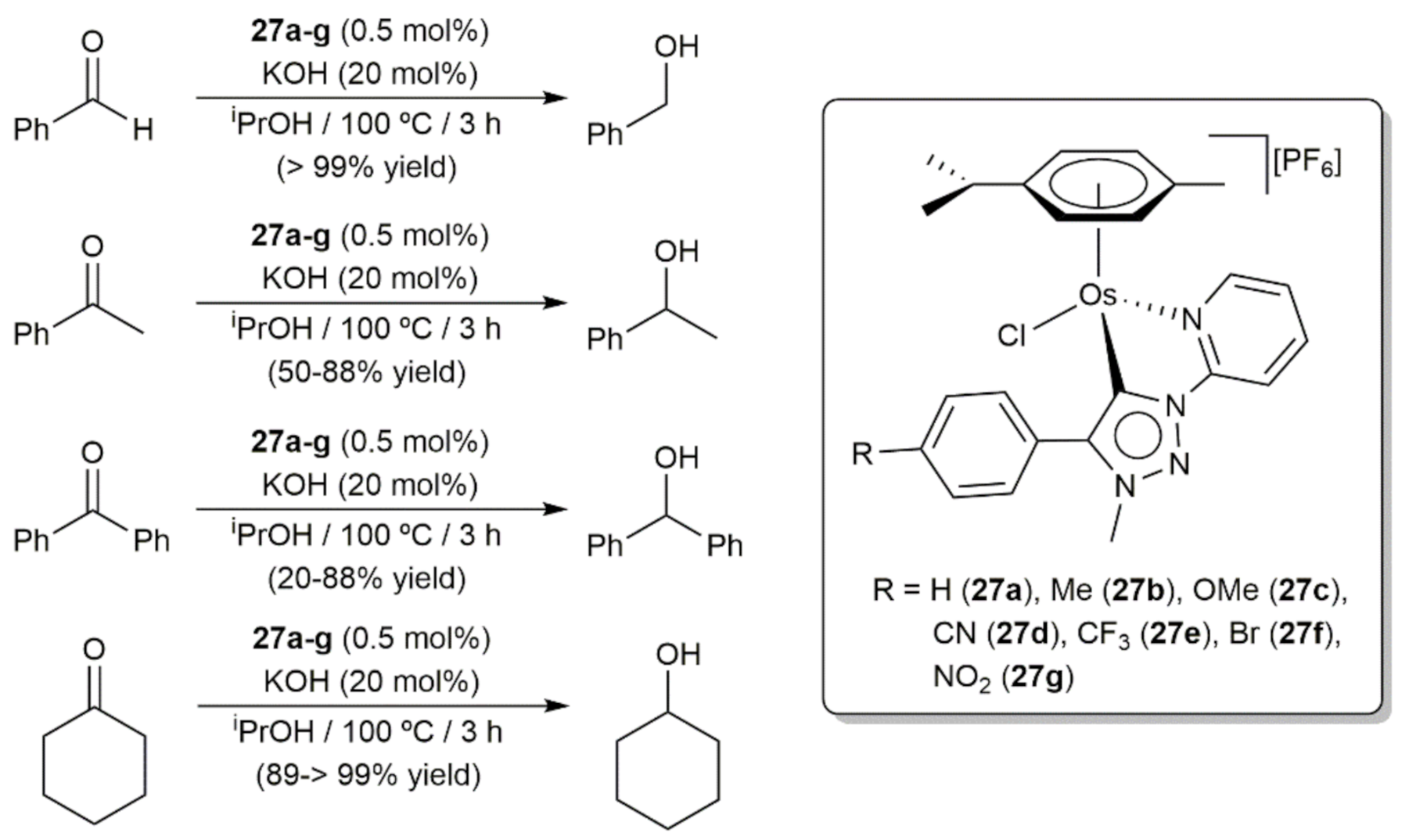
Transfer hydrogenation reactions from 2-propanol to carbonyl compounds were described by Košmrlj, Sarkar, and co-workers employing the cymene-osmium(II) complexes 27a–g, featuring chelating pyridyl-mesoionic carbene ligands, in conjunction with KOH (Scheme 9) [72]. As a general trend, those complexes bearing electron donating substituents on the ligand backbone, i.e., 27b and 27c, showed superior performances in comparison to the non-substituted one 27a and those substituted with electron-withdrawing groups, i.e., 27d–g. The same catalytic reactions were simultaneous studied, under identical conditions, with analogous cymene-ruthenium(II) complexes, observing for the reduction of benzaldehyde almost identical results. However, in the case of acetophenone, benzophenone, and cyclohexanone, the ruthenium complexes led in general to higher yields.
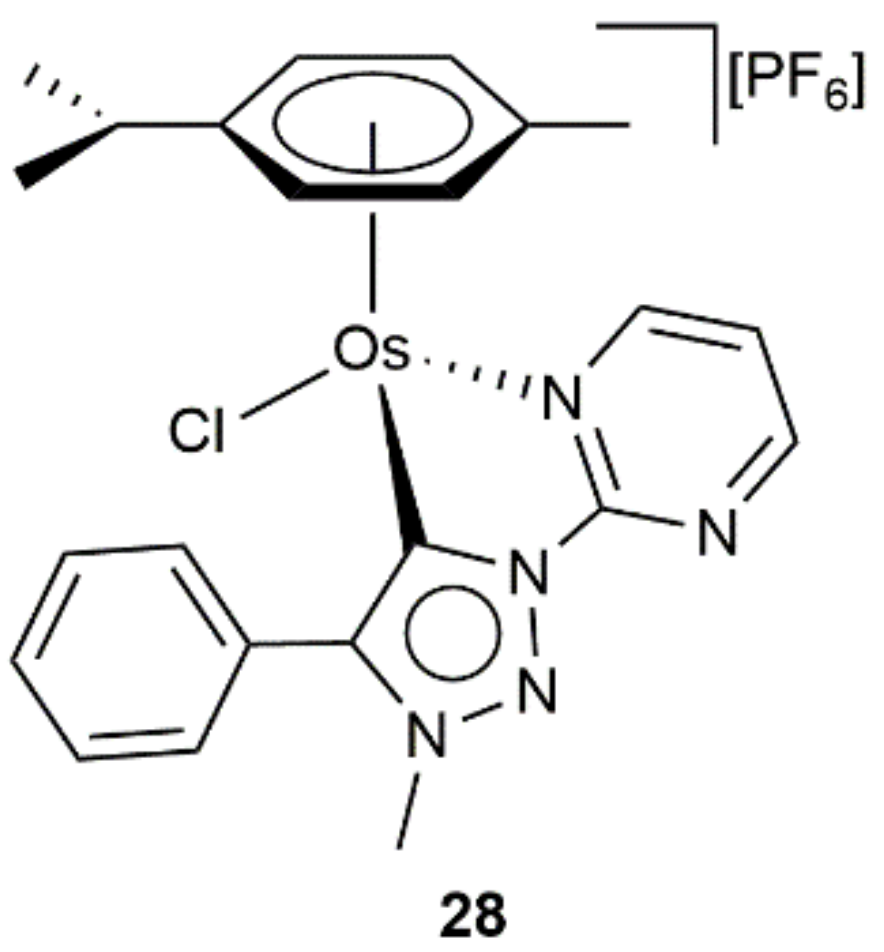
In a subsequent study, the same group described the synthesis and catalytic behavior of the related pyrimidine-based mesoionic carbene Os(II) complex 28 (see Figure 8), which showed an effectiveness comparable to that of 27b–c in the transfer hydrogenation of benzaldehyde, acetophenone, benzophenone, and cyclohexanone [73]. In addition, in the same study, the utility of compounds 27 and 28 to promote the transfer hydrogenation of olefins, imines and nitroaromatics was also briefly explored, the results obtained being strongly dependent on the nature of the substrates employed and inferior in all cases to those reached with the corresponding ruthenium(II) analogues.
3. Oxidation Reactions
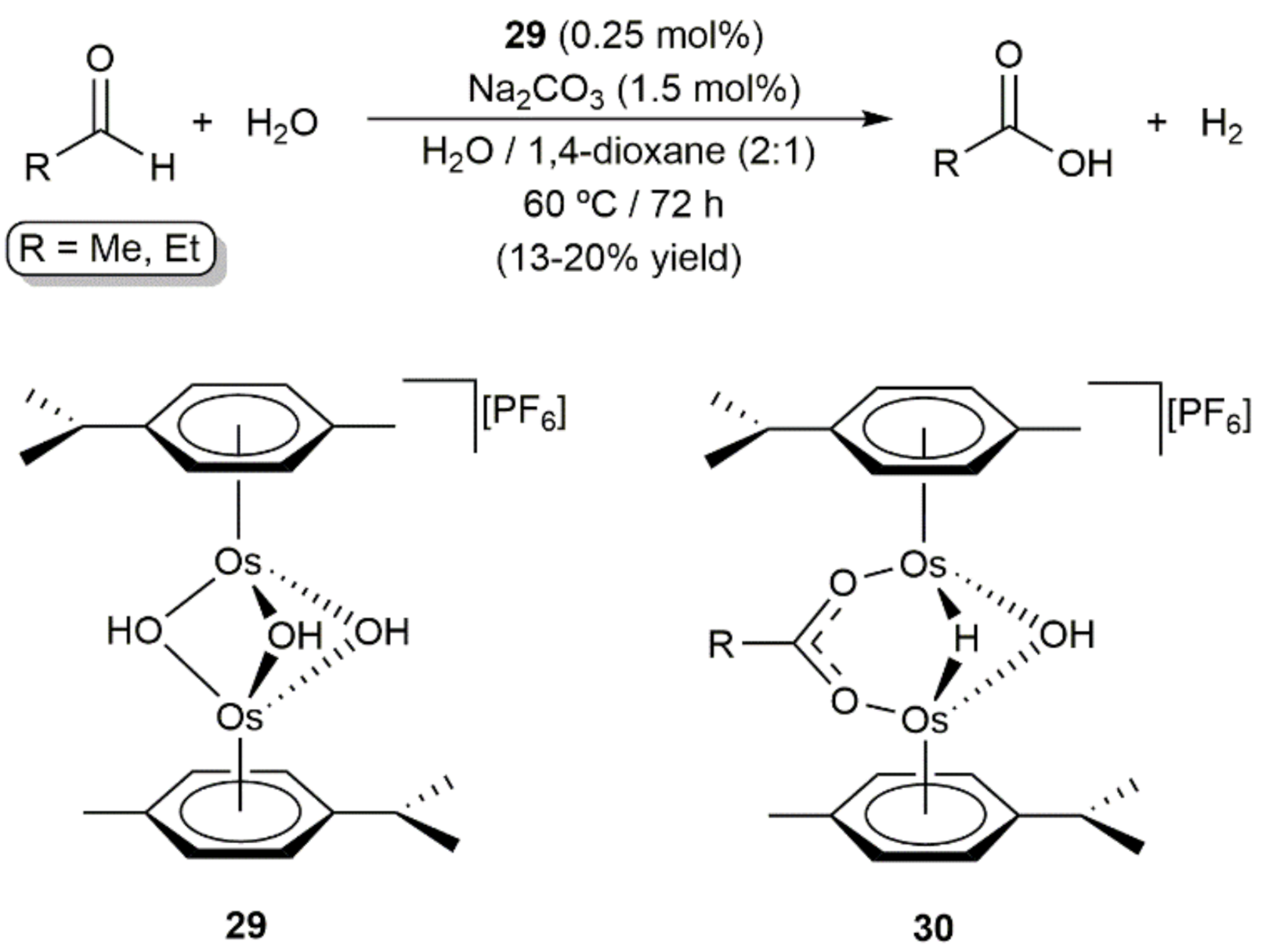
Despite the fact that osmium compounds are widely employed to promote oxidation processes [74,75,76,77,78], examples involving low-valent arene-osmium(II) complexes are still very scarce. In this context, in an early report, Maitlis and co-workers showed that the cationic dinuclear tri-μ-hydroxo derivative [{Os(η6-p-cymene)}2(μ-OH)3][PF6] (29) is able to catalyze the oxidation of acetaldehyde and propionaldehyde by water to generate the corresponding carboxylic acids, reactions in which molecular hydrogen is also produced (Scheme 10) [79]. Although the yields were very poor and comparatively lower than those obtained with its ruthenium counterpart, i.e., [{Ru(η6-p-cymene)}2(μ-OH)3][PF6], complex 29 showed a greater selectivity towards the formation of the carboxylic acids since ruthenium also catalyzes the competing disproportionation of the aldehydes into the respective alcohols and carboxylic acids. In the same work, the reactivity of [{Os(η6-p-cymene)}2(μ-OH)3][PF6] (29) towards acetaldehyde and propionaldehyde under stoichiometric conditions was also explored, the reactions performed in water allowing the isolation of compounds [{Os(η6-p-cymene)}2(μ-H)(μ-OH)(μ-O2CR)][PF6] (30) which probably participate as intermediates in the catalytic reactions.
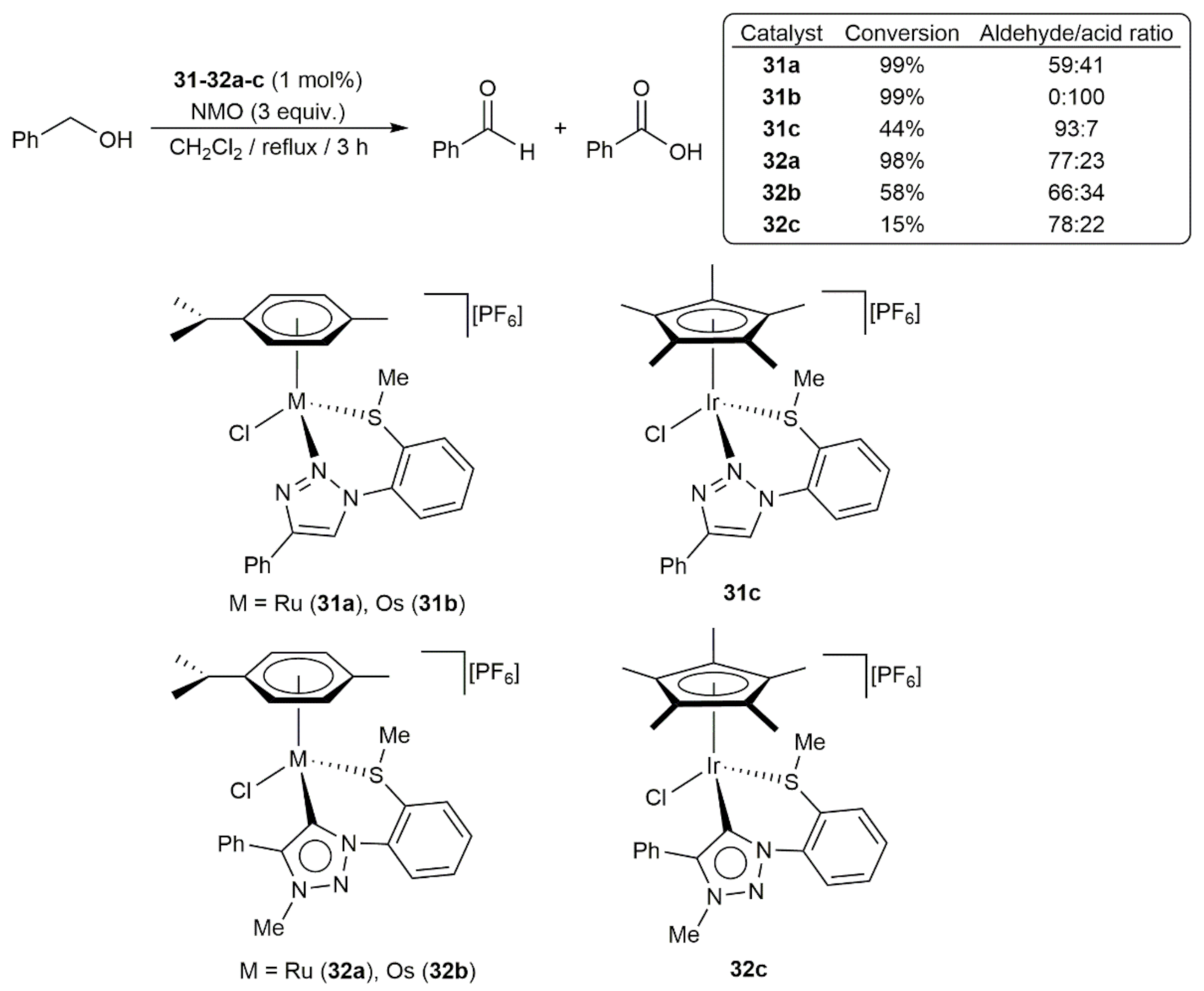
In a more recent study, Sarkar and co-workers explored the catalytic behavior of a series of mononuclear half-sandwich Ru(II), Os(II), and Ir(III) complexes containing mixed thioether/1,2,3-triazole and 1,2,3-triazol-5-ylidene ligands, i.e., compounds 31a-c and 32a-c, respectively, in the oxidation of benzyl alcohol using an excess of N-methylmorpholine N-oxide (NMO) as sacrificial oxidant (Scheme 11) [80]. As a general trend the 1,2,3-triazole derivatives 31a-c were more active than their 1,2,3-triazol-5-ylidene counterparts 32a-c, with the osmium complex 31b showing a remarkable selectivity towards the overoxidation to benzoic acid. Employing the same experimental conditions, oxidation of diphenylmethanol into benzophenone was also achieved in high yields (≥ 94%) and short time (3 h) with low metal loadings (0.01–0.2 mol%) of 31b.
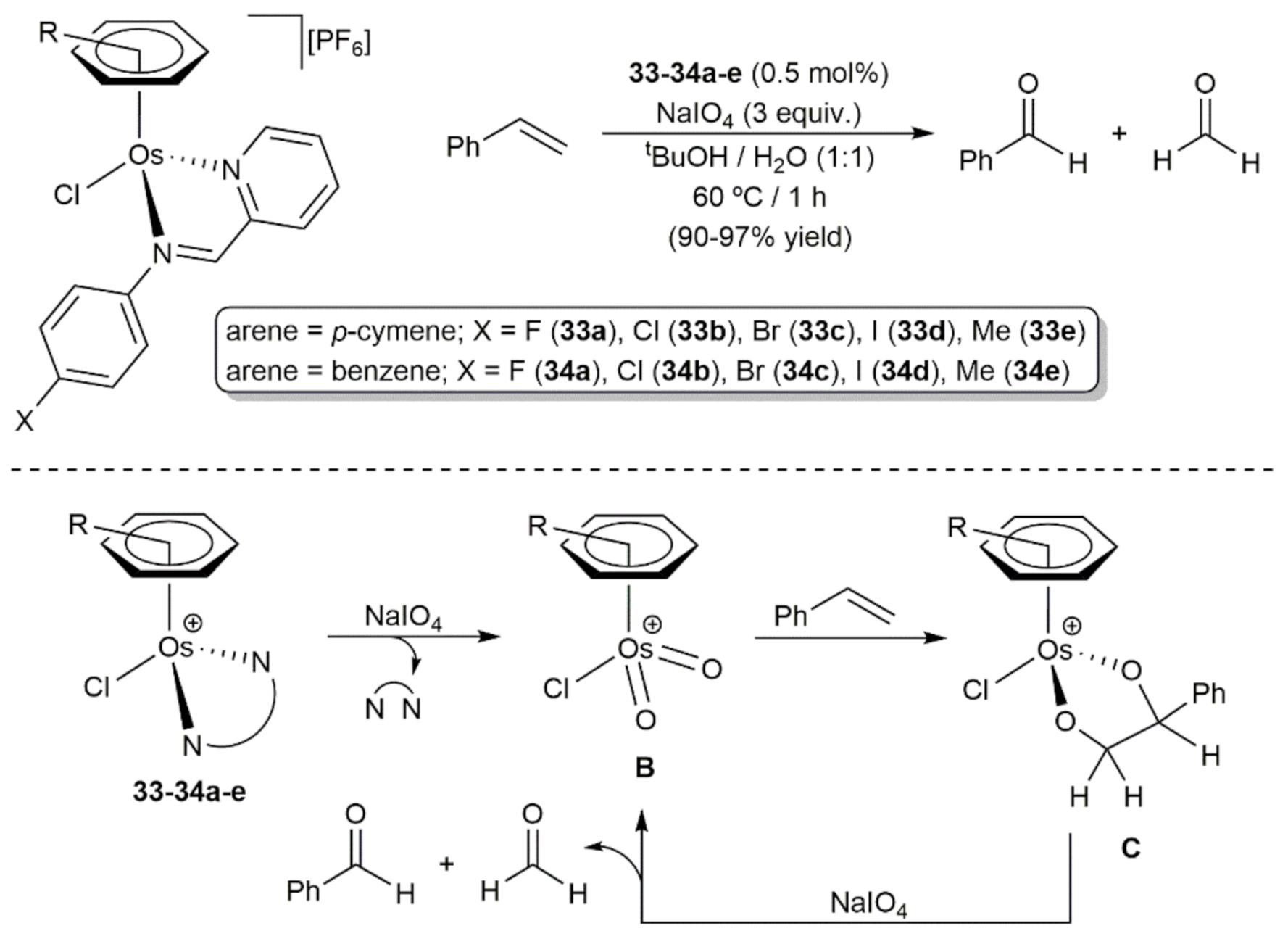
The group of Friedrich synthesized a series of cationic arene-osmium(II) complexes 33–34a-e with bidentate imino-pyridine ligands which catalyzed efficiently the selective oxidation of styrene to benzaldehyde with NaIO4 (Scheme 12) [81]. From a mechanistic point of view, spectroscopic evidence indicated that the reactions proceed through the initial oxidation of complexes 33–34a-e by NaIO4 to generate the dioxo arene-osmium(VI) species B with release of the N,N-donor ligands. Subsequent [3 + 2] cycloaddition between B and the C = C bond of the styrene molecule leads to the Os(IV) intermediates C from which benzaldehyde, as well as the secondary product formaldehyde, are liberated after reaction with NaIO4. On the other hand, the generality of this oxidation process was conveniently confirmed with complex 33d, which was able to convert 4-methoxystyrene, 4-methylstyrene, 4-clorostyrene, trans-stilbene, and 1-octene into the corresponding aldehydes in equally good yields (85–99%) and short times (0.5–1 h).
On the other hand, the mononuclear complexes [OsCl2(η6-p-cymene)(py)] (py = pyridine), [OsCl(η6-p-cymene)(bipy)][PF6] (bipy = 2,2´-bipyridine) and [Os(η6-p-cymene)(NCMe)(bipy)][PF6]2, and the dinuclear derivatives [{OsCl(μ-Cl)(η6-p-cymene)}2] and [{Os(η6-p-cymene)}2(μ-H)3][PF6], proved to be active catalysts for the C-H bond oxidation of alkanes (cyclohexane, methylcyclohexane, cis- and trans-1,2-dimethylcyclohexane, isooctane, and n-heptane) with hydrogen peroxide in air [82,83]. The reactions, performed in acetonitrile at 60 °C, led to the corresponding alkyl hydroperoxides ROOH through the intermediation of hydroxyl radicals, which were rapidly converted into alcohols ROH by PPh3. Among them, dimer [{OsCl(μ-Cl)(η6-p-cymene)}2] was the most active, allowing to reach an impressive turnover number (TON) of 200200 in the oxidation of cyclohexane (maximum conversion of 24% after 24 h using a catalyst loading of 10−5 mol%). Some of these compounds were also active in the oxidation of benzene and 1-phenylethanol to give phenol and acetophenone, respectively, under identical experimental conditions (yields up to 88%) [83].
4. C-C Bond Forming Reactions
Involvement of arene-osmium(II) complexes in catalytic C-C bond forming processes was described for the first time by Demonceau and co-workers in 1996 [84]. As shown in Scheme 13, they found that dimer [{OsCl(μ-Cl)(η6-p-cymene)}2] was able to promote efficiently the cyclopropanation of styrene derivatives with ethyl diazoacetate (EDA) at 80 °C with stereoselectivities (cis/trans ratios) comparable to those previously reported for more classical rhodium- and ruthenium-based systems. They also explored with little success the behavior of [{OsCl(μ-Cl)(η6-p-cymene)}2] towards non-activated linear and cyclic olefins. For example, cyclopropanation of cyclooctene by EDA under identical experimental conditions led to the desired cyclopropane product in only 25% yield along with minor amounts (ca. 3%) of the corresponding polymer resulting from the ring-opening metathesis polymerization (ROMP) of the substrate [84]. This result contrast with that obtained with [{RuCl(μ-Cl)(η6-p-cymene)}2] which showed to catalyze preferentially the ROMP process.
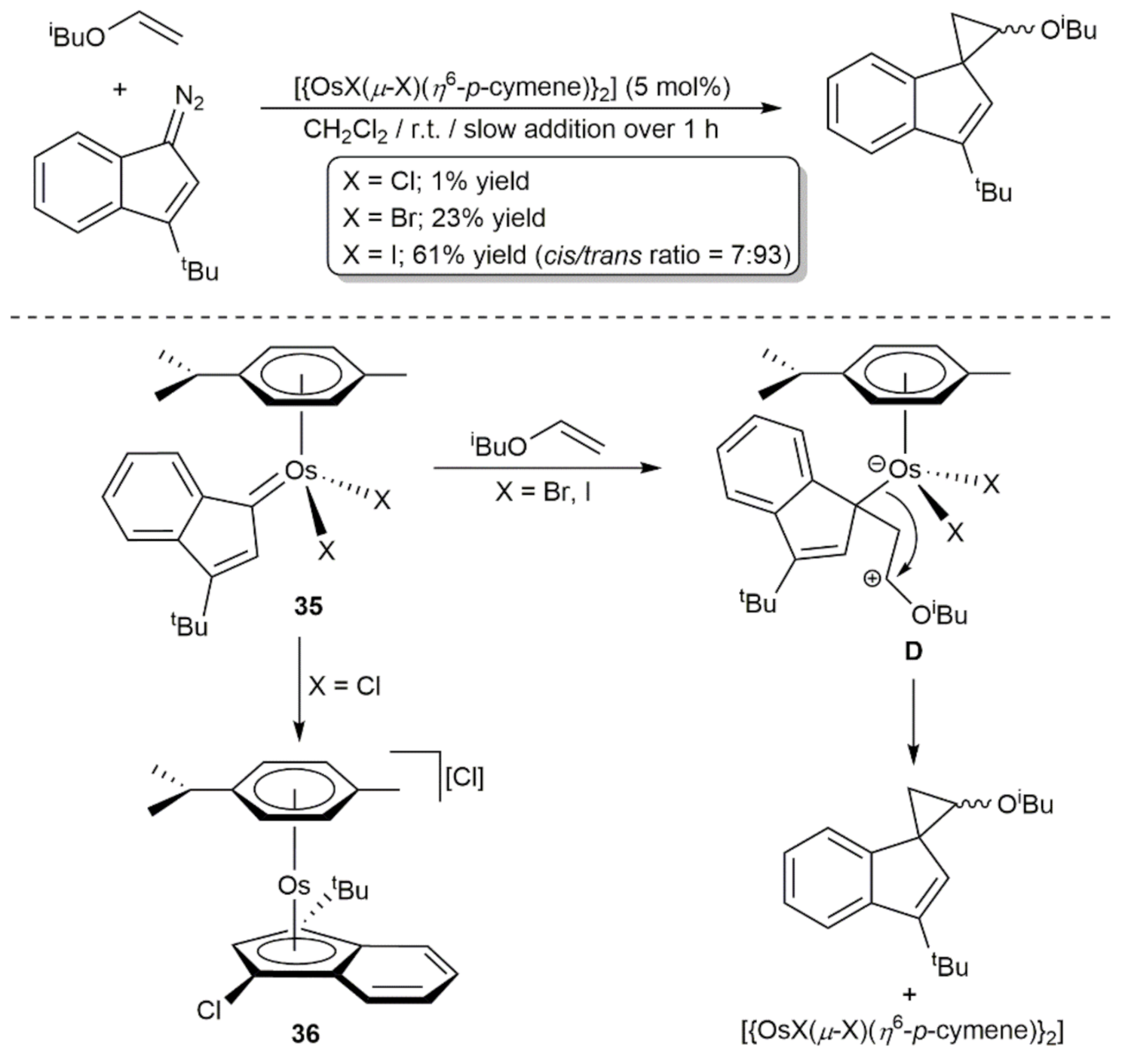
More recently, the cyclopropanation of isobutyl vinyl ether with 3-tert-butyldiazoindene was studied using dimers [{OsX(μ-X)(η6-p-cymene)}2] (X = Cl, Br, I) as catalysts (Scheme 14) [85]. A marked effect of the halides on the effectiveness of the process was observed and positive results were only obtained with [{OsBr(μ-Br)(η6-p-cymene)}2] and [{OsI(μ-I)(η6-p-cymene)}2]. DFT calculations combined with stoichiometric reactions confirmed the intermediacy of the osmium-indenylidene complexes 35, which could be isolated and fully characterized by reacting [{OsX(μ-X)(η6-p-cymene)}2] (X = Br, I) with the diazoindene. Subsequent attack of the olefin to the osmium-carbene bond gives the cyclopropane product via the zwitterionic species D. The almost null catalytic activity observed for [{OsCl(μ-Cl)(η6-p-cymene)}2] was related to the greater tendency of the corresponding indenylidene intermediate 35 to rearrange into the catalytically inactive η5-chloroindenyl complex 36, which could also be isolated and structurally characterized. Of note is the fact that [{RuI(μ-I)(η6-p-cymene)}2], [CpRuCl(PPh3)2] (Cp = cyclopentadienyl), [{Ir(μ-Cl)(cod)}2] (cod = 1,5-cyclooctadiene), Cu(OAc)2 and [Rh2(μ-OAc)4] were completely inoperative in this particular cyclopropanation reaction.
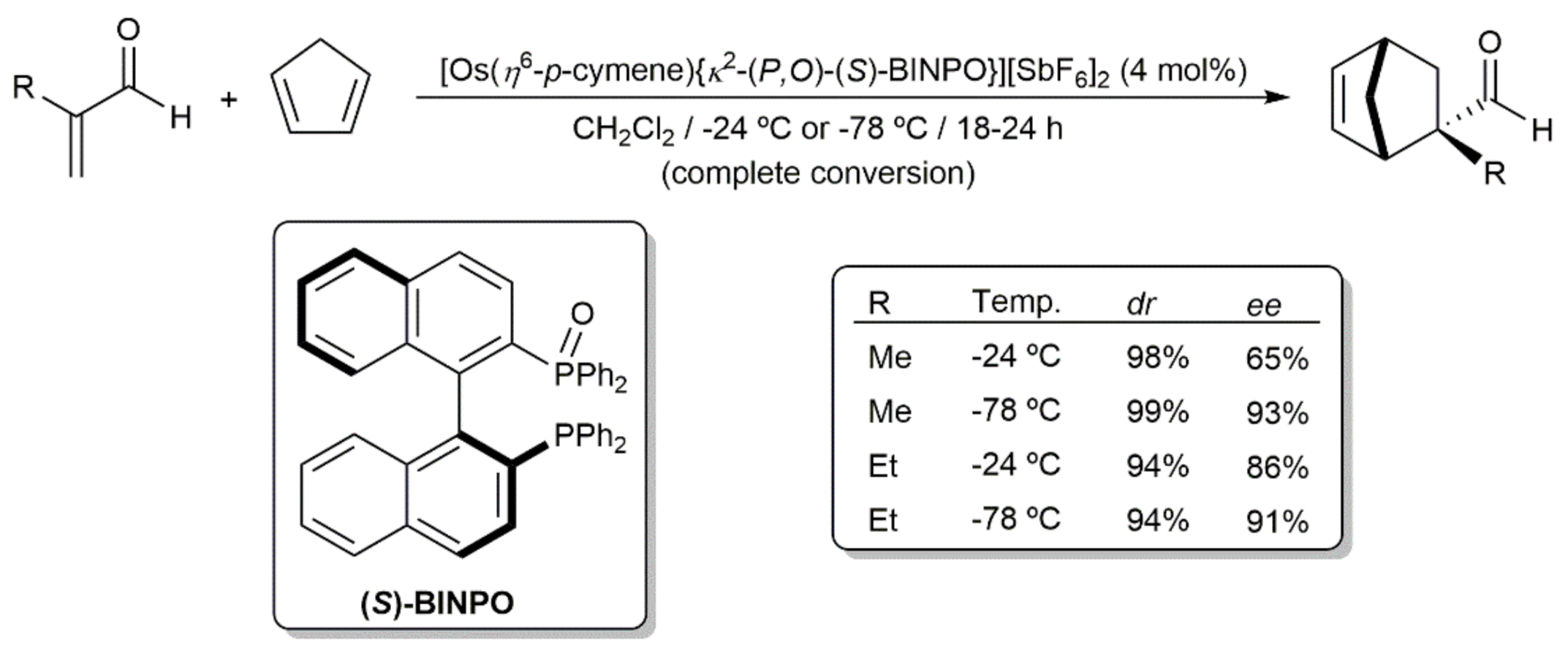
In the context of their studies on the coordination of bidentate bisphosphine monoxide ligands to half-sandwich ruthenium(II) and osmium(II) fragments [86,87,88], Faller and Parr synthesized the dicationic 16-electron derivative [Os(η6-p-cymene){κ2-(P,O)-(S)-BINPO}][SbF6]2 ((S)-BINPO = monoxide of the chiral diphosphine (S)-BINAP) which proved to be an excellent Lewis acid catalyst for the asymmetric Diels–Alder reaction of cyclopentadiene with methacrolein and ethylacrolein [87]. As shown in Scheme 15, the exo adducts were almost exclusively formed (dr ≥ 94%) with high enantioselectivity levels. Remarkably, the ee values observed (65–93%) surpassed those obtained with [Ru(η6-p-cymene){κ2-(P,O)-(S)-BINPO}][SbF6]2 (ee up to 50%). In addition, when using the ruthenium catalyst, the addition of a base (lutidine) was required, the ee values dropping to 5–27% in its absence [88].
The catalytic behavior of a series of Os(II) complexes of general composition [Os(η6-p-cymene)(PN)(H2O)][X]2 (PN = chelated chiral phosphinooxazoline ligand; X- = SbF6-, BF4- or TfO-) in the Diels–Alder reaction of cyclopentadiene with methacrolein was additionally explored by Carmona, Lamata, and co-workers [89]. They led to the expected cycloadduct product in high yields (73–93%) and with a good exo:endo selectivity (dr = 90–94%), but with a low enantioselectivity (ee values up to 39%).
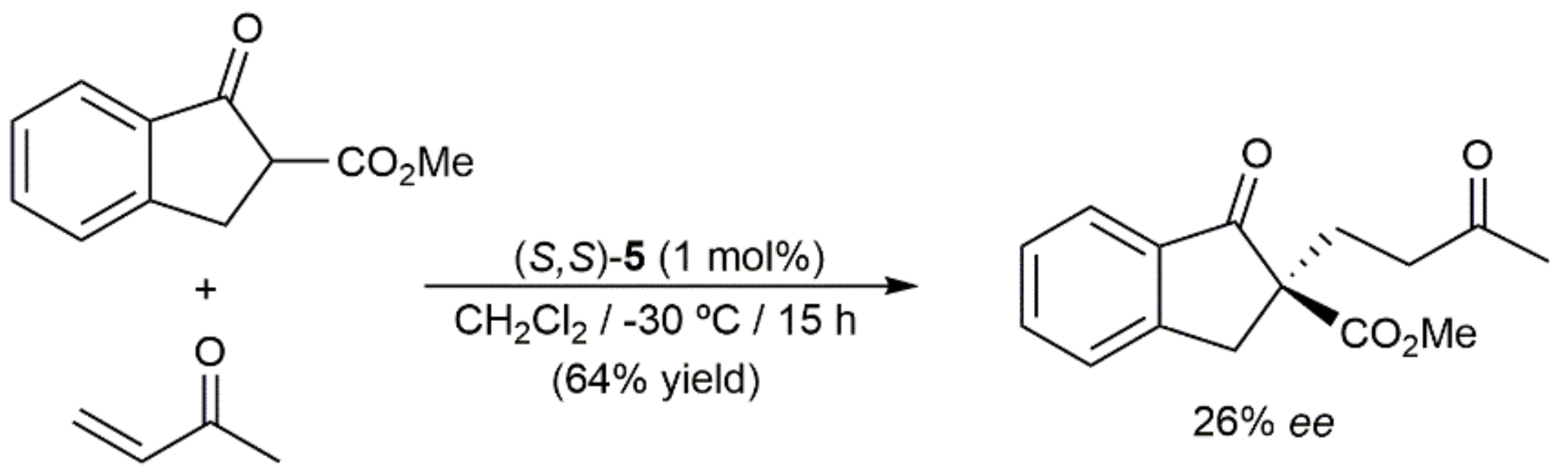
Within the field of asymmetric synthesis, Suzuki and Torii also described the use of the chiral 16-electron complex (S,S)-5 (see Scheme 2) in the catalytic Michael addition of 2-methoxycarbonyl-1-indanone to 3-buten-2-one (Scheme 16) [90]. The desired Michael adduct was obtained in 64% yield and 26% ee, values comparable to those reached with the analogous ruthenium(II) complex (69% yield; 39% ee) but clearly inferior to those achieved when related Cp*Rh(III) systems were employed as catalysts (up to 99% yield and 75% ee).
The participation of arene-osmium(II) complexes in olefin metathesis reactions can also be found in the literature. For example, Hafner and co-workers reported that, under ultraviolet irradiation, compounds [OsCl2(η6-p-cymene)(PR3)] (R = Cy, iPr) are able to promote the rapid ROMP of norbornene without the need of a diazomethane activator [91]. The presence of a sterically demanding phosphine ligand seems to play a key role in the photoactivation of these compounds since very slow or no polymerization was observed with the related species [OsCl2(η6-p-cymene)(PR3)] (R = Me, nBu, Ph). However, it should be noted that the reactivity of [OsCl2(η6-p-cymene)(PR3)] (R = Cy, iPr) in this polymerization process was much lower to that of complexes [RuCl2(η6-p-cymene)(PR3)] (R = Cy, iPr).
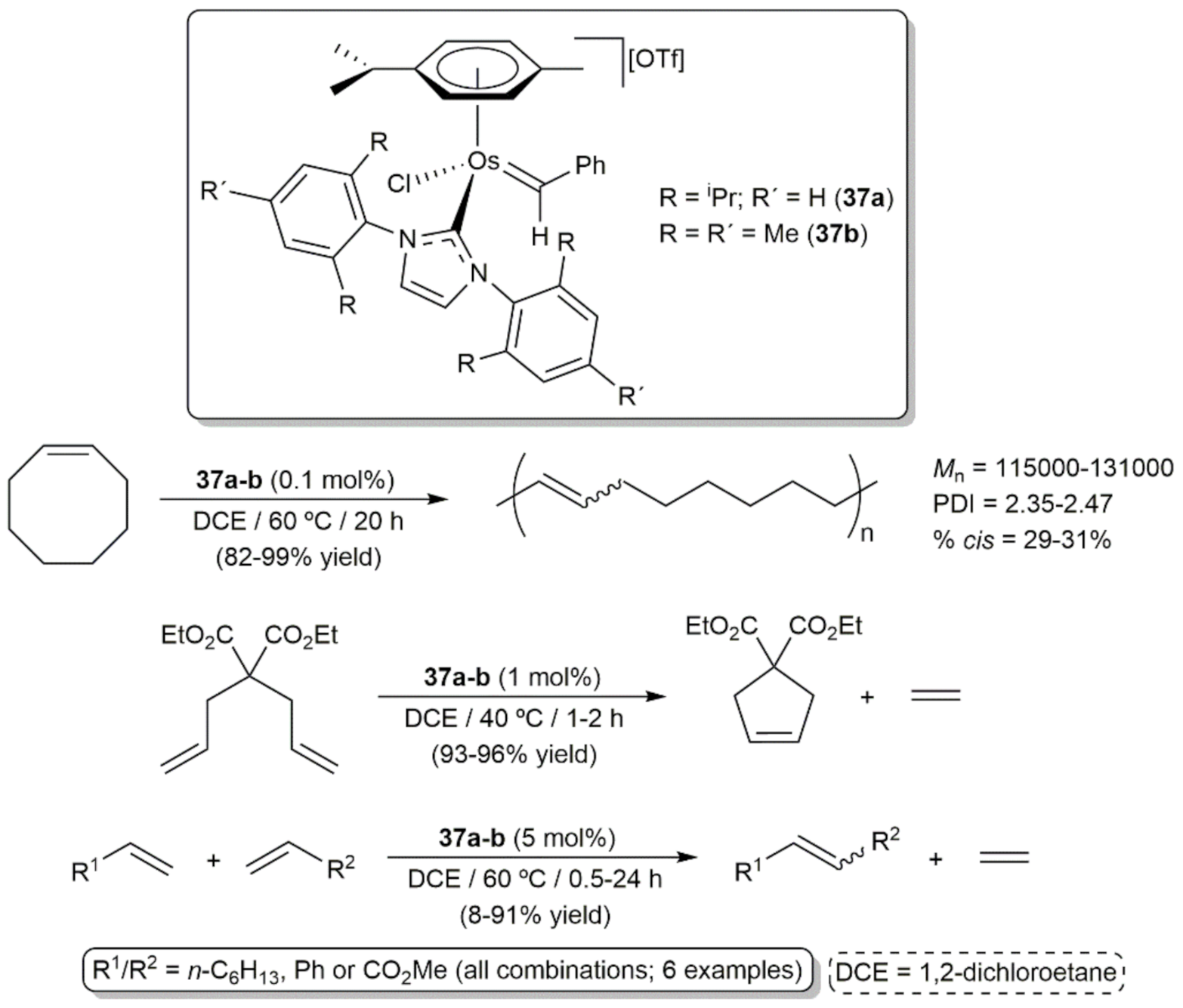
In a more general study, Esteruelas and co-workers demonstrated the utility of the benzylidene-osmium(II) complexes 37a-b, synthesized by reaction of the corresponding 16-electron precursors [OsCl(η6-p-cymene)(NHC)][OTf] (NHC = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene (IPr) or 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene (IMes)) with phenyldiazomethane, as catalysts for the ROMP of cyclooctene, the ring-closing metathesis (RCM) of diethyl diallylmalonate and the self- and cross-metathesis (CM) of a variety of alkenes (Scheme 17) [92]. At the beginning of the reactions the presence of free p-cymene was observed by gas chromatography-mass spectrometry (GC-MS) in the catalytic solutions, indicating that complexes 37a-b are transformed into the 12-electron species [OsCl(=CHPh)(NHC)]+ which are the real initiators in these metathesis processes. Although 37a-b were not as efficient as the standard ruthenium-based Grubbs catalysts, they displayed a decent activity that should be taken into account when designing future osmium-based olefin metathesis catalysts.
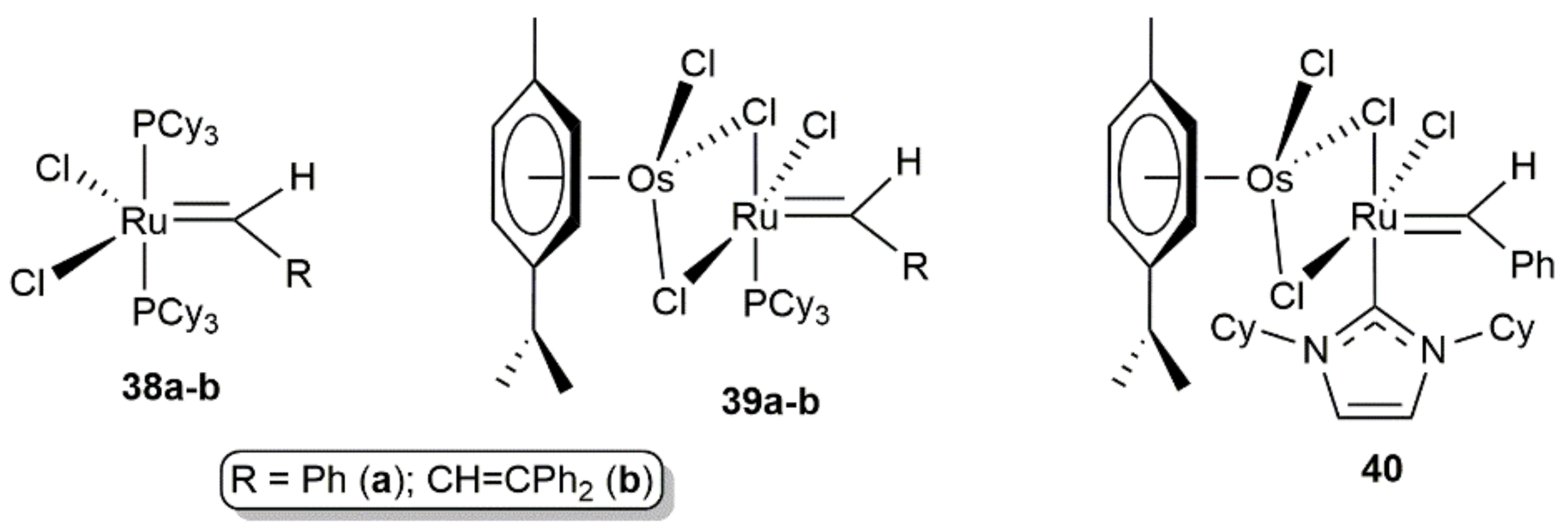
It is also worth mentioning that Dias and Grubbs found that the effectiveness of the classical ruthenium-alkylidene catalysts [RuCl2(=CHR)(PCy3)2] (R = Ph (38a), CH = CPh2 (38b) in olefin metathesis processes can be increased if they are made to react with dimers [{RuCl(μ-Cl)(η6-p-cymene)}2] and [{OsCl(μ-Cl)(η6-p-cymene)}2], reactions that lead to the generation of bridged-chloride bimetallic species [93]. In particular, with regard to complexes 39a-b generated from the cymene-osmium dimer (Figure 9), the reaction rates observed in the ROMP of 1,5-cyclooctadiene surpassed 30–40 times that of 38a-b (ca. 20 times with the analogous ruthenium-based bimetallic systems). The enhanced reactivity of 39a-b vs 38a-b was reasoned in terms of the greater lability of the [OsCl2(η6-p-cymene)] unit compared to the PCy3 ligand which makes it easier to generate the required vacant site for substrate binding. The related heterobimetallic complex 40 (see Figure 9) was synthesized by Herrmann and co-workers by reacting the bis-NHC derivative [RuCl2(=CHPh)(NHC)2] (NHC = 1,3-dicyclohexylimidazol-2-ylidene) with [{OsCl(μ-Cl)(η6-p-cymene)}2] and successfully employed in the ROMP of 5-norbornenyl-2-acetate (95% yield after 3 min in CH2Cl2 at r.t. with 5000:1 monomer/catalyst ratio; identical result to that obtained when the cymene-osmium(II) fragment was replaced by the cymene-ruthenium(II) one) [94,95].
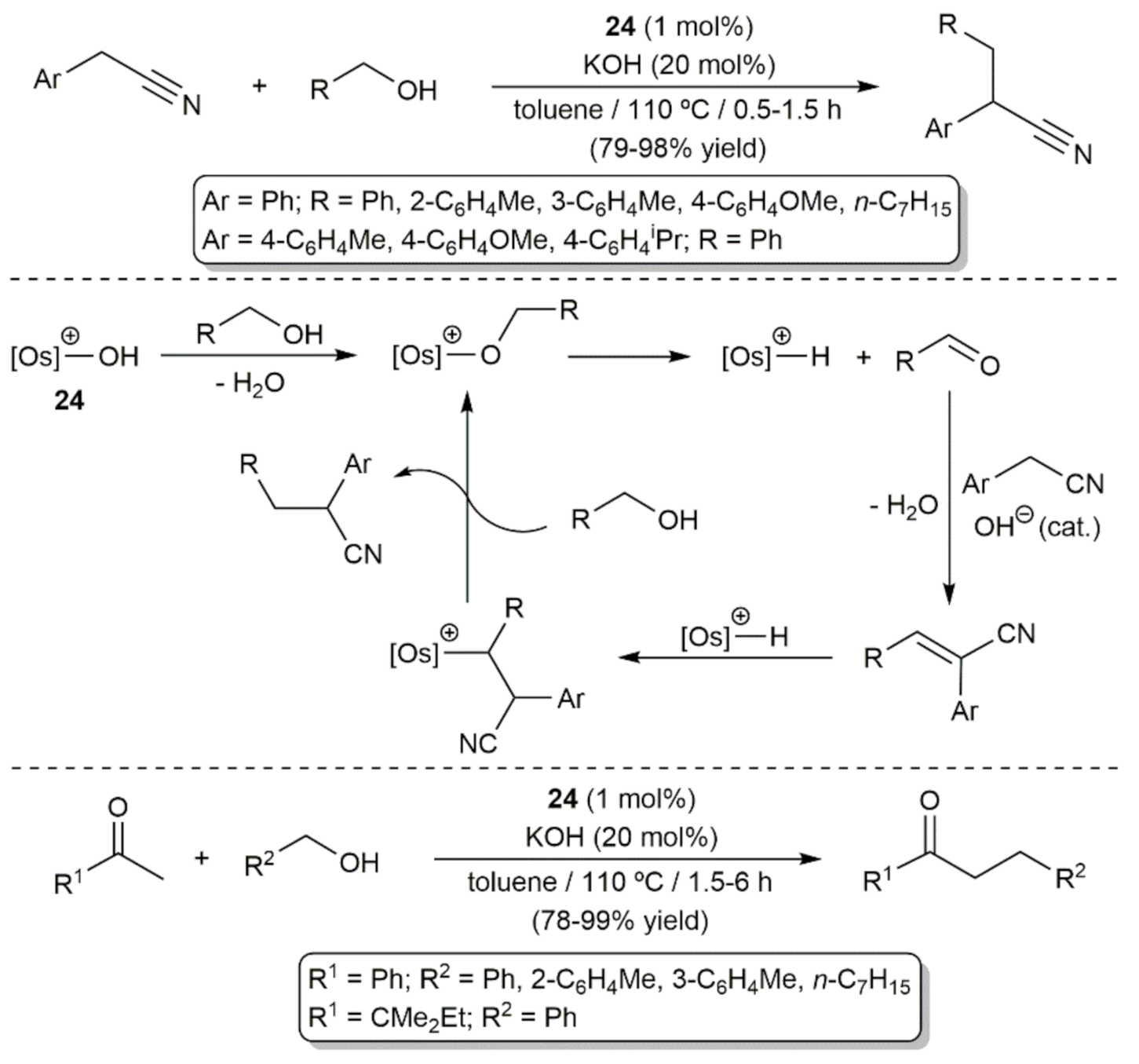
On the other hand, the use of primary alcohols as alkylating agents through the so-called borrowing hydrogen methodology (also referred to as hydrogen autotransfer) has attracted enormous interest during the last two decades due to its economic and environmental benefits (water is the only byproduct formed) [96,97,98]. In 2013, Esteruelas, Yus and co-workers demonstrated the utility of osmium in this type of transformations (Scheme 18) [99]. In particular, they found that, in conjunction with KOH, the 16-electron hydroxo complex [Os(OH)(η6-p-cymene)(IPr)][OTf] (24; see Scheme 8) is able to promote the α-alkylation of a series of arylacetonitriles by removing azeotropically the generated H2O to avoid the competing hydration of the nitrile, a process for which 24 is also an efficient catalyst (see below). A classical reaction mechanism involving the dehydrogenation of the alcohol into the corresponding aldehyde and generation a metal-hydride species, responsible for the reduction of the Knoevenagel adduct formed by the base-catalyzed condensation of the arylacetonitrile substrate and the aldehyde, was proposed. As shown in Scheme 18, complex [Os(OH)(η6-p-cymene)(IPr)][OTf] (24) proved to be also competent in the α-alkylation of methyl ketones, featuring for both processes a greater effectiveness to that previously reported with Ru, Ir, and Pd catalysts.
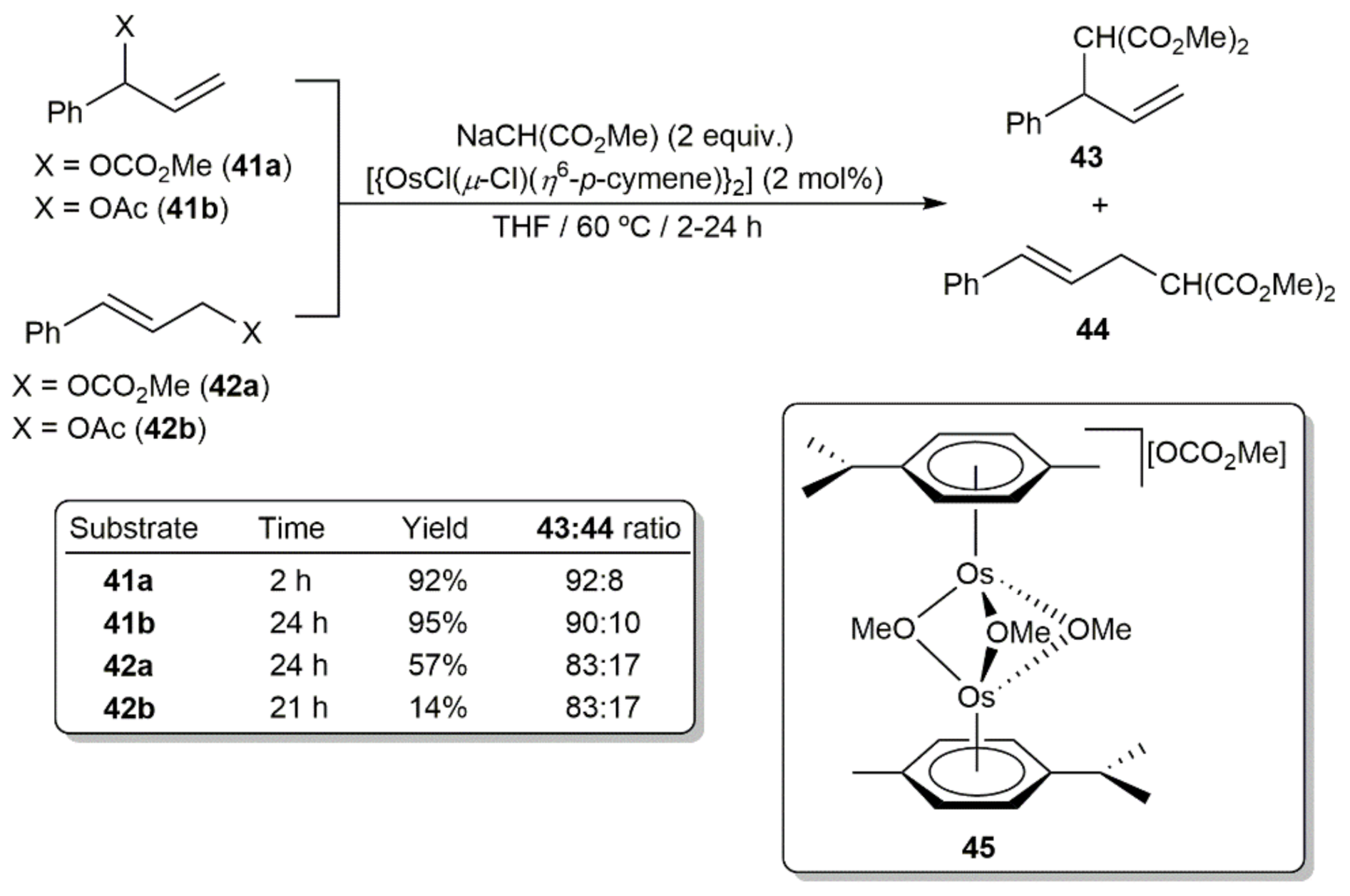
Esteruelas and co-workers also demonstrated that arene-osmium(II) complexes can promote more classical allylic alkylation C-C bond forming reactions [100]. In particular, they screened the catalytic behavior of dimer [{OsCl(μ-Cl)(η6-p-cymene)}2], and a series of mononuclear derivatives [OsCl2(η6-p-cymene)(L)] (L = phosphite or phosphoramidite ligand), in the alkylation of 1-phenylallyl methylcarbonate (41a), 1-phenylallyl acetate (41b), cinnamyl methylcarbonate (42a) and cinnamyl acetate (42b) with sodium dimethylmalonate. The efficiency of the process was strongly dependent on the leaving group and the branched or linear nature of the substrates, with dimer [{OsCl(μ-Cl)(η6-p-cymene)}2] showing the best catalytic performances. Thus, as shown in Scheme 19, the alkylation was more effective for carbonates vs acetates and for branched vs linear substrates, with the formation of the branched product 43 being in all the cases preferred. Mechanistic investigations on the alkylation reaction of 41a with NaCH(CO2Me)2 indicated that the dinuclear derivative [{Os(η6-p-cymene)}2(µ-OMe)3][OCO2Me] (45), which could be isolated and fully characterized by reacting [{OsCl(μ-Cl)(η6-p-cymene)}2] with excess of 41a, acts the real catalytically active species in the process.
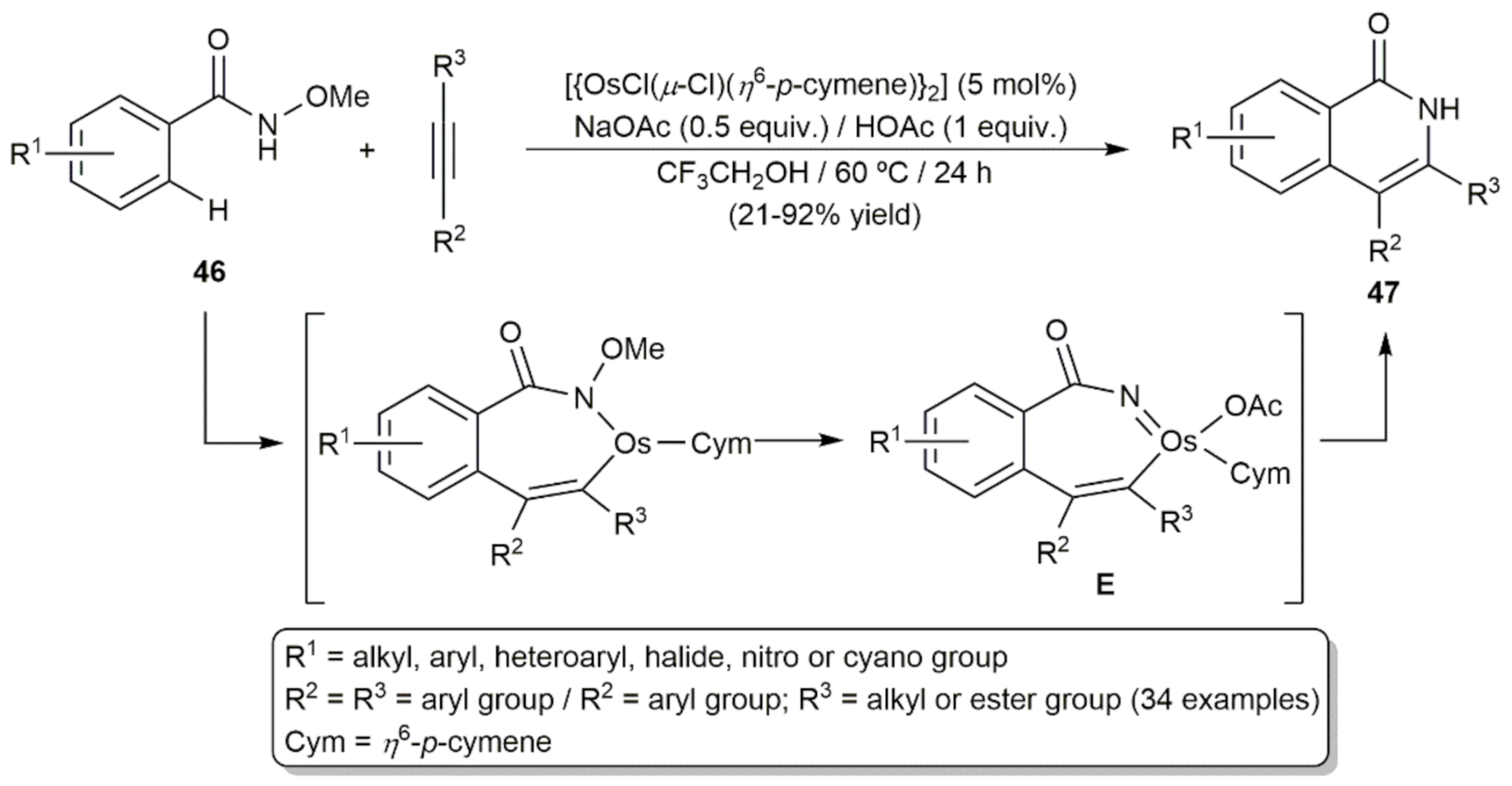
Driven by the enormous success of the carboxylate-assisted ruthenium-catalyzed annulation reactions of arenes with alkynes, via tandem C-H/Het-H functionalization, for the atom-economical assembly of heterocyclic compounds [101,102], Zhuo, Yi, and co-workers explored the potential use of osmium in this type of transformations [103]. Employing the [{OsCl(μ-Cl)(η6-p-cymene)}2]/NaOAc/HOAc combination, they found that isoquinolones 47 can be generated by [4 + 2] annulation of N-methoxybenzamides 46 with internal alkynes in comparable yields to those previously described with related ruthenium-based catalytic systems (Scheme 20) [104,105]. However, it should be noted that while ruthenium is capable of operating at room temperature [105], in the case of osmium, heating at 60 °C and longer reaction times were needed. DFT calculations revealed that the annulation process follows an Os(II)-Os(IV)-Os(II) reaction pathway with the imido-Os(IV) derivative E, generated by HOAc-assisted oxidative addition of the substrate N-O bond to the metal, as key intermediate.
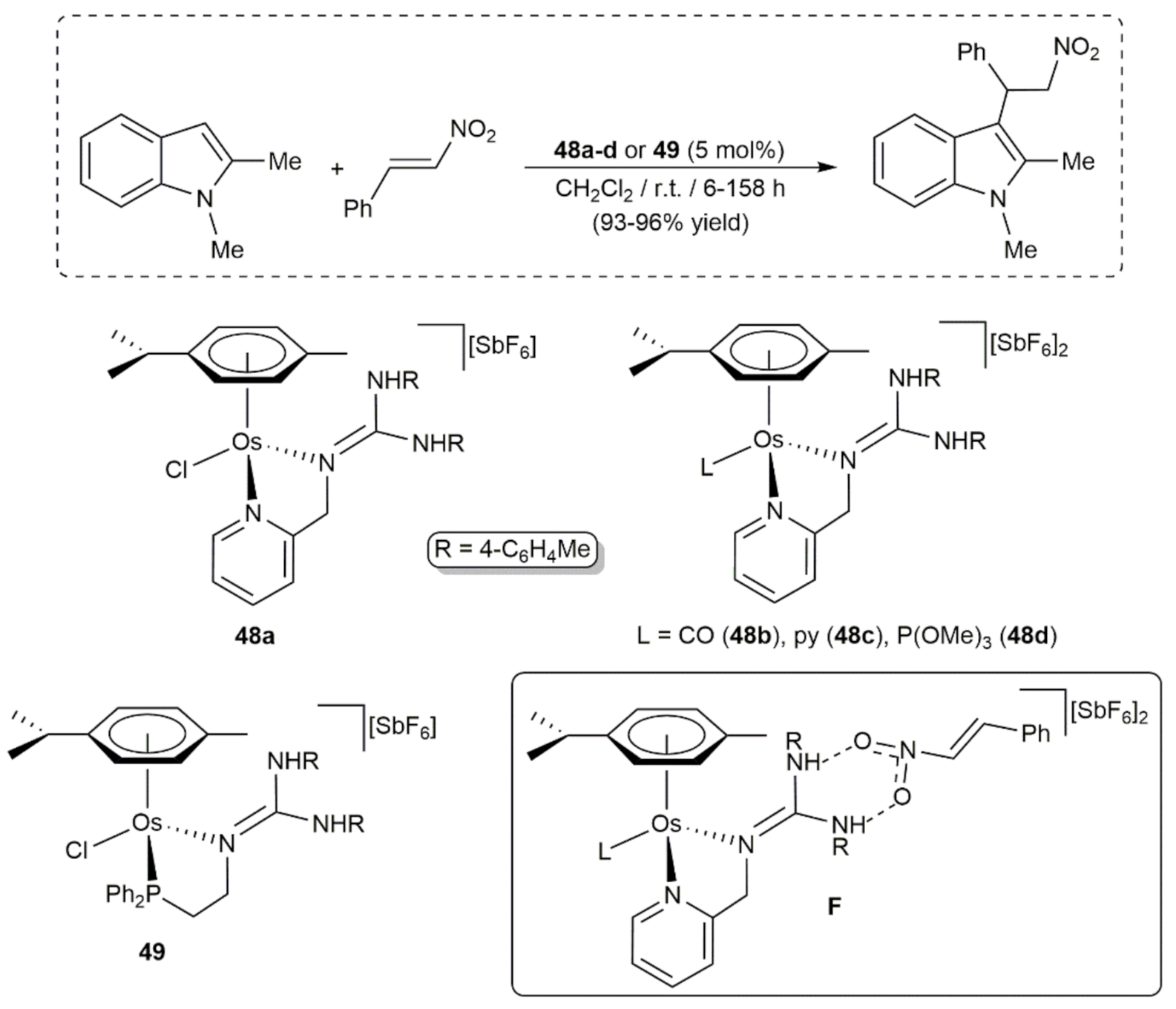
Very recently, Lamata, Carmona and co-workers synthesized different cymene-osmium(II) complexes containing chelating pyridinyl- and phosphino-guanidine ligands, i.e., compounds 48a-d and 49 respectively, which proved to be active catalysts for the Friedel-Crafts reaction between trans-β-nitrostyrene and N-methyl-2-methylindole (Scheme 21) [106]. Although excellent yields were obtained with all of them, the reaction times varied considerably depending on their mono- or dicationic nature, with the dicationic species 48b-d leading to the fastest transformations. 1H NMR studies supported a mechanism in which the substrates do not interact directly with the metal. Instead, activation of the trans-β-nitrostyrene molecule through H-bonding with the NH protons of the coordinated guanidine ligands (intermediates F) facilitates the nucleophilic addition of the indole C-3 carbon to the C=C bond. Blank experiments employing the free ligands led to the alkylated indole product in less than 20% yield, thus evidencing that coordination to the osmium center enhances the acidity of the guanidine NH protons.
5. Nitrile Hydration Reactions
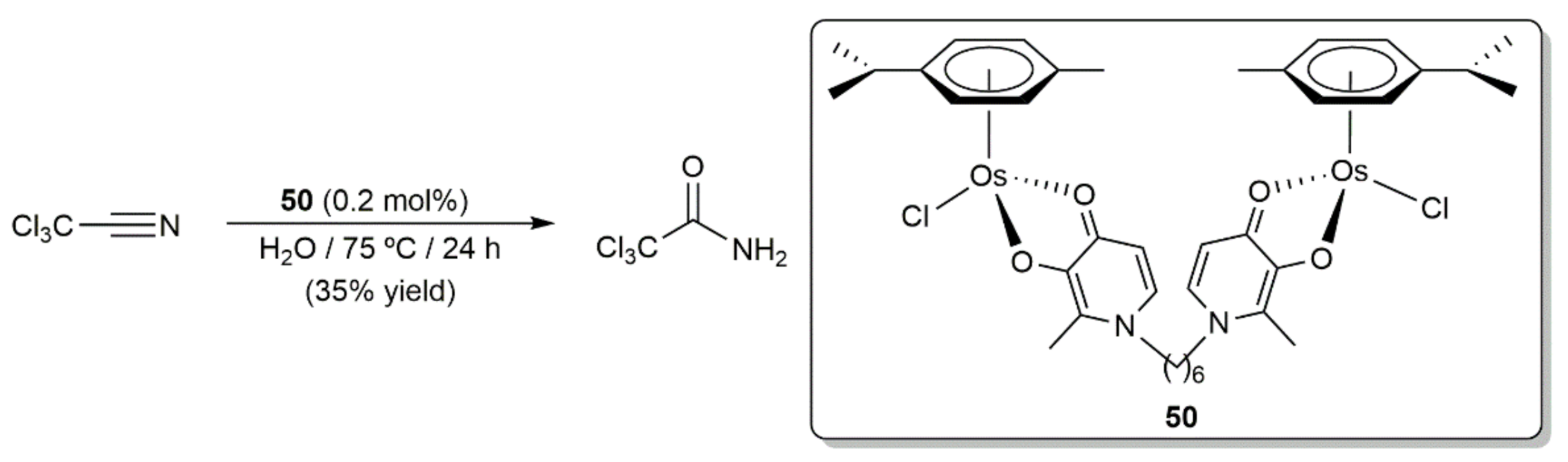
The catalytic hydration of nitriles represents an efficient and atom economical route to synthesize primary amides, that are important structural elements in organic chemistry. A huge number of catalytic systems have been developed during the last decades, with those involving ruthenium being by far the most abundant since they usually feature an outstanding activity and functional group compatibility [107,108,109,110,111]. Although osmium has been comparatively much less studied, some arene-osmium(II) derivatives have been found to promote these reactions with activities comparable, or even superior, to those of related ruthenium-based systems [111]. This was evidenced for the first time by Nazarov, Hartinger, and co-workers in 2008 with complex 50, which managed to convert trichloroacetonitrile into trichloroacetamide with the same effectiveness as its ruthenium analog performing the hydration process in pure water at 75 °C in the absence of any additive (Scheme 22) [112]. However, we must note that the yield reached was relatively low (35%).
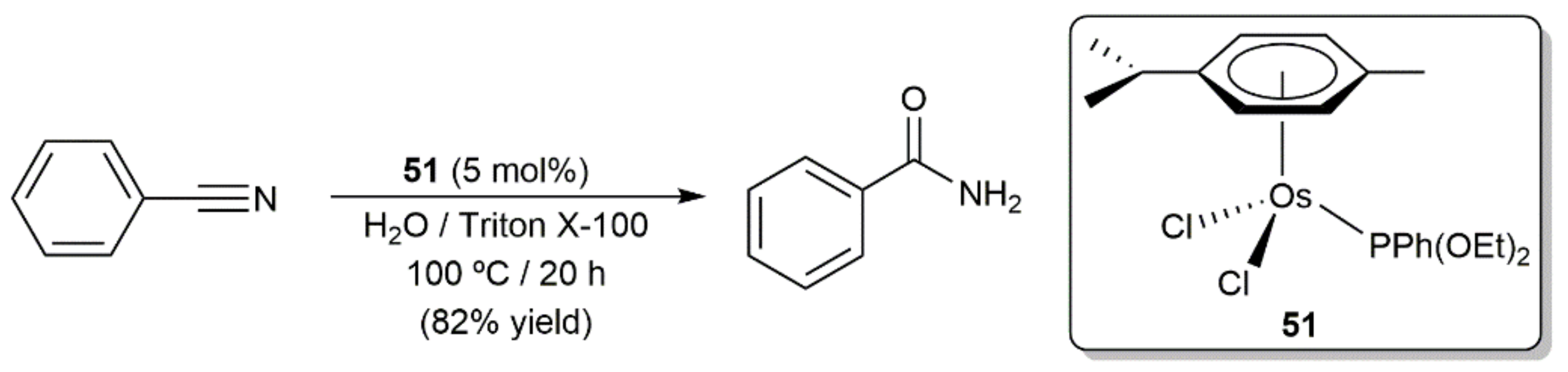
In the context of a broad study on the applicability of arene-ruthenium(II) complexes as catalysts for nitriles hydration in aqueous micellar media, Scarso, Strukul, and co-workers examined in 2010 the behavior of the osmium derivative [OsCl2(η6-p-cymene){PPh(OEt)2}] (51) in the hydration of the model benzonitrile substrate [113]. As shown in Scheme 23, employing 5 mol% of complex 51 and Triton X-100 (polyethylene glycol iso-octylphenyl ether) as the surfactant, benzamide was selectively formed in 82% yield after 20 h of heating at 100 °C, a result clearly superior in this case to that obtained with the homologous Ru(II) species [RuCl2(η6-p-cymene){PPh(OEt)2}] (37% yield under identical reaction conditions). Unfortunately, the generality of this catalyst was not explored since more active ruthenium derivatives were identified.
In combination with KOH, the 16-electron hydroxo complex [Os(OH)(η6-p-cymene)(IPr)][OTf] (24) was later employed by Esteruelas and co-workers to hydrate a wide range of organonitriles (Scheme 24) [114]. Performing the reactions in a water/2-propanol mixture at 120 °C, the corresponding amides were selectively generated in good to excellent yields with TON and TOF values up to 33 and 66 h−1, respectively. DFT calculations along with experimental observations allowed the authors to propose the following catalytic cycle: After coordination of the substrate to osmium, a cationic κ2-imidinate G is formed by intramolecular attack of the OH ligand to the nitrile carbon atom, intermediate species that in some cases could be isolated and fully characterized by performing the reactions under stoichiometric conditions. The subsequent reaction of G with a hydroxide ion (from the KOH present in the medium) generates the neutral κ1-imidinate complex H from which the primary amide product is liberated by hydrolysis, with concomitant regeneration of the catalyst.
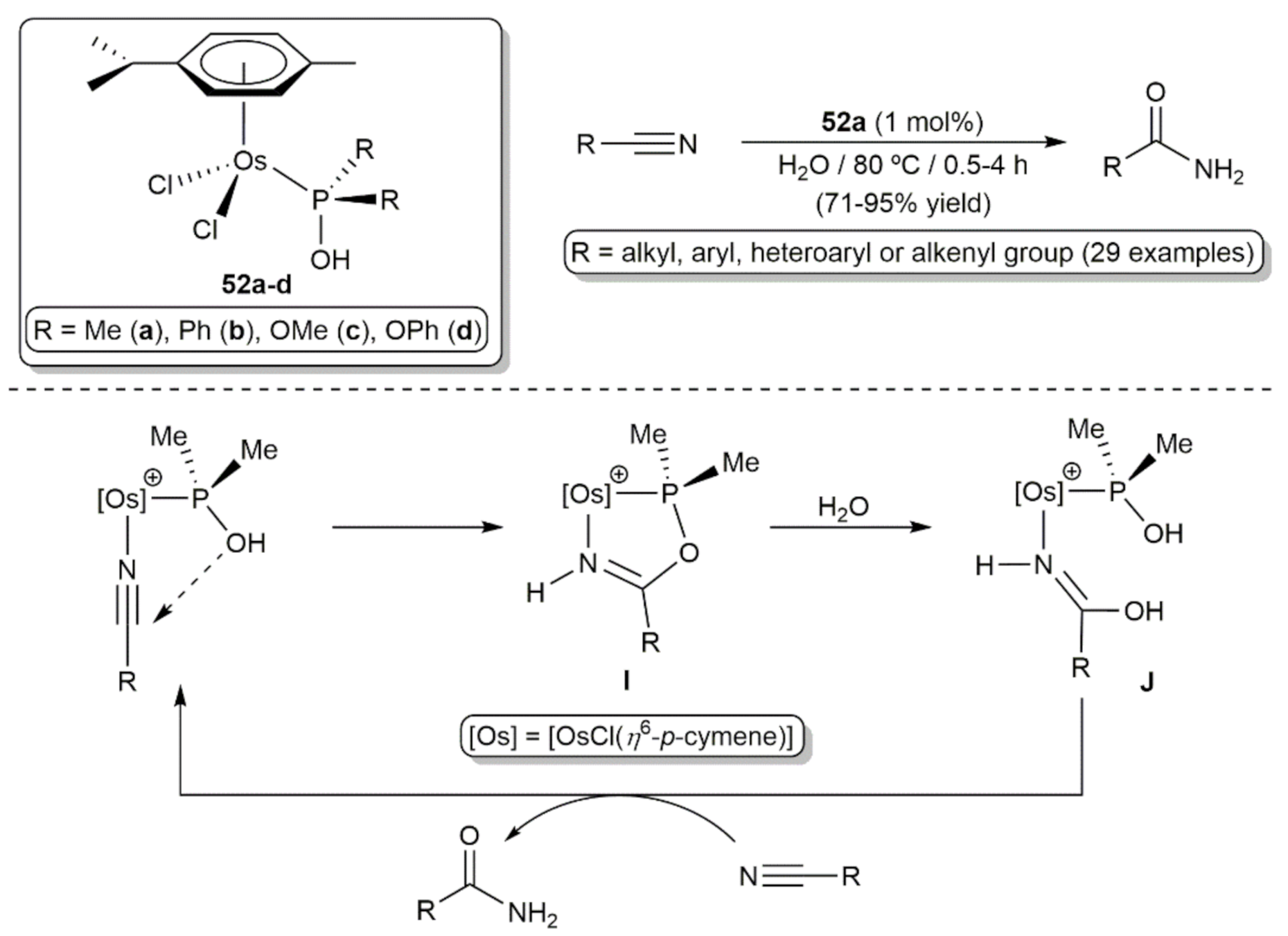
Extending previous studies with ruthenium [115], Cadierno, López, and co-workers explored the catalytic potential of a series of cymene-osmium(II) complexes with phosphinous acid-type ligands, i.e., compounds [OsCl2(η6-p-cymene)(PR2OH)] (52a-d) (Scheme 25) [116]. All of them were able to operate in pure water without the assistance of any acidic or basic additive, with [OsCl2(η6-p-cymene)(PMe2OH)] (52a) featuring the best performances (TOF values up to 200 h−1) and a broad substrate scope. Thus, employing a metal loading of only 1 mol% and a working temperature of 80 °C, 52a selectively hydrated the C≡N bond of a large variety of aliphatic, aromatic, heteroaromatic and α,β-unsaturated nitriles in high yields and short times, with an efficiency comparable to that of its ruthenium analogue [RuCl2(η6-p-cymene)(PMe2OH)]. Concerning the mechanism of the process, DFT calculations indicated that the reactions proceed, as previously established for [RuCl2(η6-p-cymene)(PMe2OH)] [115], through the initial formation of a five-membered metallacyclic intermediate I generated by intramolecular addition of the P-OH group of the dimethylphosphinous acid ligand to the osmium-coordinated nitrile. The subsequent hydrolysis of the metallacycle leads to an iminol complex J from which the primary amide product is liberated after the iminol-to-amide tautomerization and displacement with a new molecule of the nitrile.
Complex [OsCl2(η6-p-cymene)(PMe2OH)] (52a) was also successfully employed in the hydration of cyanamide derivatives (Scheme 26) [117]. The reactions, performed again in pure water and in the absence of additives, allowed the high yield access to a broad family of substituted ureas under relatively mild conditions (40–70 °C) and using low metal loadings (1–3 mol%). For these particular substrates, the effectiveness of complex 52a was superior to that of [RuCl2(η6-p-cymene)(PMe2OH)] and the Parkins catalyst [PtH{(PMe2O)2H}(PMe2OH)]. In this sense, it should be noted that, despite being recognized as the most versatile nitrile hydration catalyst reported to date in the literature [118], the latter resulted completely inoperative.
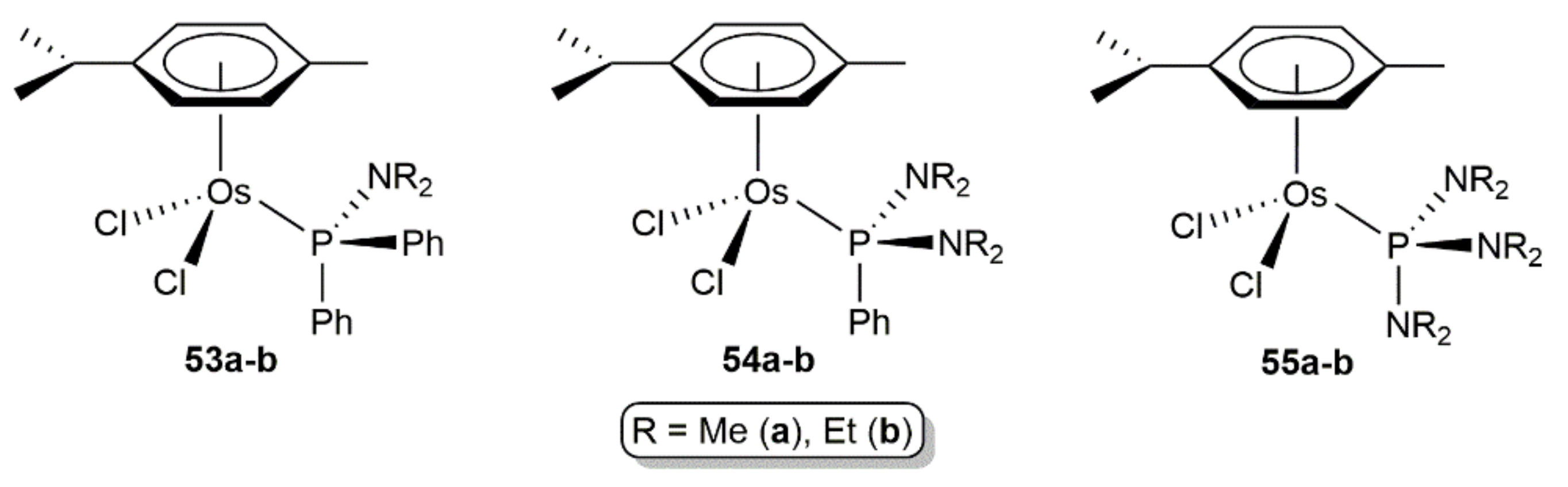
The amino-phosphine complexes [OsCl2(η6-p-cymene){PPh3-n(NR2)n}] (R = Me, Et; n = 1, 2, 3; 53–55a-b in Figure 10) are additional examples of arene-osmium(II) derivatives catalytically active in the hydration of organonitriles [119]. Among them, the best results were obtained with [OsCl2(η6-p-cymene){PPh2(NMe2)}] (53a) which was able to convert selectively a large variety of aliphatic, aromatic, heteroaromatic, and α,β-unsaturated nitriles (26 examples) into the corresponding primary amides working in pure water at 100 °C, with a metal loading of 1 mol% (TOF up to 200 h−1), and in the absence of additives. In aqueous solution, complex 53a evolves into the phosphinous acid derivative [OsCl2(η6-p-cymene)(PPh2OH)] (52b) by hydrolysis of the P-N bond of the amino-phosphine ligand PPh2(NMe2), thus suggesting that 52b probably acts as the real hydration catalyst.
6. Other Catalytic Transformations
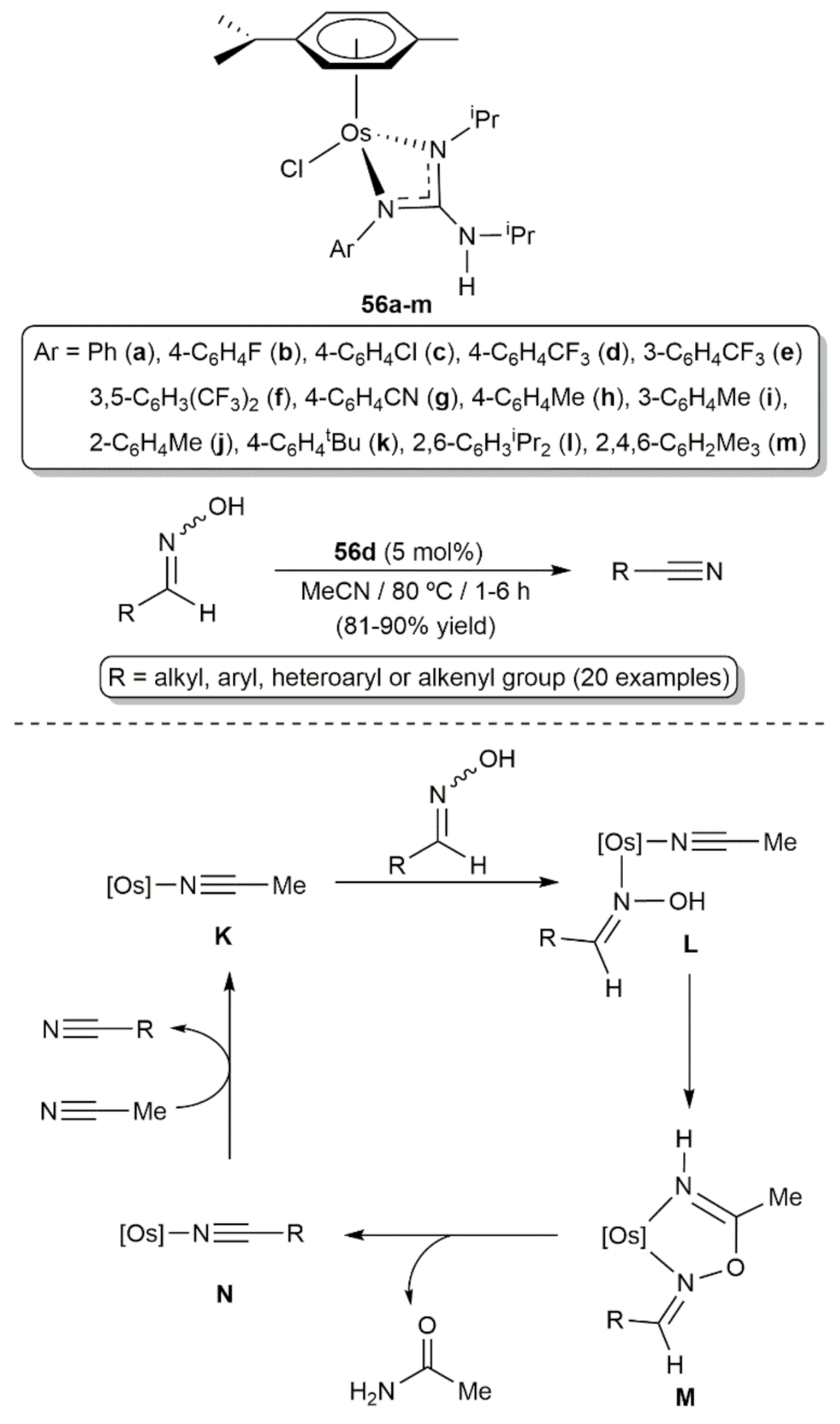
Cadierno, Antiñolo and co-workers explored the behavior of the guanidinate complexes [OsCl{κ2-(N,N´)-C(NAr)(NiPr)NHiPr}(η6-p-cymene)] (56a-m) in the catalytic dehydration of aldoximes to generate nitriles (Scheme 27) [120]. All of them proved to be active, although the electronic nature of the N-aryl unit of the guanidinate ligand was found to play a significant role in both the efficiency and selectivity of the process, with compounds 56d-g featuring strong electron-withdrawing substituents, i.e., CF3 and C≡N, in the aromatic ring showing a superior effectiveness. In particular, the best results were obtained with [OsCl{κ2-(N,N´)-C(N-4-C6H4CF3)(NiPr)NHiPr}(η6-p-cymene)] (56d) which was capable to convert a large variety of aromatic, heteroaromatic, α,β-unsaturated and aliphatic aldoximes into the corresponding nitriles, in high yields and short times, performing the reactions in acetonitrile at 80 °C with an osmium loading of 5 mol%. The selectivity of the process was very high and only trace amounts (1–3%) of the corresponding primary amides, resulting from the rearrangement of the aldoxime [121,122,123], were formed as byproducts. From a mechanistic point of view, these dehydration reactions seem to proceed through the initial coordination of the acetonitrile solvent to the osmium center to generate the catalytically active species K. Subsequent coordination of the aldoxime substrate to K leads to the intermediate species L from which the aldoxime is dehydrated, and the acetonitrile molecule converted into acetamide, via the five membered metallacycle M. The nitrile product is finally released from the intermediate complex N by displacement with the solvent. In favor of this proposal is the fact that an equimolar amount of acetamide with respect to the nitrile product was systematically observed by NMR spectroscopy in all the reaction crudes. It should also be mentioned that compounds 56a-m are the only osmium-based catalysts for the dehydration of aldoximes described to date in the literature.
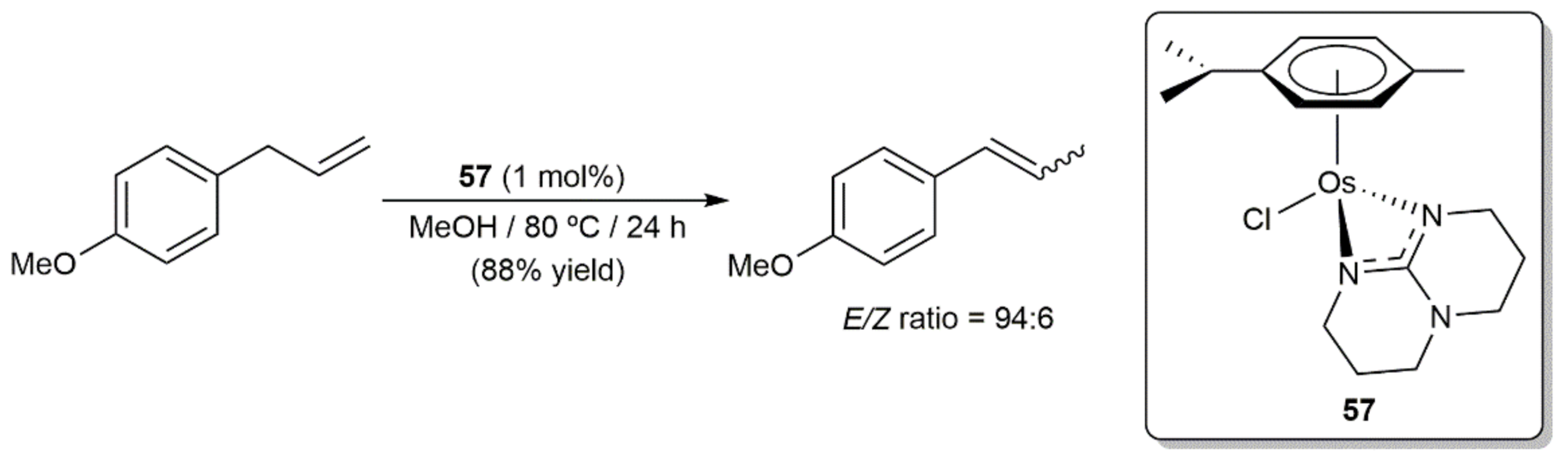
The related guanidinate-osmium(II) complex 57 was used to promote the isomerization of 1-allyl-4-methoxybenzene (estragole) into 1-methoxy-4-(1-propenyl)benzene (anethole) [124], a process of industrial relevance since trans-anethole is commonly used as a flavoring material (anise flavor) in food, beverages, oral hygiene products, and cosmetics [125,126]. As shown in Scheme 28, performing the catalytic reaction in methanol at 80 °C with 1 mol% of 57, the desired anethole was generated in 88% yield after 24 h with a very high trans-selectivity (E/Z ratio = 94:6). However, we must note that this result was clearly inferior to that obtained when the analogous ruthenium complex was used as catalyst under identical conditions (full conversion after only 3 h with a trans-selectivity of 95%) [124].
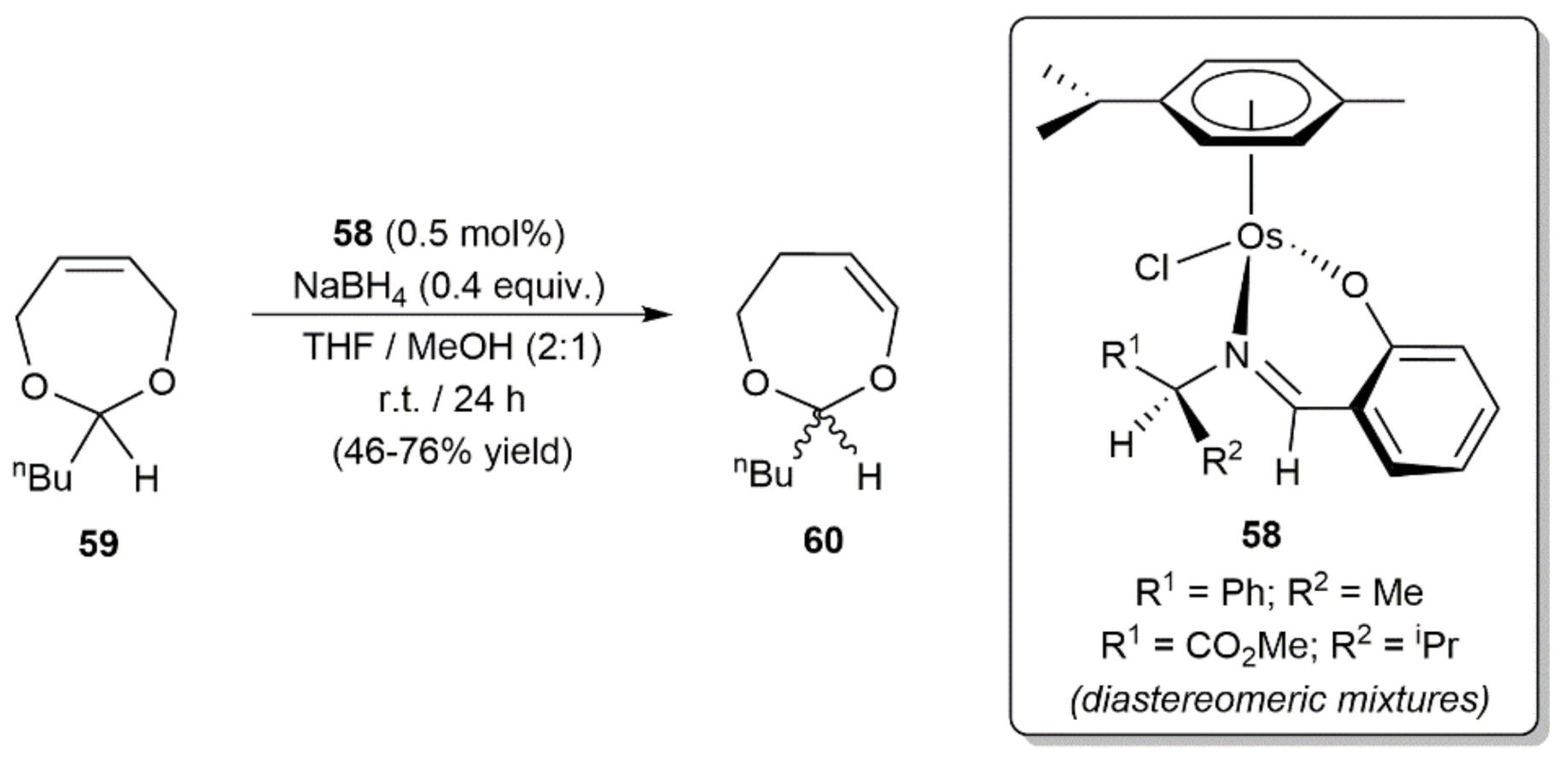
Another carbon–carbon double bond migration reaction promoted by an (η6-arene)-osmium(II) complex reported in the literature is that of 2-n-butyl-4,7-dihydro-1,3-dioxepin 59 to give 2-n-butyl-4,5-dihydro-1,3-dioxepin 60 (Scheme 29) [127]. Employing as catalysts diastereomeric mixtures of the chiral-at-metal complexes 58, containing optically pure salicylaldiminate ligands, Brunner and co-workers obtained the isomerized product 60 in moderate to good yield but without observing any chiral induction, a result that contrast with that obtained with analogous ruthenium complexes with which ee values up to 64% were achieved.
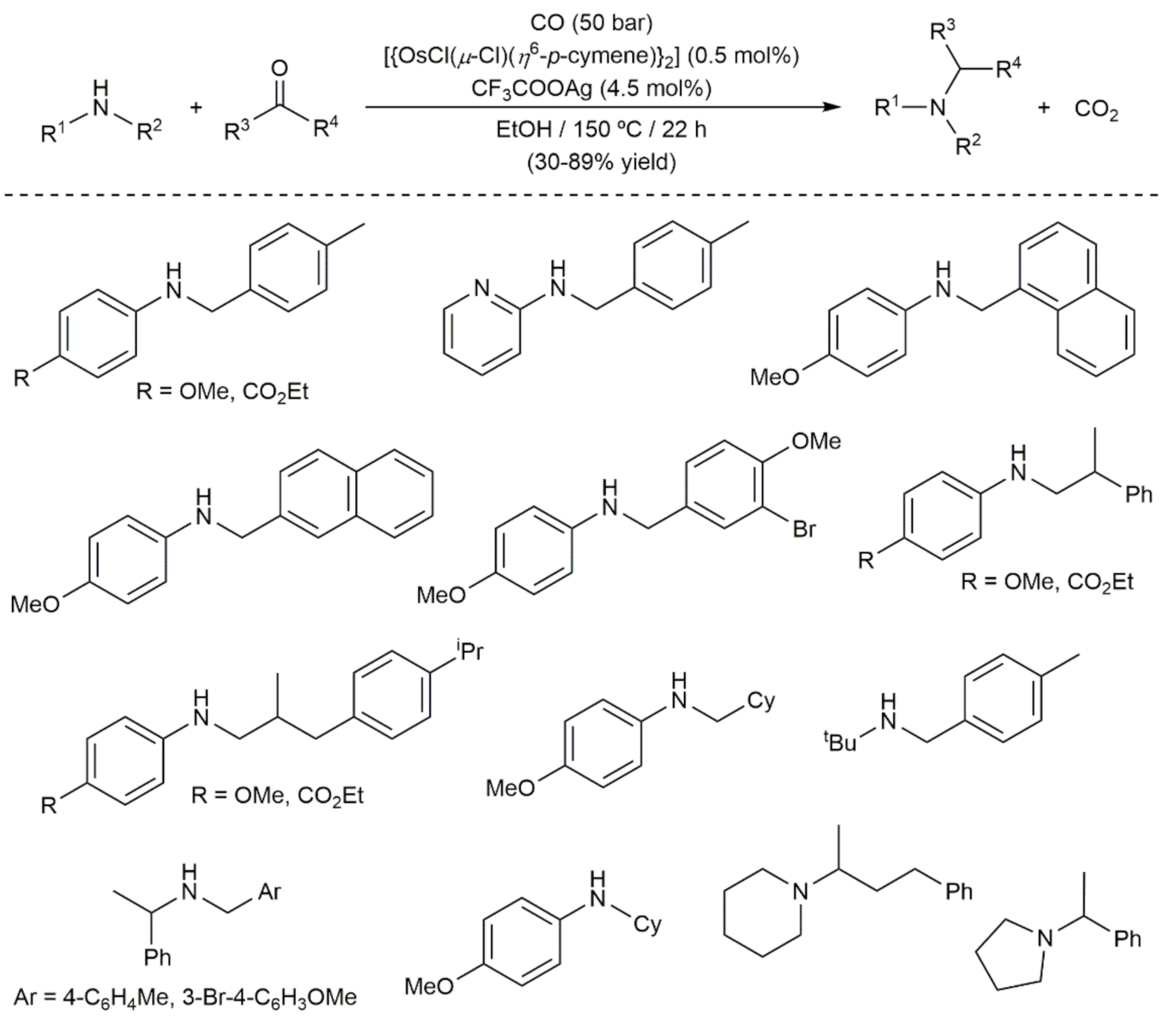
Finally, in a very recent study, Chusov and co-workers evaluated the catalytic potential of a series of osmium complexes in the reductive amination of carbonyl compounds, and they found that dimer [{OsCl(μ-Cl)(η6-p-cymene)}2], in conjunction with silver trifluoroacetate and carbon monoxide as the reducing agent, efficiently catalyzes the process [128]. As shown in Scheme 30, different combinations of primary/secondary amines and aldehydes/ketones were well tolerated, and the corresponding products could be isolated in moderate to good yields. According to DFT calculations, the reactions proceed through the deoxygenation of an hemiaminal intermediate, generated by addition of the amine to the carbonyl compound, after coordination to the unsaturated species [Os(CO)3Cl2] resulting from the reaction of [{OsCl(μ-Cl)(η6-p-cymene)}2] with CO. It is noteworthy that the effectiveness shown by this catalytic system was found to be superior to that previously described with iridium complexes, but far from that of Ru- and Rh-based systems which are the benchmarks for this transformation.
7. Conclusions
The slow ligand exchange kinetics in its complexes, and the higher price in comparison to other platinum-group metals, have been for long time considered as major drawbacks for the application of osmium derivatives in homogeneous catalysis. However, although the catalytic potential of this metal has been underestimated, numerous studies can be found in the literature showing that osmium-based catalysts can feature an effectiveness and selectivity comparable to those shown by the more commonly employed ruthenium, rhodium and iridium ones [35,40,41,42,43]. Some examples have been discussed in this contribution, in which the application of (η6-arene)-osmium(II) complexes in homogenous catalysis has been comprehensively reviewed. Such derivatives have been successfully screened in different catalytic transformations, including transfer hydrogenation, hydrogenation, oxidation and nitrile hydration reactions, as well as several C-C bond forming processes, thus giving solid evidences of the enormous potential of osmium in homogeneous catalysis. Although there is already a body of work in the field, there are countless catalytic processes that have not been yet addressed and in which this type of catalysts, whose steric and electronic properties can be easily tuned by selecting appropriate auxiliary ligands, could lead to remarkable results. We hope that this contribution serves to stimulate future research in this direction.
Author Contributions
Both authors participated equally to the conceptualization and redaction of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO project CTQ2016-75986-P) is gratefully acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Seyferth, D. Bis(benzene)chromium. Its discovery by E.O. Fischer and W. Hafner and subsequent work by the research groups of E.O. Fischer, H.H. Zeiss, F. Hein, C. Elschenbroich, and others. Organometallics 2002, 21, 2800–2820. [Google Scholar] [CrossRef] [Green Version]
- Gastinger, R.G.; Klabunde, K.J. π-Arene complexes of the group VIII transition metals. Transit. Met. Chem. 1979, 4, 1–13. [Google Scholar] [CrossRef]
- Muetterties, E.L.; Bleeke, J.R.; Wucherer, E.J.; Albright, T. Structural, stereochemical, and electronic features of arene-metal complexes. Chem. Rev. 1982, 82, 499–525. [Google Scholar] [CrossRef]
- Kündig, P.E. Transition Metal Arene π-Complexes in Organic Synthesis and Catalysis; Springer: Berlin, Germany, 2004. [Google Scholar]
- Rosillo, M.; Domínguez, G.; Pérez-Castells, J. Chromium arene complexes in organic synthesis. Chem. Soc. Rev. 2007, 36, 1589–1604. [Google Scholar] [CrossRef]
- Pampaloni, G. Aromatic hydrocarbons as ligands. Recent advances in the synthesis, the reactivity and the applications of bis(η6-arene) complexes. Coord. Chem. Rev. 2010, 254, 402–419. [Google Scholar] [CrossRef]
- Takemoto, S.; Matsuzaka, H. Recent topics on catalytic transformations of aromatic molecules via η6 arene transition metal complexes. Tetrahedron Lett. 2018, 59, 697–703. [Google Scholar] [CrossRef]
- Walton, J.W.; Wilkinson, L. π-Coordinated arene metal complexes and catalysis. Organomet. Chem. 2018, 42, 125–171. [Google Scholar] [CrossRef]
- Shvydkiy, N.V.; Perekalin, D.S. Reactions of arene replacement in transition metal complexes. Coord. Chem. Rev. 2020, 411, 213238. [Google Scholar] [CrossRef]
- Winkhaus, G.; Singer, H. Ruthen(II)-komplexe mit zweizähnigem cycloheptatrien und benzol. J. Organomet. Chem. 1967, 7, 487–491. [Google Scholar] [CrossRef]
- Le Bozec, H.; Touchard, D.; Dixneuf, P.H. Organometallic Chemistry of Arene Ruthenium and Osmium Complexes. Adv. Organomet. Chem. 1989, 29, 163–247. [Google Scholar] [CrossRef]
- Bennett, M.A. Recent advances in the chemistry of arene complexes of ruthenium(0) and ruthenium(II). Coord. Chem. Rev. 1997, 166, 225–254. [Google Scholar] [CrossRef]
- Pigge, F.C.; Coniglio, J.J. Stoichiometric applications of η6-arene ruthenium(II) complexes in organic chemistry. Curr. Org. Chem. 2001, 5, 757–784. [Google Scholar] [CrossRef]
- Geldbach, T.J.; Pregosin, P.S. η1 to η6 Ru-arene π complexation: New bonding modes and P-C cleavage chemistry. Eur. J. Inorg. Chem. 2002, 2002, 1907–1918. [Google Scholar] [CrossRef]
- Therrien, B. Functionalised η6-arene ruthenium complexes. Coord. Chem. Rev. 2009, 253, 493–519. [Google Scholar] [CrossRef]
- Süss-Fink, G. Water-soluble arene ruthenium complexes: From serendipity to catalysis and drug design. J. Organomet. Chem. 2014, 751, 2–19. [Google Scholar] [CrossRef]
- Singh, S.K.; Pandey, D.S. Multifaceted half-sandwich arene-ruthenium complexes: Interactions with biomolecules, photoac-tivation, and multinuclearity approach. RSC Adv. 2014, 4, 1819–1840. [Google Scholar] [CrossRef]
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Sadler, P.J. Organometallic chemistry, biology and medicine: Ruthenium arene anticancer complexes. Chem. Commun. 2005, 38, 4764–4776. [Google Scholar] [CrossRef]
- Severin, K. Supramolecular chemistry with organometallic half-sandwich complexes. Chem. Commun. 2006, 37, 3859–3867. [Google Scholar] [CrossRef] [PubMed]
- Therrien, B. Arene ruthenium cages: Boxes full of surprises. Eur. J. Inorg. Chem. 2009, 2009, 2445–2453. [Google Scholar] [CrossRef] [Green Version]
- Süss-Fink, G. Arene ruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef]
- Mishra, A.; Kang, S.C.; Chi, K.-W. Coordination-Driven Self-Assembly of Arene-Ruthenium Compounds. Eur. J. Inorg. Chem. 2013, 2013, 5222–5232. [Google Scholar] [CrossRef]
- Nazarov, A.A.; Hartinger, C.; Dyson, P. Opening the lid on piano-stool complexes: An account of ruthenium(II)–arene complexes with medicinal applications. J. Organomet. Chem. 2014, 751, 251–260. [Google Scholar] [CrossRef]
- Singh, A.K.; Pandey, D.S.; Xu, Q.; Braunstein, P. Recent advances in supramolecular and biological aspects of arene ruthe-nium(II) complexes. Coord. Chem. Rev. 2014, 270, 31–56. [Google Scholar] [CrossRef]
- Murray, B.; Babak, M.; Hartinger, C.; Dyson, P. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Therrien, B. Arene Ruthenium Complexes in Supramolecular Chemistry. Med. Chem. 2018, 71, 379–402. [Google Scholar] [CrossRef]
- Zaki, M.; Hairat, S.; Aazam, E.S. Scope of organometallic compounds based on transition metal-arene systems as anticancer agents: Starting from the classical paradigm to targeting multiple strategies. RSC Adv. 2019, 9, 3239–3278. [Google Scholar] [CrossRef] [Green Version]
- Therrien, B. Unmasking Arene Ruthenium Building Blocks. Chem. Rec. 2021, 21, 460–468. [Google Scholar] [CrossRef]
- Delaude, L.; Demonceau, A. Retracing the evolution of monometallic ruthenium–arene catalysts for C–C bond formation. Dalton Trans. 2012, 41, 9257. [Google Scholar] [CrossRef]
- Crochet, P.; Cadierno, V.; Cadierno-Menéndez, V. Arene-ruthenium(ii) complexes with hydrophilic P-donor ligands: Versatile catalysts in aqueous media. Dalton Trans. 2014, 43, 12447. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Gupta, R.K.; Pandey, D.S. Half-sandwich arene ruthenium complexes: Synthetic strategies and relevance in catalysis. Chem. Soc. Rev. 2014, 43, 707–733. [Google Scholar] [CrossRef]
- Ackermann, L. Robust ruthenium(II)-catalyzed C-H arylations: Carboxylate assistance for the efficient synthesis of angiotensin-II-receptor blockers. Org. Process Res. Dev. 2015, 19, 260–269. [Google Scholar] [CrossRef]
- Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalysed functionalization of C-H bonds with alkenes: Alkenylation versus alkylation. Top. Organomet. Chem. 2016, 55, 137–188. [Google Scholar]
- Manikandan, R.; Jeganmohan, M. Recent advances in the ruthenium(II)-catalyzed chelation-assisted C-H olefination of substituted aromatics, alkenes and heteroaromatics with alkenes via the deprotonation pathway. Chem. Commun. 2017, 53, 8931–8947. [Google Scholar] [CrossRef] [PubMed]
- Gichumbi, J.M.; Friedrich, H.B. Half-sandwich complexes of platinum group metals (Ir, Rh, Ru and Os) and some recent bilogical and catalytic applications. J. Organomet. Chem. 2018, 866, 123–143. [Google Scholar] [CrossRef]
- Adams, R.D.; Selegue, J.P. Comprehensive Organometallic Chemistry; Wilkinson, G., Stone, F.G.A., Abel, E.W., Eds.; Pergamon Press: Oxford, UK, 1982; Volume 4, pp. 1020–1022. [Google Scholar]
- Bennett, M.A. Comprehensive Organometallic Chemistry II; Abel, E.W., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon Press: Oxford, UK, 1995; Volume 7, pp. 556–587. [Google Scholar]
- Gimeno, J.; Cadierno, V.; Crochet, P. Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier Science: Oxford, UK, 2007; Volume 6, pp. 516–540. [Google Scholar]
- Gimeno, J.; Cadierno, V. Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier Science: Oxford, UK, 2007; Volume 6, pp. 600–619. [Google Scholar]
- Sánchez-Delgado, R.A.; Rosales, M.; Esteruelas, M.A.; Oro, L.A. Homogeneous catalysis by osmium complexes: A review. J. Mol. Catal. A Chem. 1995, 96, 231–243. [Google Scholar] [CrossRef]
- Chelucci, G.; Baldino, S.; Baratta, W. Recent Advances in Osmium-Catalyzed Hydrogenation and Dehydrogenation Reactions. Acc. Chem. Res. 2015, 48, 363–379. [Google Scholar] [CrossRef]
- Chelucci, G.; Pinna, G.A.; Pinna, G.; Solinas, M.; Sechi, B. Osmium complexes in catalysis of olefin hydrogenation and isomerization. Chin. J. Catal. 2016, 37, 1824–1836. [Google Scholar] [CrossRef]
- Chelucci, G. Ruthenium and osmium complexes in C-C bond-forming reactions by borrowing hydrogen catalysis. Coord. Chem. Rev. 2017, 331, 1–36. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Seo, C.S.G.; Morris, R.H. Catalytic Homogeneous Asymmetric Hydrogenation: Successes and Opportunities. Organometallics 2019, 38, 47–65. [Google Scholar] [CrossRef]
- Farrar-Tobar, R.A.; Dell’Acqua, A.; Tin, S.; de Vries, J.G. Metal-catalysed selective transfer hydrogenation of α,β-unsaturated carbonyl compounds to allylic alcohols. Green Chem. 2020, 22, 3323–3357. [Google Scholar] [CrossRef]
- Blacker, A.J. The Handbook of Homogeneous Hydrogenation; de Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp. 1215–1244. [Google Scholar]
- Ikariya, T.; Blacker, A.J. Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid−Triethylamine Mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Accounts Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Coverdale, J.; Sanchez-Cano, C.; Clarkson, G.J.; Soni, R.; Wills, M.; Sadler, P.J. Easy To Synthesize, Robust Organo-osmium Asymmetric Transfer Hydrogenation Catalysts. Chem. A Eur. J. 2015, 21, 8043–8046. [Google Scholar] [CrossRef] [Green Version]
- Coverdale, J.P.C.; Romero-Canelón, I.; Sanchez-Cano, C.; Clarkson, G.J.; Habtemariam, A.; Wills, M.; Sadler, P.J. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 2018, 10, 347–354. [Google Scholar] [CrossRef]
- Bolitho, E.M.; Coverdale, J.P.C.; Bridgewater, H.E.; Clarkson, G.J.; Quinn, P.D.; Sanchez-Cano, C.; Sadler, P.J. Tracking reactions of asymmetric organo-osmium transfer hydrogenation catalysts in cancer cells. Angew. Chem. Int. Ed. 2021, 60, 6462–6472. [Google Scholar] [CrossRef]
- Ngo, A.H.; Do, L.H. Structure–activity relationship study of half-sandwich metal complexes in aqueous transfer hydrogenation catalysis. Inorg. Chem. Front. 2020, 7, 583–591. [Google Scholar] [CrossRef]
- Infante-Tadeo, S.; Rodríguez-Fanjul, V.; Habtemariam, A.; Pizarro, A.M. Osmium(II) tethered half-sandwich complexes: pH-dependent aqueous speciation and transfer hydrogenation in cells. Chem. Sci. 2021, in press. [Google Scholar] [CrossRef]
- Wang, W.; Yang, X. Mechanistic insights into asymmetric transfer hydrogenation of pyruvic acid catalysed by chiral osmium complexes with formic assisted proton transfer. Chem. Commun. 2019, 55, 9633–9636. [Google Scholar] [CrossRef]
- Faller, J.W.; Lavoie, A.R. Enantioselective Routes to Both Enantiomers of Aryl Alcohols with a Single Catalyst Antipode: Ru and Os Transfer Hydrogenation Catalysts. Org. Lett. 2001, 3, 3703–3706. [Google Scholar] [CrossRef]
- Palmer, M.; Walsgrove, A.T.; Wills, M. (1R,2S)-(+)-cis-1-Amino-2-indanol: An Effective Ligand for Asymmetric Catalysis of Transfer Hydrogenations of Ketones. J. Org. Chem. 1997, 62, 5226–5228. [Google Scholar] [CrossRef]
- Faller, J.W.; Lavoie, A.R. Enantioselective Syntheses of Nonracemic Benzyl-α-dAlcohols via Catalytic Transfer-Hydrogenation with Ru, Os, Rh, and Ir Catalysts. Organometallics 2002, 21, 3493–3495. [Google Scholar] [CrossRef]
- Carmona, D.; Lamata, M.P.; Viguri, F.; Dobrinovich, I.; Lahoz, F.J.; Oro, L.A. On the Sense of the Enantioselection in Hydrogen Transfer Reactions from 2-Propanol to Ketones. Adv. Synth. Catal. 2002, 344, 499–502. [Google Scholar] [CrossRef]
- Carmona, D.; Lahoz, F.J.; García-Orduña, P.; Oro, L.A.; Lamata, M.P.; Viguri, F. Half-sandwich complexes of osmium(II) with l-α-amino carboxylate ligands as asymmetric transfer hydrogenation catalysts. On the origin of the enantioselectivity. Organometallics 2012, 31, 3333–3345. [Google Scholar] [CrossRef]
- Sarfraz, R.A.; Kazi, T.G.; Iqbal, S.; Afridi, H.I.; Jamali, M.K.; Jalbani, N.; Arain, M.B. Synthesis, structure determination and chemoselective catalytic studies of amino acids complexes of osmium(II). Appl. Organomet. Chem. 2008, 22, 187–192. [Google Scholar] [CrossRef]
- Fu, Y.; Soni, R.; Romero, M.J.; Pizarro, A.M.; Salassa, L.; Clarkson, G.J.; Hearn, J.M.; Habtemariam, A.; Wills, M.; Sadler, P.J. Mirror-Image Organometallic Osmium Arene Iminopyridine Halido Complexes Exhibit Similar Potent Anticancer Activity. Chem. A Eur. J. 2013, 19, 15199–15209. [Google Scholar] [CrossRef] [Green Version]
- Gichumbi, J.; Omondi, B.; Friedrich, H. Half-Sandwich Osmium(II) Complexes with Bidentate N,N-Chelating Ligands and Their Use in the Transfer Hydrogenation of Ketones. Eur. J. Inorg. Chem. 2017, 2017, 915–924. [Google Scholar] [CrossRef]
- Müller, A.L.; Bleith, T.; Roth, T.; Wadepohl, H.; Gade, L.H. Iridium Half-Sandwich Complexes with Di- and Tridentate Bis(pyridylimino)isoindolato Ligands: Stoichiometric and Catalytic Reactivity. Organometallics 2014, 34, 2326–2342. [Google Scholar] [CrossRef]
- Schreiber, D.F.; O.´Connor, C.; Grave, C.; Müller-Bunz, H.; Scopelliti, R.; Dyson, P.J.; Phillips, A.D. Synthesis, characterization, and reactivity of the first osmium β-diketiminato complexes and application in catalysis. Organometallics 2013, 32, 7345–7356. [Google Scholar] [CrossRef]
- Albertin, G.; Antoniutti, S.; Castro, J.; Paganelli, S. Preparation and reactivity of p-cymene complexes of ruthenium and osmium incorporating 1,3-triazenide ligands. J. Organomet. Chem. 2010, 695, 2142–2152. [Google Scholar] [CrossRef]
- Castarlenas, R.; Esteruelas, M.A.; Oñate, E. Preparation, X-ray structure, and reactivity of an osmium-hydroxo complex stabi-lized by an N-heterocyclic carbene ligand: A base-free catalytic precursor for hydrogen transfer from 2-propanol to aldehydes. Organometallics 2008, 27, 3240–3247. [Google Scholar] [CrossRef]
- Wylie, W.N.; Lough, A.J.; Morris, R.H. Mechanistic Investigation of the Hydrogenation of Ketones Catalyzed by a Ruthenium(II) Complex Featuring an N-Heterocyclic Carbene with a Tethered Primary Amine Donor: Evidence for an Inner Sphere Mechanism. Organometallics 2011, 30, 1236–1252. [Google Scholar] [CrossRef]
- Castañón, E.B.; Kaposi, M.; Reich, R.M.; Kühn, F.E. Water-soluble transition metal complexes of ruthenium(II), osmium(II), rhodium(III) and iridium(III) with chelating N-heterocyclic carbene ligands in hydrogenation and transfer hydrogenation catalysis. Dalton Trans. 2018, 47, 2318–2329. [Google Scholar] [CrossRef] [PubMed]
- Bolje, A.; Hohloch, S.; van der Meer, M.; Košmrlj, J.; Sarkar, B. RuII, OsII, and IrIII complexes with chelating pyridyl-mesoionic carbene ligands: Structural characterization and applications in transfer hydrogenation catalysis. Chem. Eur. J. 2015, 21, 6756–6764. [Google Scholar] [CrossRef]
- Bolje, A.; Hohloch, S.; Košmrlj, J.; Sarkar, B. RuII, IrIII and OsII mesoionic carbene complexes: Efficient catalysts for transfer hydrogenation of selected functionalities. Dalton Trans. 2016, 45, 15983–15993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Muñiz, K. The development of asymmetric deamination of alkenes with imido-osmium reagents. New J. Chem. 2005, 29, 1371–1385. [Google Scholar] [CrossRef]
- Christie, S.D.R.; Warrington, A.D. Osmium and palladium: Complementary metals in alkene activation and oxidation. Synthesis 2008, 1325–1341. [Google Scholar] [CrossRef]
- Pilgrim, B.S.; Donohoe, T.J. Osmium-Catalyzed Oxidative Cyclization of Dienes and Their Derivatives. J. Org. Chem. 2013, 78, 2149–2167. [Google Scholar] [CrossRef]
- Shul´pin, G.B.; Vinogradov, M.M.; Shul´pina, L.S. Oxidative functionalization of C-H compounds induced by extremely effi-cient osmium catalysts. Catal. Sci. Technol. 2018, 8, 4287–4313. [Google Scholar] [CrossRef]
- Cabeza, J.A.; Smith, A.J.; Adams, H.; Maitlis, P.M. The reactions of the tri-μ-hydroxo-bis[η6-p-cymeneosmium(II)] cation with aldehydes and acids and the homogeneously catalysed oxidation of acetaldehyde and propionaldehyde with water. X-ray structure of [(p-MeC6H4CHMe2)2Os2(μ-HCO2)(μ-OH)(μ-H)]][PF6]. J. Chem. Soc. Dalton Trans. 1986, 17, 1155–1160. [Google Scholar] [CrossRef]
- Hohloch, S.; Hettmanczyk, L.; Sarkar, B. Introducing Potential Hemilability into “Click” Triazoles and Triazolylidenes: Synthesis and Characterization of d6-Metal Complexes and Oxidation Catalysis. Eur. J. Inorg. Chem. 2014, 2014, 3164–3171. [Google Scholar] [CrossRef]
- Gichumbi, J.; Omondi, B.; Friedrich, H.B. Oxidation of olefins catalyzed by half-sandwich osmium(II) arene complexes. J. Organomet. Chem. 2018, 856, 56–62. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Kozlov, Y.N.; Nesterov, D.S.; Shul´pina, L.S.; Pombeiro, A.J.L.; Shul´pin, G.B. Oxidation of hydrocarbons with H2O2/O2 catalyzed by osmium complexes containing p-cymene ligands in acetonitrile. Catal. Sci. Technol. 2014, 4, 3214–3226. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Shul´pina, L.S.; Kozlov, Y.N.; Kudinov, A.R.; Ikonnikov, N.S.; Shul´pin, G.B. Oxidation of hydrocarbons and alcohols with peroxides catalyzed by new π-cymene osmium complexes. J. Organomet. Chem. 2015, 784, 52–61. [Google Scholar] [CrossRef]
- Demonceau, A.; Lemoine, C.; Noels, A. Osmium-catalysed cyclopropanation of olefins. Tetrahedron Lett. 1996, 37, 1025–1026. [Google Scholar] [CrossRef]
- Cui, M.; Guo, X.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Halide Effects on the Stability of Osmium Indenylidene Complexes: Isolation, Characterization, and Reactivities. Organometallics 2020, 39, 2142–2151. [Google Scholar] [CrossRef]
- Faller, J.; Parr, J. Diastereoselectivity in Chiral Osmium Complexes of a Bidentate Bisphosphine Monoxide Ligand. Organometallics 2000, 19, 3556–3561. [Google Scholar] [CrossRef]
- Faller, J.W.; Parr, J. Utility of Osmium(II) in the Catalysis of Asymmetric Diels−Alder Reactions. Organometallics 2001, 20, 697–699. [Google Scholar] [CrossRef]
- Faller, J.W.; Parr, J. Synthesis, Characterization, Diastereoselectivity, and Catalytic Activity of Complexes of Ruthenium with BINAP Monoxide. Organometallics 2000, 19, 1829–1832. [Google Scholar] [CrossRef]
- Carmona, D.; Vega, C.; García, N.; Lahoz, F.J.; Elipe, S.; Oro, L.A.; Lamata, M.P.; Viguri, F.; Borao, R. Chiral phosphinooxa-zoline-ruthenium(II) and –osmium(II) complexes as catalysts in Diels-Alder reactions. Organometallics 2006, 23, 1592–1606. [Google Scholar] [CrossRef]
- Suzuki, T.; Torii, T. Catalytic asymmetric Michael reactions using a chiral rhodium complex. Tetrahedron Asymmetry 2001, 12, 1077–1081. [Google Scholar] [CrossRef]
- Hafner, A.; Mühlebach, A.; van der Schaaf, P.A. One-component catalysts for thermal and photoinduced ring opening metathesis polymerization. Angew. Chem. Int. Ed. Engl. 1997, 36, 2121–2124. [Google Scholar] [CrossRef]
- Castarlenas, R.; Esteruelas, M.A.; Oñate, E. N-Heterocyclic Carbene−Osmium Complexes for Olefin Metathesis Reactions. Organometallics 2005, 24, 4343–4346. [Google Scholar] [CrossRef]
- Dias, E.L.; Grubbs, R.H. Synthesis and investigation of homo- and heterobimetallic ruthenium olefin metathesis catalysts exhibiting increased activities. Organometallics 1998, 17, 2758–2767. [Google Scholar] [CrossRef]
- Weskamp, T.; Kohl, F.J.; Herrmann, A.W. N-heterocyclic carbenes: Novel ruthenium–alkylidene complexes. J. Organomet. Chem. 1999, 582, 362–365. [Google Scholar] [CrossRef]
- Frenzel, U.; Weskamp, T.; Kohl, F.J.; Schattenmann, W.C.; Nuyken, O.; Herrmann, A.W. N-Heterocyclic carbenes: Application of ruthenium–alkylidene complexes in ring-opening metathesis polymerization. J. Organomet. Chem. 1999, 586, 263–265. [Google Scholar] [CrossRef]
- Guillena, G.; Ramón, D.J.; Yus, M. Alcohols as electrophiles in C-C bond-forming reactions: The hydrogen autotransfer process. Angew. Chem. Int. Ed. 2007, 46, 2358–2364. [Google Scholar] [CrossRef]
- Nixon, T.D.; Whittlesey, M.K.; Williams, J.M.J. Transition metal catalysed reactions of alcohols using borrowing hydrogen methodology. Dalton Trans. 2009, 40, 753–762. [Google Scholar] [CrossRef]
- Obora, Y. Recent Advances in α-Alkylation Reactions using Alcohols with Hydrogen Borrowing Methodologies. ACS Catal. 2014, 4, 3972–3981. [Google Scholar] [CrossRef]
- Buil, M.L.; Esteruelas, M.A.; Herrero, J.; Izquierdo, S.; Pastor, I.M.; Yus, M. Osmium Catalyst for the Borrowing Hydrogen Methodology: α-Alkylation of Arylacetonitriles and Methyl Ketones. ACS Catal. 2013, 3, 2072–2075. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; García-Yebra, C.; Oliván, M.; Oñate, E.; Valencia, M. Osmium-Catalyzed Allylic Alkylation. Organometallics 2008, 27, 4892–4902. [Google Scholar] [CrossRef]
- Ackermann, L. Carboxylate-Assisted Ruthenium-Catalyzed Alkyne Annulations by C–H/Het–H Bond Functionalizations. Acc. Chem. Res. 2013, 47, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.S. Recent Advances in C–H Bond Functionalization with Ruthenium-Based Catalysts. Catalysts 2019, 9, 173. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wu, L.; Xu, H.; Gao, H.; Zhou, Z.; Yi, W. Redox-Neutral [4 + 2] Annulation ofN-Methoxybenzamides with Alkynes Enabled by an Osmium(II)/HOAc Catalytic System. Org. Lett. 2019, 21, 9904–9908. [Google Scholar] [CrossRef]
- Li, B.; Feng, H.; Xu, S.; Wang, B. Ruthenium-Catalyzed Isoquinolone Synthesis through C-H Activation Using an Oxidizing Directing Group. Chem. A Eur. J. 2011, 17, 12573–12577. [Google Scholar] [CrossRef]
- Ackermann, L.; Fenner, S. Ruthenium-Catalyzed C–H/N–O Bond Functionalization: Green Isoquinolone Syntheses in Water. Org. Lett. 2011, 13, 6548–6551. [Google Scholar] [CrossRef]
- Parker, A.; Lamata, P.; Viguri, F.; Rodríguez, R.; López, J.A.; Lahoz, F.J.; García-Orduña, P.; Carmona, D. Half-sandwich complexes of osmium containing guanidine-derived ligands. Dalton Trans. 2020, 49, 13601–13617. [Google Scholar] [CrossRef]
- Ahmed, T.J.; Knapp, S.M.; Tyler, D.R. Frontiers in catalytic nitrile hydration: Nitrile and cyanohydrin hydration catalyzed by homogeneous organometallic complexes. Coord. Chem. Rev. 2011, 255, 949–974. [Google Scholar] [CrossRef]
- García-Álvarez, R.; Crochet, P.; Cadierno, V.; Cadierno-Menéndez, V. Metal-catalyzed amide bond forming reactions in an environmentally friendly aqueous medium: Nitrile hydrations and beyond. Green Chem. 2012, 15, 46–66. [Google Scholar] [CrossRef]
- García-Álvarez, R.; Francos, J.; Tomás-Mendivil, E.; Crochet, P.; Cadierno, V. Metal-catalyzed nitrile hydration reactions: The specific contribution of ruthenium. J. Organomet. Chem. 2014, 771, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Sarbaina, A.; Bera, J.K. Bifunctional organometallic catalysts for selective hydration of nitriles to amides. J. Indian Chem. Soc. 2018, 95, 853–861. [Google Scholar]
- González-Fernández, R.; Crochet, P.; Cadierno, V. Arene-ruthenium(II) and osmium(II) complexes as catalysts for nitrile hydration and aldoxime rearrangement reactions. Inorg. Chim. Acta 2021, 517, 120180. [Google Scholar] [CrossRef]
- Ashraf, S.M.; Kandioller, W.; Mendoza-Ferri, M.-G.; Nazarov, A.A.; Hartinger, C.G.; Keppler, B.K. The hydration of chloroacetonitriles catalyzed by mono- and dinuclear RuII- and OsII-arene complexes. Chem. Biodivers. 2008, 5, 2060–2066. [Google Scholar] [CrossRef]
- Cavarzan, A.; Scarso, A.; Strukul, G. Efficient nitrile hydration mediated by RuII catalysts in micellar media. Green Chem. 2010, 12, 790–794. [Google Scholar] [CrossRef]
- Buil, M.L.; Cadierno, V.; Esteruelas, M.A.; Gimeno, J.; Herrero, J.; Izquierdo, S.; Oñate, E.; Cadierno-Menéndez, V. Selective Hydration of Nitriles to Amides Promoted by an Os–NHC Catalyst: Formation and X-ray Characterization of κ2-Amidate Intermediates. Organometallics 2012, 31, 6861–6867. [Google Scholar] [CrossRef]
- Tomás-Mendivil, E.; Cadierno, V.; Menéndez, M.I.; López, R.; Cadierno-Menéndez, V. Unmasking the Action of Phosphinous Acid Ligands in Nitrile Hydration Reactions Catalyzed by Arene-Ruthenium(II) Complexes. Chem. A Eur. J. 2015, 21, 16874–16886. [Google Scholar] [CrossRef]
- González-Fernández, R.; Crochet, P.; Cadierno, V.; Menéndez, M.I.; López, R. Phosphinous Acid-Assisted Hydration of Nitriles: Understanding the Controversial Reactivity of Osmium and Ruthenium Catalysts. Chem. A Eur. J. 2017, 23, 15210–15221. [Google Scholar] [CrossRef]
- González-Fernández, R.; Álvarez, D.; Crochet, P.; Cadierno, V.; Menéndez, M.I.; López, R. Catalytic hydration of cyanamides with phosphinous acid-based ruthenium(ii) and osmium(ii) complexes: Scope and mechanistic insights. Catal. Sci. Technol. 2020, 10, 4084–4098. [Google Scholar] [CrossRef]
- Cadierno, V. Synthetic applications of the Parkins nitrile hydration catalyst [PtH{(PMe2O)2H}(PMe2OH)]: A review. Appl. Sci. 2015, 5, 380. [Google Scholar] [CrossRef] [Green Version]
- González-Fernández, R.; Crochet, P.; Cadierno, V. Cymene-osmium(II) complexes with amino-phosphane ligands as precat-alysts for nitrile hydration reactions. ChemistrySelect 2018, 3, 4324–4329. [Google Scholar] [CrossRef]
- Francos, J.; González-Liste, P.J.; Menéndez-Rodríguez, L.; Crochet, P.; Cadierno, V.; Borge, J.; Antiñolo, A.; Fernández-Galán, R.; Carrillo-Hermosilla, F. Half-sandwich guanidinate-osmium(II) complexes: Synthesis and application in the selective de-hydration of aldoximes. Eur. J. Inorg. Chem. 2016, 2016, 393–402. [Google Scholar] [CrossRef]
- Crochet, P.; Cadierno, V. Catalytic synthesis of amides via aldoximes rearrangement. Chem. Commun. 2015, 51, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Debnath, P. Recent Advances in the Synthesis of Amides via Oxime Rearrangements and its Applications. Curr. Org. Synth. 2018, 15, 666–706. [Google Scholar] [CrossRef]
- Reddy, T.N.; Beatriz, A.; Rao, V.J.; de Lima, D.P. Carbonyl compounds´ journey to amide bond formation. Chem. Asian, J. 2019, 14, 344–388. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Rivera, S.A.; Francos, J.; Borge, J.; Cadierno, V. Mononuclear Ruthenium and Osmium Complexes with a Bicyclic Guanidinate Ligand: Synthesis and Catalytic Behavior in Olefin Isomerization Processes. Eur. J. Inorg. Chem. 2017, 2017, 4138–4146. [Google Scholar] [CrossRef]
- Lastra-Barreira, B.; Francos, J.; Crochet, P.; Cadierno, V. Ruthenium(IV) catalysts for the selective estragole to trans-anethole isomerization in environmentally friendly media. Green Chem. 2011, 13, 307–313. [Google Scholar] [CrossRef]
- Hassam, M.; Taher, A.; Arnott, G.; Green, I.R.; Van Otterlo, W.A.L. Isomerization of Allylbenzenes. Chem. Rev. 2015, 115, 5462–5569. [Google Scholar] [CrossRef]
- Brunner, H.; Zwack, T.; Zabel, M.; Beck, W.; Böhm, A. Optically active transition metal complexes. Synthesis, crystal structures, and catalytic properties of chiral-at-metal (η6-arene)ruthenium(II) and (η6-arene)osmium(II) half-sandwich com-plexes. Crystallization of pure diastereoisomers versus diastereomer mixtures in a 1:1 ratio. Organometallics 2003, 22, 1741–1750. [Google Scholar]
- Vinogradov, M.M.; Afanasyev, O.I.; Nelyubina, Y.V.; Denisov, G.L.; Loginov, D.A.; Chusov, D. Osmium catalysis in the re-ductive amination using carbon monoxide as a reducing agent. Mol. Catal. 2020, 498, 111260. [Google Scholar] [CrossRef]
Scheme 1.
Simplified catalytic cycle for the transfer hydrogenation reactions catalyzed by the Noyori´s catalyst (S,S)-1.
Scheme 1.
Simplified catalytic cycle for the transfer hydrogenation reactions catalyzed by the Noyori´s catalyst (S,S)-1.

Figure 1.
Transition state for the transfer hydrogenation of ketones with the Noyori´s catalyst (S,S)-1.
Figure 1.
Transition state for the transfer hydrogenation of ketones with the Noyori´s catalyst (S,S)-1.

Scheme 2.
Asymmetric transfer hydrogenation of ketones with the chiral Os(II) complexes 4 and 5.

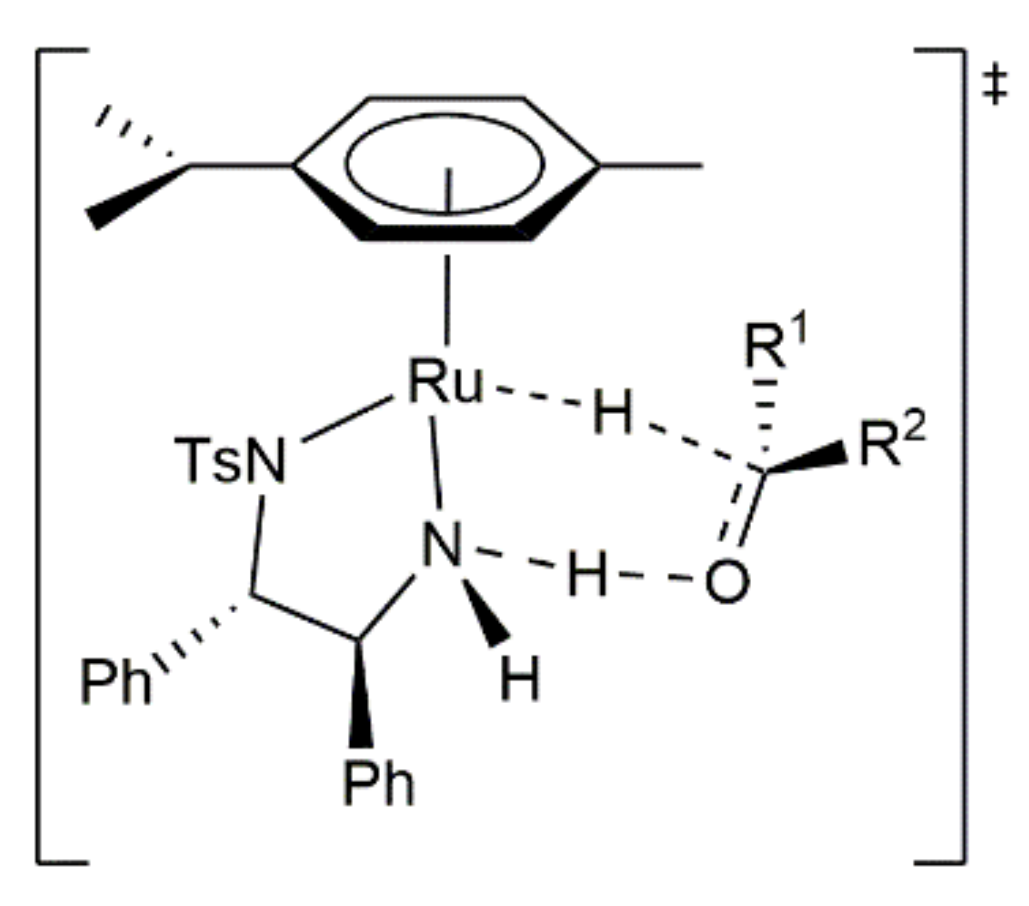
Figure 2.
Structure of the 16-electron complexes (R,R)/(S,S)-6 and the transition state A.

Scheme 3.
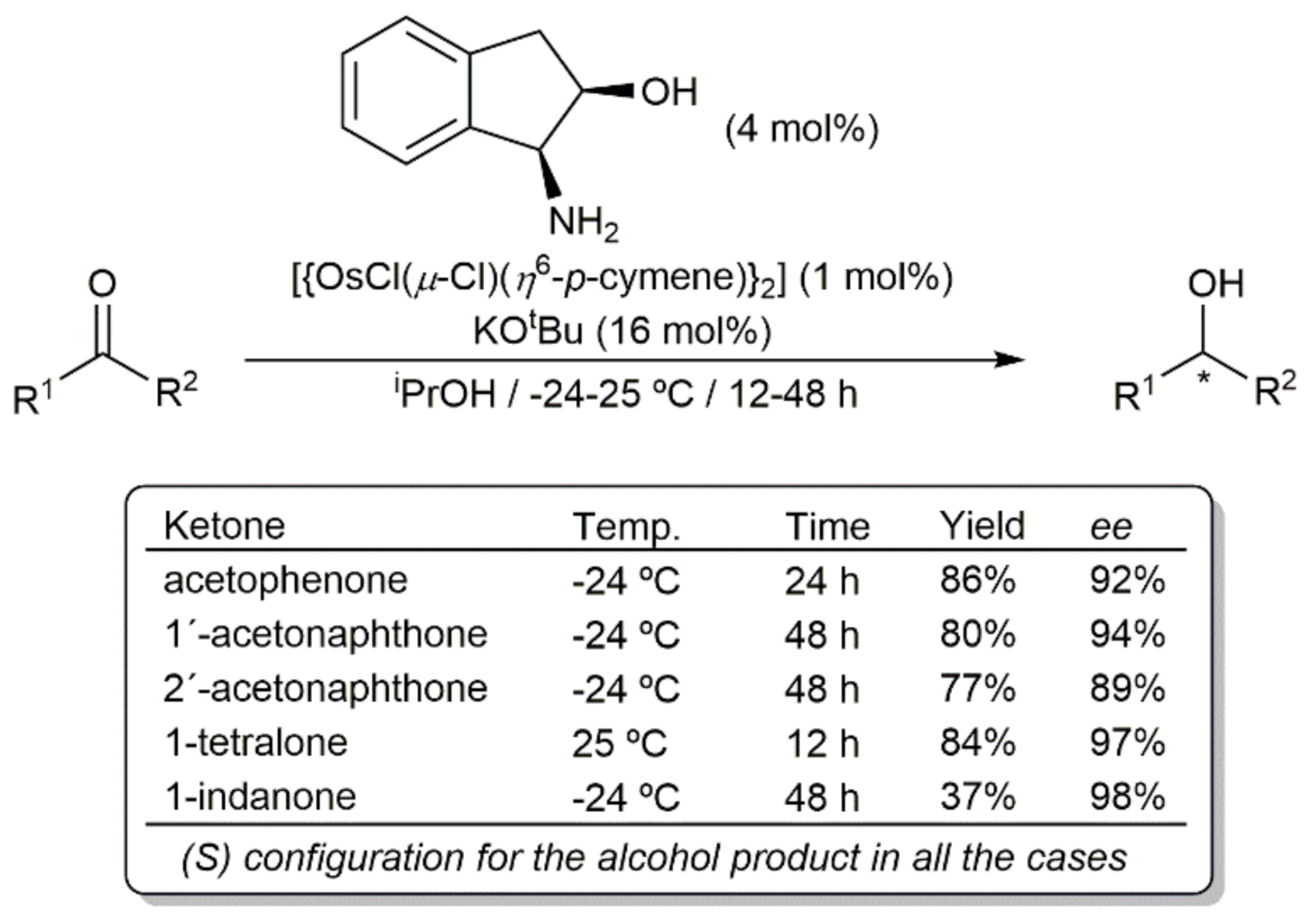
Os-catalyzed asymmetric transfer hydrogenation of ketones by 2-propanol.

Scheme 4.
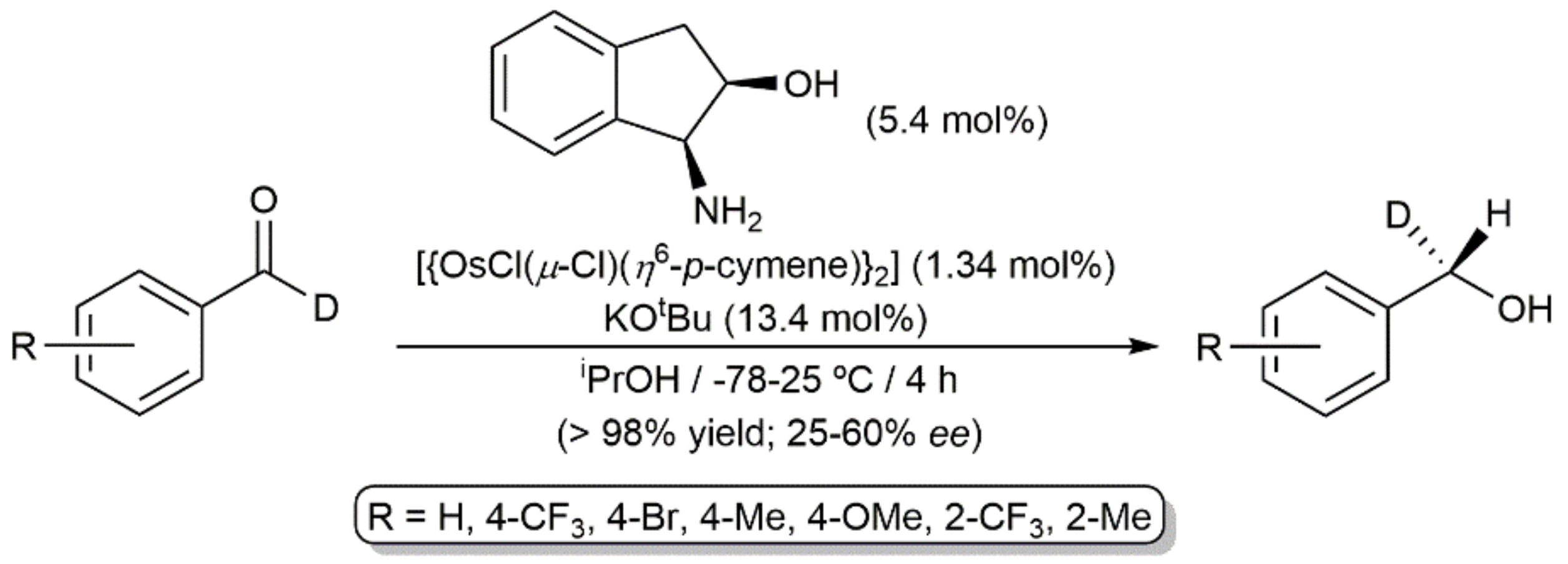
Os-catalyzed asymmetric transfer hydrogenation of benzaldehyde-α-d derivatives.

Figure 3.
Structures of the arene-osmium(II) complexes 7–8a-h with l-α-amino carboxylate ligands.

Figure 4.
Structure of the amino acid derived Os(II) complexes 9 and 10.

Scheme 5.
Os(II)-catalyzed asymmetric transfer hydrogenation of a cyclic imine.

Figure 5.
Structure of arene-osmium(II) complexes with chelated N,N-donor ligands active in the transfer hydrogenation of ketones.
Figure 5.
Structure of arene-osmium(II) complexes with chelated N,N-donor ligands active in the transfer hydrogenation of ketones.

Figure 6.
Structure of the osmium(II) and iridium(III) complexes 18 and 19.

Scheme 6.
Hydrogenation of limonene catalyzed by the β-diketiminato complexes 20–21a-b.

Scheme 7.
Hydrogenation of styrene catalyzed by the triazenide-Os(II) complexes 22–23.

Scheme 8.
Base-free transfer hydrogenation of aldehydes catalyzed by the Os(II) complex 24.

Figure 7.
Structure of the arene complexes 25a-b and 26a-b containing chelating NHC ligands.

Scheme 9.
Transfer hydrogenation of carbonyl compounds catalyzed by the Os(II) complexes 27a–g.

Figure 8.
Structure of the arene-osmium(II) complex 28.

Scheme 10.
Osmium-catalyzed oxidation of acetaldehyde and propionaldehyde with water.

Scheme 11.
Catalytic oxidation of benzyl alcohol using NMO as sacrificial oxidant.

Scheme 12.
Oxidation of styrene catalyzed by arene-osmium(II) complexes.

Scheme 13.
Osmium-catalyzed cyclopropanation of styrene derivatives.

Scheme 14.
Osmium-catalyzed cyclopropanation of isobutyl vinyl ether with 3-tert-butyldiazoindene.

Scheme 15.
Osmium-catalyzed asymmetric Diels–Alder reactions.

Scheme 16.
Os-catalyzed asymmetric Michael addition of 2-methoxycarbonyl-1-indanone to 3-buten-2-one.
Scheme 16.
Os-catalyzed asymmetric Michael addition of 2-methoxycarbonyl-1-indanone to 3-buten-2-one.

Scheme 17.
Olefin metathesis reactions catalyzed by the benzylidene-osmium(II) complexes 37a-b.

Figure 9.
Structure of the ruthenium-alkylidene complexes 38–39a-b and 40.

Scheme 18.
Os-catalyzed α-alkylation of arylacetonitriles and methyl ketones with alcohols.

Scheme 19.
Osmium-catalyzed allylic alkylation processes.

Scheme 20.
Osmium-catalyzed [4 + 2] annulation of N-methoxybenzamides with alkynes.

Scheme 21.
Os-catalyzed Friedel-Crafts reaction between trans-β-nitrostyrene and N-methyl-2-methylindole.
Scheme 21.
Os-catalyzed Friedel-Crafts reaction between trans-β-nitrostyrene and N-methyl-2-methylindole.

Scheme 22.
Osmium-catalyzed hydration of trichloroacetonitrile.

Scheme 23.
Osmium-catalyzed hydration of benzonitrile in an aqueous micellar medium.

Scheme 24.
Catalytic hydration of nitriles employing the 16-electron osmium(II) complex 24.

Scheme 25.
Hydration of nitriles catalyzed by the phosphinous acid-osmium(II) complex 52a.

Scheme 26.
Osmium-catalyzed hydration of cyanamides to ureas.

Figure 10.
Structure of the cymene-osmium(II) complexes 53–55a-b with amino-phosphine ligands.

Scheme 27.
Osmium-catalyzed dehydration of aldoximes.

Scheme 28.
Isomerization of estragole into anethole using a guanidinate-Os(II) catalyst.

Scheme 29.
Catalytic isomerization of 2-n-butyl-4,7-dihydro-1,3-dioxepin 59.

Scheme 30.
Osmium-catalyzed reductive amination of carbonyl compounds.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Crochet, P.; Cadierno, V. Arene-Osmium(II) Complexes in Homogeneous Catalysis. Inorganics 2021, 9, 55. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9070055
AMA Style
Crochet P, Cadierno V. Arene-Osmium(II) Complexes in Homogeneous Catalysis. Inorganics. 2021; 9(7):55. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9070055
Chicago/Turabian StyleCrochet, Pascale, and Victorio Cadierno. 2021. "Arene-Osmium(II) Complexes in Homogeneous Catalysis" Inorganics 9, no. 7: 55. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9070055
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.