
Triiodothyronine or Antioxidants Block the Inhibitory Effects of BDE-47 and BDE-49 on Axonal Growth in Rat Hippocampal Neuron-Glia Co-Cultures


Abstract
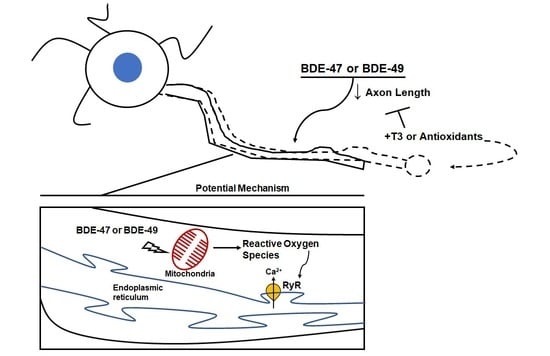
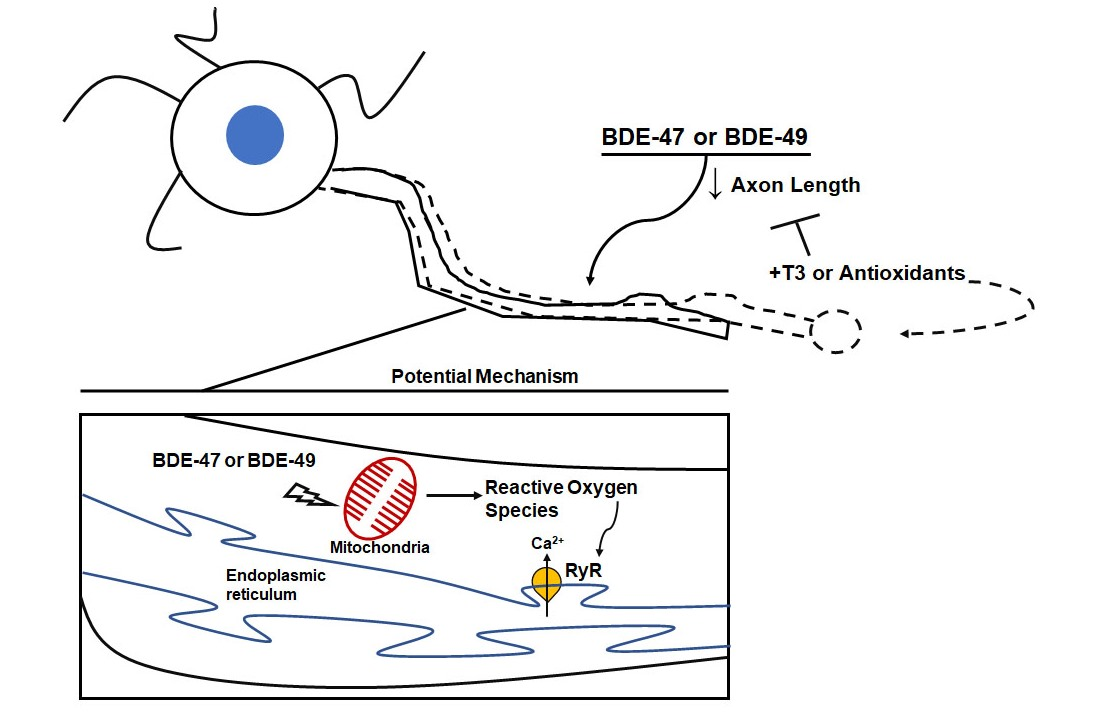
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Cell Culture
2.4. Quantification of Axonal Outgrowth
2.5. Quantitative Polymerase Chain Reaction (qPCR)
2.6. ROS Measurements
2.7. Mitochondrial Metabolism Kinetics
2.8. Statistics
3. Results
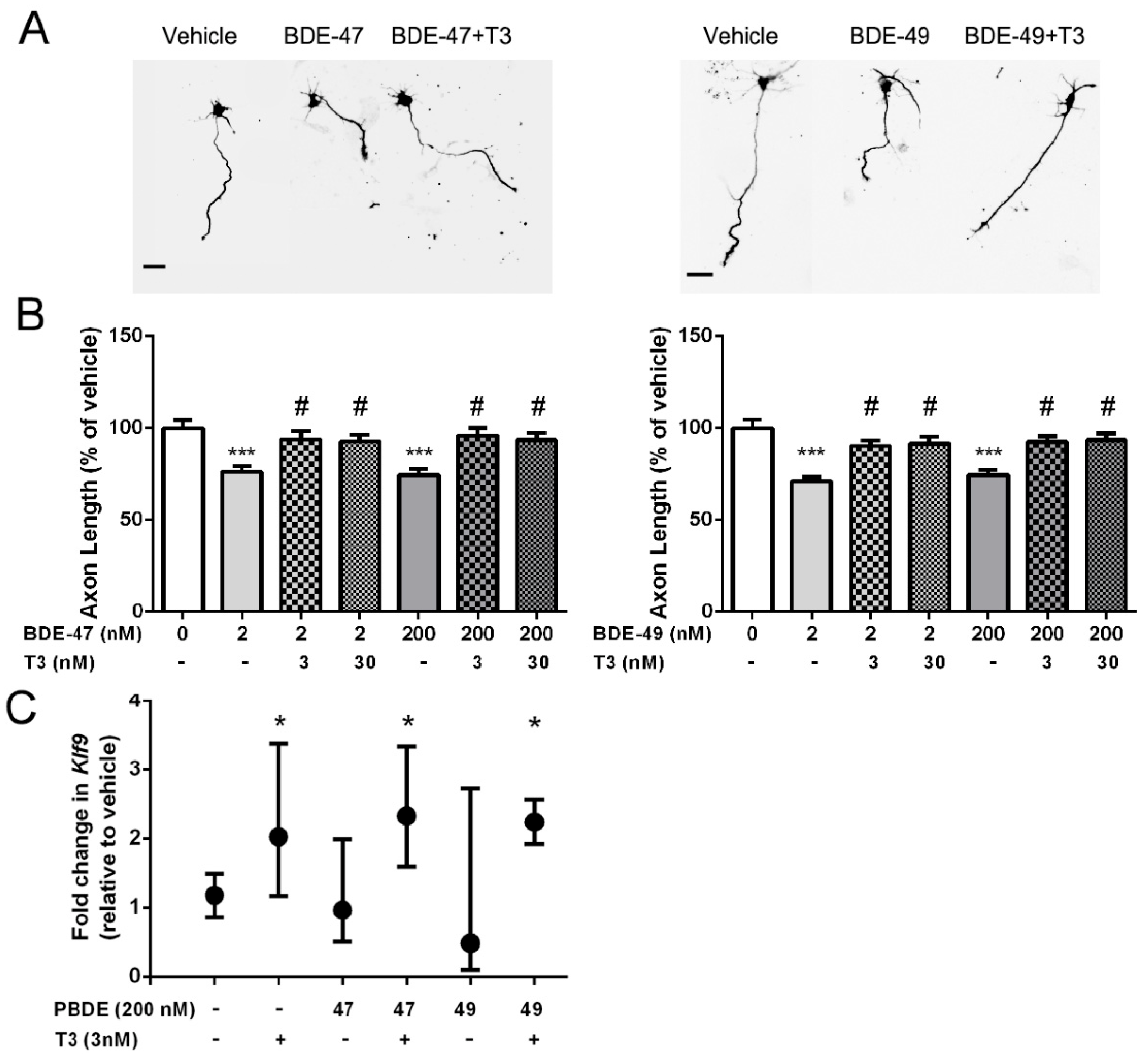
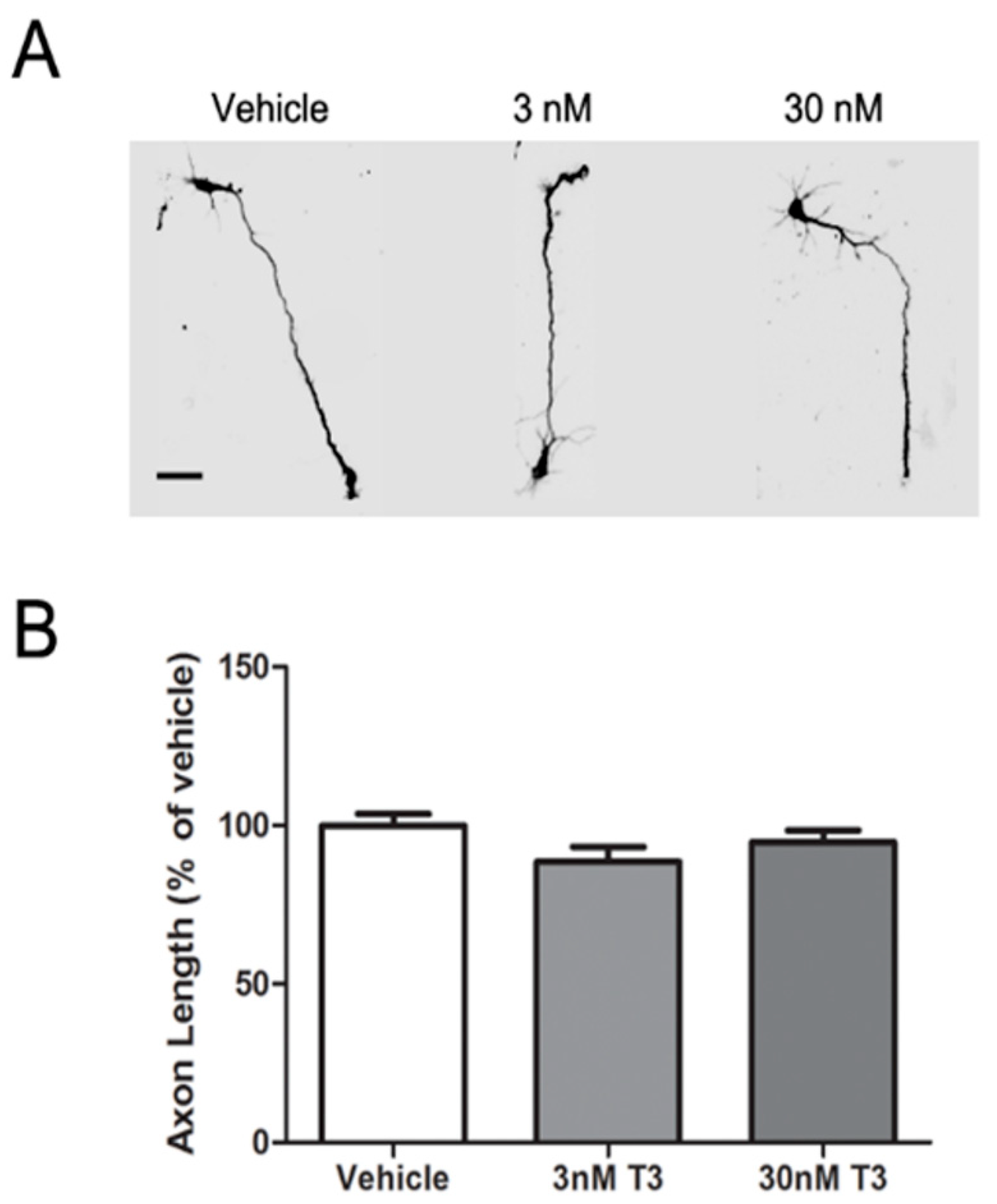
3.1. T3 Blocked the Axon Inhibitory Effects of BDE-47 and BDE-49
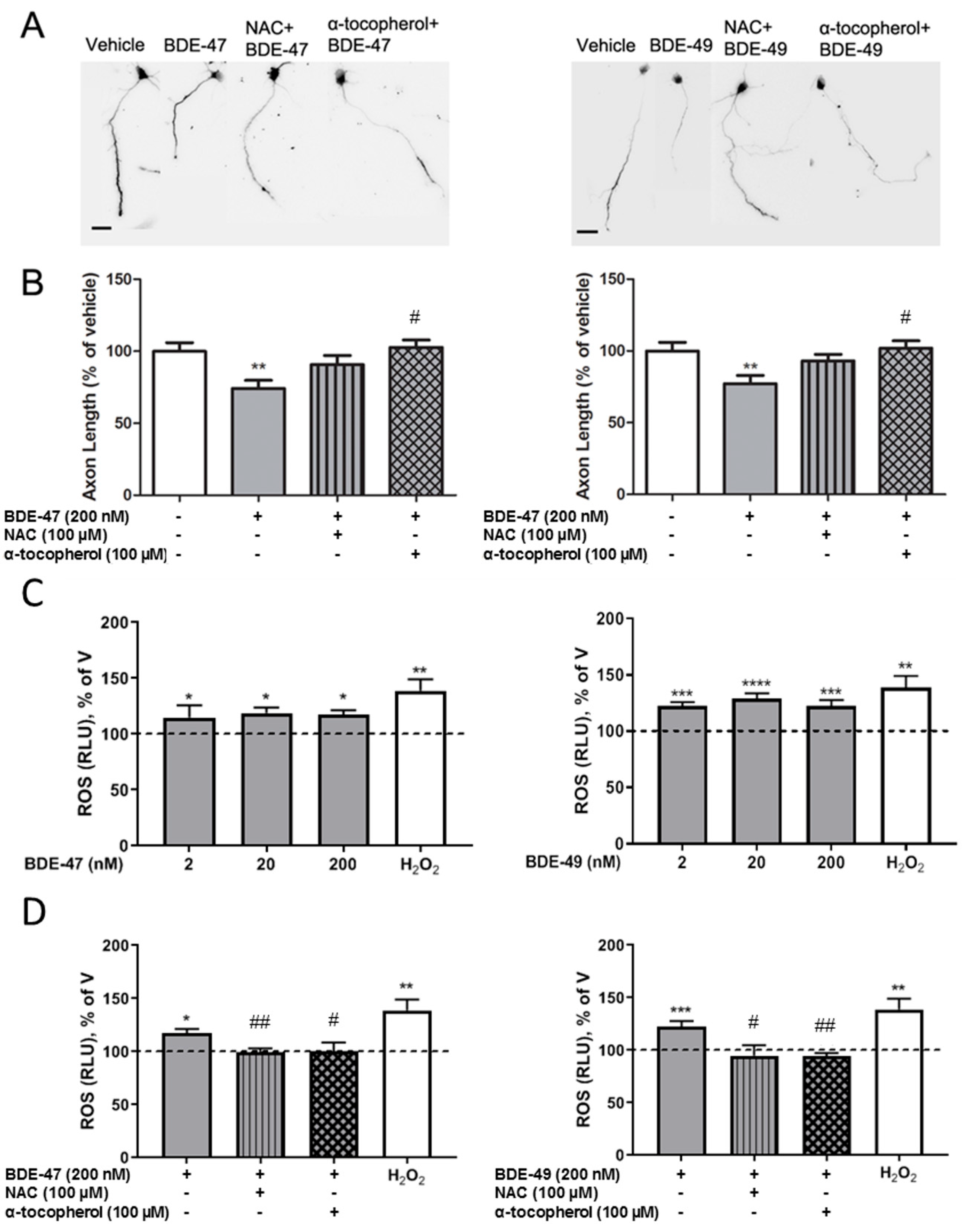
3.2. Antioxidants Blocked PBDE Inhibition of Axonal Growth
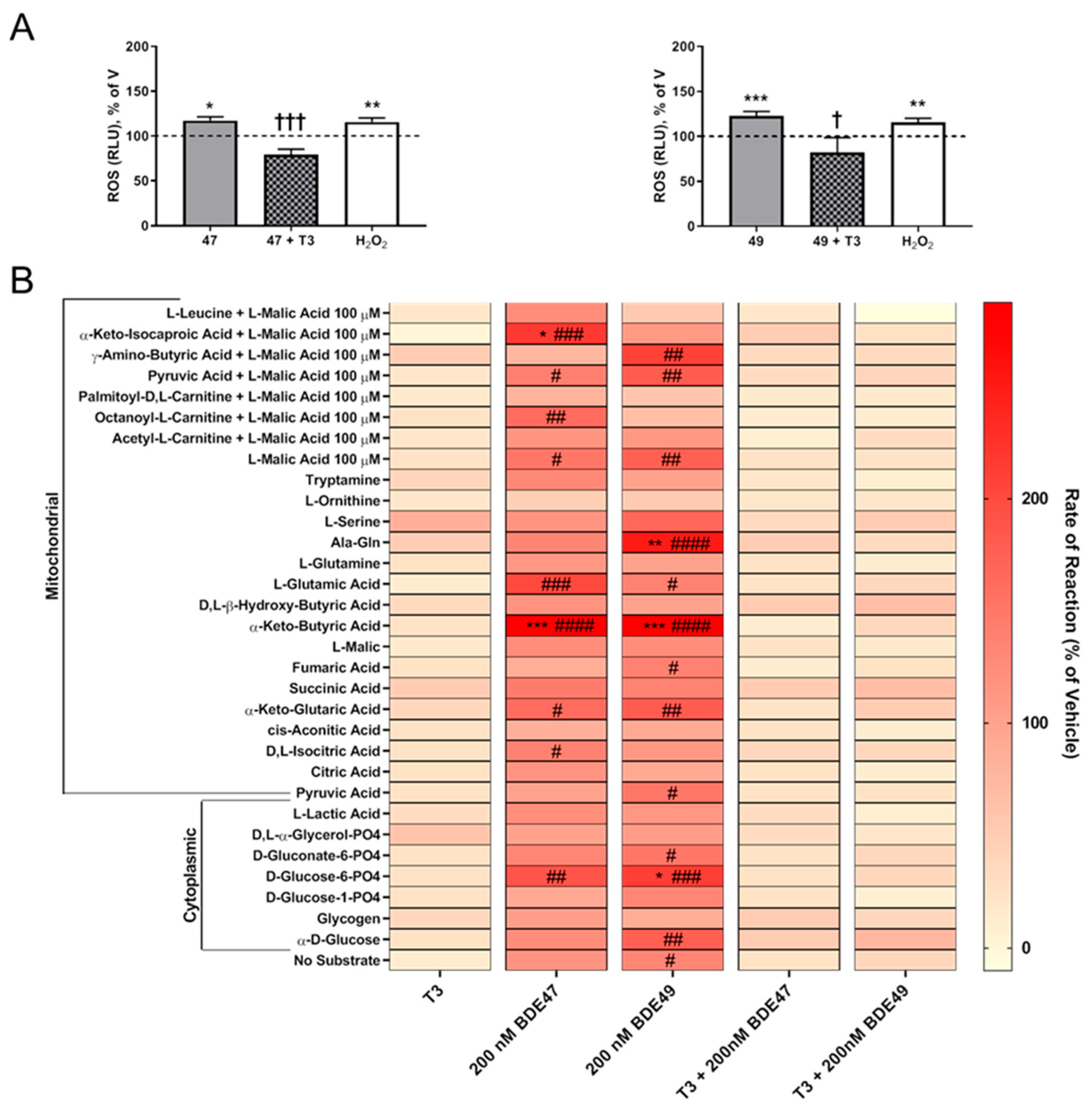
3.3. T3 Blocked PBDE Axon Inhibition by Blocking PBDE-Induced ROS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lam, J.; Lanphear, B.P.; Bellinger, D.; Axelrad, D.A.; McPartland, J.; Sutton, P.; Davidson, L.; Daniels, N.; Sen, S.; Woodruff, T.J. Developmental PBDE Exposure and IQ/ADHD in Childhood: A Systematic Review and Meta-analysis. Environ. Health Perspect. 2017, 125, 086001. [Google Scholar] [CrossRef]
- Vuong, A.M.; Braun, J.M.; Webster, G.M.; Thomas Zoeller, R.; Hoofnagle, A.N.; Sjödin, A.; Yolton, K.; Lanphear, B.P.; Chen, A. Polybrominated Diphenyl Ether (PBDE) Exposures and Thyroid Hormones in Children at Age 3 Years. Environ. Int. 2018, 117, 339–347. [Google Scholar] [CrossRef]
- Gibson, E.A.; Siegel, E.L.; Eniola, F.; Herbstman, J.B.; Factor-Litvak, P. Effects of Polybrominated Diphenyl Ethers on Child Cognitive, Behavioral, and Motor Development. Int. J. Environ. Res. Public Health 2018, 15, 1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorman, D.C.; Chiu, W.; Hales, B.F.; Hauser, R.; Johnson, K.J.; Mantus, E.; Martel, S.; Robinson, K.A.; Rooney, A.A.; Rudel, R.; et al. Polybrominated Diphenyl Ether (PBDE) Neurotoxicity: A Systematic Review and Meta-Analysis of Animal Evidence. J. Toxicol. Environ. Health B Crit. Rev. 2018, 21, 269–289. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.R.; Tsou, T.C.; Huang, H.L.; Chang-Chien, G.P. Levels of Breast Milk Pbdes from Southern Taiwan and Their Potential Impact on Neurodevelopment. Pediatr. Res. 2011, 70, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskenazi, B.; Chevrier, J.; Rauch, S.A.; Kogut, K.; Harley, K.G.; Johnson, C.; Trujillo, C.; Sjodin, A.; Bradman, A. In Utero and Childhood Polybrominated Diphenyl Ether (PBDE) Exposures and Neurodevelopment in the CHAMACOS Study. Environ. Health Perspect. 2013, 121, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Berghuis, S.A.; Bos, A.F.; Sauer, P.J.; Roze, E. Developmental Neurotoxicity of Persistent Organic Pollutants: An Update on Childhood Outcome. Arch. Toxicol. 2015, 89, 687–709. [Google Scholar] [CrossRef]
- Cowell, W.J.; Lederman, S.A.; Sjodin, A.; Jones, R.; Wang, S.; Perera, F.P.; Wang, R.; Rauh, V.A.; Herbstman, J.B. Prenatal Exposure to Polybrominated Diphenyl Ethers and Child Attention Problems at 3–7 Years. Neurotoxicol. Teratol. 2015, 52, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Linares, V.; Belles, M.; Domingo, J.L. Human Exposure to PBDE and Critical Evaluation of Health Hazards. Arch. Toxicol. 2015, 89, 335–356. [Google Scholar] [CrossRef]
- Tsai, M.H.; Chao, H.R.; Hsu, W.L.; Tsai, C.C.; Lin, C.W.; Chen, C.H. Analysis of Polybrominated Diphenyl Ethers and Lipid Composition in Human Breast Milk and Their Correlation with Infant Neurodevelopment. Int. J. Environ. Res. Public Health 2021, 18, 11501. [Google Scholar] [CrossRef]
- Azar, N.; Booij, L.; Muckle, G.; Arbuckle, T.E.; Seguin, J.R.; Asztalos, E.; Fraser, W.D.; Lanphear, B.P.; Bouchard, M.F. Prenatal Exposure to Polybrominated Diphenyl Ethers (PBDEs) and Cognitive Ability in Early Childhood. Environ. Int. 2021, 146, 106296. [Google Scholar] [CrossRef] [PubMed]
- de Water, E.; Curtin, P.; Zilverstand, A.; Sjodin, A.; Bonilla, A.; Herbstman, J.B.; Ramirez, J.; Margolis, A.E.; Bansal, R.; Whyatt, R.M.; et al. A Preliminary Study on Prenatal Polybrominated Diphenyl Ether Serum Concentrations and Intrinsic Functional Network Organization and Executive Functioning in Childhood. J. Child Psychol. Psychiatry 2019, 60, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Schecter, A.; Papke, O.; Tung, K.C.; Joseph, J.; Harris, T.R.; Dahlgren, J. Polybrominated Diphenyl Ether Flame Retardants in the U.S. Population: Current Levels, Temporal Trends, and Comparison with Dioxins, Dibenzofurans, and Polychlorinated Biphenyls. J. Occup. Environ. Med. 2005, 47, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, M.; Vorkamp, K.; Thomsen, M.; Knudsen, L.E. Human Internal and External Exposure to Pbdes—A Review of Levels and Sources. Int. J. Hyg. Environ. Health 2009, 212, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, P.R.; Curras-Collazo, M.C. Neuroendocrine Actions of Organohalogens: Thyroid Hormones, Arginine Vasopressin, and Neuroplasticity. Front. Neuroendocrinol. 2010, 31, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Stamou, M.; Streifel, K.M.; Goines, P.E.; Lein, P.J. Neuronal Connectivity as A Convergent Target of Gene X Environment Interactions that Confer Risk for Autism Spectrum Disorders. Neurotoxicol. Teratol. 2013, 36, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Berger-Sweeney, J.; Hohmann, C.F. Behavioral Consequences of Abnormal Cortical Development: Insights into Developmental Disabilities. Behav. Brain Res. 1997, 86, 121–142. [Google Scholar] [CrossRef]
- Cremer, H.; Chazal, G.; Carleton, A.; Goridis, C.; Vincent, J.D.; Lledo, P.M. Long-Term but not Short-Term Plasticity at Mossy Fiber Synapses is Impaired in Neural Cell Adhesion Molecule-Deficient Mice. Proc. Natl. Acad. Sci. USA 1998, 95, 13242–13247. [Google Scholar] [CrossRef] [Green Version]
- Barone, S., Jr.; Das, K.P.; Lassiter, T.L.; White, L.D. Vulnerable Processes of Nervous System Development: A Review of Markers and Methods. Neurotoxicology 2000, 21, 15–36. [Google Scholar]
- Geschwind, D.H.; Levitt, P. Autism Spectrum Disorders: Developmental Disconnection Syndromes. Curr. Opin. Neurobiol. 2007, 17, 103–111. [Google Scholar] [CrossRef]
- Engle, E.C. Human Genetic Disorders of Axon Guidance. Cold Spring Harb. Protoc. 2010, 2, a001784. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.F.; Chernyak, S.M.; Batterman, S.; Loch-Caruso, R. Polybrominated Diphenyl Ethers in Human Gestational Membranes from Women in Southeast Michigan. Environ. Sci. Technol. 2009, 43, 3042–3046. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Streifel, K.M.; Singh, V.; Yang, D.; Mangini, L.; Wulff, H.; Lein, P.J. From the Cover: BDE-47 and BDE-49 Inhibit Axonal Growth in Primary Rat Hippocampal Neuron-Glia Co-Cultures via Ryanodine Receptor-Dependent Mechanisms. Toxicol. Sci. 2017, 156, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Bose, D.D.; Ghogha, A.; Riehl, J.; Zhang, R.; Barnhart, C.D.; Lein, P.J.; Pessah, I.N. Para- and Ortho-Substitutions are Key Determinants of Polybrominated Diphenyl Ether Activity toward Ryanodine Receptors and Neurotoxicity. Environ. Health Perspect. 2011, 119, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, L.G.; de Laat, R.; Tagliaferri, S.; Pellacani, C. A Mechanistic View of Polybrominated Diphenyl Ether (PBDE) Developmental Neurotoxicity. Toxicol. Lett. 2014, 230, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, H.S.; Westerink, R.H. Neurotoxicity and Risk Assessment of Brominated and Alternative Flame Retardants. Neurotoxicol. Teratol. 2015, 52, 248–269. [Google Scholar] [CrossRef]
- Pessah, I.N.; Cherednichenko, G.; Lein, P.J. Minding the calcium store: Ryanodine Receptor Activation as a Convergent Mechanism of PCB Toxicity. Pharmacol. Ther. 2010, 125, 260–285. [Google Scholar] [CrossRef] [Green Version]
- Batistuzzo, A.; Ribeiro, M.O. Clinical and Subclinical Maternal Hypothyroidism and their Effects on Neurodevelopment, Behavior and Cognition. Arch. Endocrinol. Metab. 2020, 64, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Terzi, A.; Suter, D.M. The Role of NADPH Oxidases in Neuronal Development. Free Radic. Biol. Med. 2020, 154, 33–47. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Brewer, G.J. Isolation and Culture of Adult Rat Hippocampal Neurons. J. Neurosci. Methods 1997, 71, 143–155. [Google Scholar] [CrossRef]
- Brewer, G.J.; Torricelli, J.R.; Evege, E.K.; Price, P.J. Optimized Survival of Hippocampal Neurons in B27-Supplemented Neurobasal, a New Serum-Free Medium Combination. J. Neurosci. Res. 1993, 35, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Stevens, B.; Chang, J.; Milbrandt, J.; Barres, B.A.; Hell, J.W. NS21: Re-Defined and Modified Supplement B27 for Neuronal Cultures. J. Neurosci. Methods 2008, 171, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Meijering, E.; Jacob, M.; Sarria, J.C.; Steiner, P.; Hirling, H.; Unser, M. Design and Validation of a Tool for Neurite Tracing and Analysis in Fluorescence Microscopy Images. Cytometry A 2004, 58, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotti, C.G.; Sullivan, C.A.; Banker, G.A. The Establishment of Polarity by Hippocampal Neurons in Culture. J. Neurosci. 1988, 8, 1454–1468. [Google Scholar] [CrossRef] [Green Version]
- Denver, R.J.; Ouellet, L.; Furling, D.; Kobayashi, A.; Fujii-Kuriyama, Y.; Puymirat, J. Basic Transcription Element-Binding Protein (BTEB) is a Thyroid Hormone-Regulated Gene in the Developing Central Nervous System. Evidence for a role in neurite outgrowth. Int. J. Biol. Chem. 1999, 274, 23128–23134. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.E.; Sanchez-Huerta, K.; Wood, C. Mild Thyroid Hormone Insufficiency during Development Compromises Activity-Dependent Neuroplasticity in the Hippocampus of Adult Male Rats. Endocrinology 2016, 157, 774–787. [Google Scholar] [CrossRef]
- Chen, J.; Liufu, C.; Sun, W.; Sun, X.; Chen, D. Assessment of the Neurotoxic Mechanisms of Decabrominated Diphenyl Ether (PBDE-209) in Primary Cultured Neonatal Rat Hippocampal Neurons Includes Alterations in Second Messenger Signaling and Oxidative Stress. Toxicol. Lett. 2010, 192, 431–439. [Google Scholar] [CrossRef]
- Tagliaferri, S.; Caglieri, A.; Goldoni, M.; Pinelli, S.; Alinovi, R.; Poli, D.; Pellacani, C.; Giordano, G.; Mutti, A.; Costa, L.G. Low Concentrations of the Brominated Flame Retardants BDE-47 and BDE-99 Induce Synergistic Oxidative Stress-Mediated Neurotoxicity in Human Neuroblastoma Cells. Toxicol. In Vitro 2010, 24, 116–122. [Google Scholar] [CrossRef]
- Costa, L.G.; Tagliaferri, S.; Roque, P.J.; Pellacani, C. Role of Glutamate Receptors in Tetrabrominated Diphenyl Ether (BDE-47) Neurotoxicity in Mouse Cerebellar Granule Neurons. Toxicol. Lett. 2016, 241, 159–166. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Yin, L.; Shang, Y.; Zhong, Y.; Zhang, X.; Wu, M.; Yu, Z.; Sheng, G.; Fu, J.; Huang, Y. The Combined Effects of BDE47 and Bap on Oxidatively Generated DNA Damage in L02 Cells and the Possible Molecular Mechanism. Mutat. Res. 2011, 721, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Park, H.R.; Kamau, P.W.; Loch-Caruso, R. Involvement of Reactive Oxygen Species in Brominated Diphenyl Ether-47-Induced Inflammatory Cytokine Release from Human Extravillous Trophoblasts in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS Generation and its Regulation: Mechanisms involved in H2O2 Signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Huang, D.; Zhang, Y. The Involvement of ROS Overproduction and Mitochondrial Dysfunction in PBDE-47-Induced Apoptosis on Jurkat Cells. Exp. Toxicol. Pathol. 2011, 63, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Pazin, M.; Pereira, L.C.; Dorta, D.J. Toxicity of Brominated Flame Retardants, BDE-47 and BDE-99 Stems from Impaired Mitochondrial Bioenergetics. Toxicol. Mech. Methods 2015, 25, 34–41. [Google Scholar] [CrossRef]
- Lefevre, P.L.; Wade, M.; Goodyer, C.; Hales, B.F.; Robaire, B. A Mixture Reflecting Polybrominated Diphenyl Ether (PBDE) Profiles Detected in Human Follicular Fluid Significantly Affects Steroidogenesis and Induces Oxidative Stress in a Female Human Granulosa Cell Line. Endocrinology 2016, 157, 2698–2711. [Google Scholar] [CrossRef]
- Napoli, E.; Hung, C.; Wong, S.; Giulivi, C. Toxicity of the Flame-Retardant BDE-49 on Brain Mitochondria and Neuronal Progenitor Striatal Cells Enhanced by a PTEN-Deficient Background. Toxicol. Sci. 2013, 132, 196–210. [Google Scholar] [CrossRef] [Green Version]
- Belles, M.; Alonso, V.; Linares, V.; Albina, M.L.; Sirvent, J.J.; Domingo, J.L.; Sanchez, D.J. Behavioral Effects and Oxidative Status in Brain Regions of Adult Rats Exposed to BDE-99. Toxicol. Lett. 2010, 194, 1–7. [Google Scholar] [CrossRef]
- Costa, L.G.; Pellacani, C.; Dao, K.; Kavanagh, T.J.; Roque, P.J. The Brominated Flame Retardant BDE-47 Causes Oxidative Stress and Apoptotic Cell Death in vitro and in vivo in Mice. Neurotoxicology 2015, 48, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Giordano, G.; Kavanagh, T.J.; Costa, L.G. Neurotoxicity of A Polybrominated Diphenyl Ether Mixture (DE-71) in Mouse Neurons and Astrocytes is Modulated by Intracellular Glutathione Levels. Toxicol. Appl. Pharmacol. 2008, 232, 161–168. [Google Scholar] [CrossRef] [Green Version]
- He, P.; He, W.; Wang, A.; Xia, T.; Xu, B.; Zhang, M.; Chen, X. PBDE-47-Induced Oxidative Stress, DNA Damage and Apoptosis in Primary Cultured Rat Hippocampal Neurons. Neurotoxicology 2008, 29, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Giordano, G.; Costa, L.G. Comparative Cytotoxicity and Intracellular Accumulation of Five Polybrominated Diphenyl Ether Congeners in Mouse Cerebellar Granule Neurons. Toxicol. Sci. 2010, 114, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kuang, G.; Zhao, G.; Wu, X.; Zhang, C.; Lei, R.; Xia, T.; Chen, J.; Wang, Z.; Ma, R.; et al. Involvement of the Mitochondrial P53 Pathway in PBDE-47-Induced SH-SY5Y Cells Apoptosis and its Underlying Activation Mechanism. Food Chem. Toxicol. 2013, 62, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Gonzalez-Billault, C. Regulation of Cytoskeletal Dynamics by Redox Signaling and Oxidative Stress: Implications for Neuronal Development and Trafficking. Front. Cell. Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olguin-Albuerne, M.; Moran, J. ROS Produced by NOX2 Control in vitro Development of Cerebellar Granule Neurons Development. ASN Neuro 2015, 7. [Google Scholar] [CrossRef]
- Wilson, C.; Munoz-Palma, E.; Henriquez, D.R.; Palmisano, I.; Nunez, M.T.; Di Giovanni, S.; Gonzalez-Billault, C. A Feed-Forward Mechanism Involving the NOX Complex and RyR-Mediated Ca2+ Release During Axonal Specification. Neurosci. Res. 2016, 36, 11107–11119. [Google Scholar] [CrossRef]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically Encoded Fluorescent Indicator for Intracellular Hydrogen Peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- Ibhazehiebo, K.; Iwasaki, T.; Kimura-Kuroda, J.; Miyazaki, W.; Shimokawa, N.; Koibuchi, N. Disruption of Thyroid Hormone Receptor-Mediated Transcription and Thyroid Hormone-Induced Purkinje Cell Dendrite Arborization by Polybrominated Diphenyl Ethers. Environ. Health Perspect. 2011, 119, 168–175. [Google Scholar] [CrossRef]
- Denver, R.J.; Williamson, K.E. Identification of A Thyroid Hormone Response Element in the Mouse Kruppel-Like Factor 9 Gene to Explain its Postnatal Expression in the Brain. Endocrinology 2009, 150, 3935–3943. [Google Scholar] [CrossRef] [Green Version]
- Walter, K.M.; Singh, L.; Singh, V.; Lein, P.J. Investigation of NH3 as A Selective Thyroid Hormone Receptor Modulator in Larval Zebrafish (Danio rerio). Neurotoxicology 2021, 84, 96–104. [Google Scholar] [CrossRef]
- Sethi, S.; Morgan, R.K.; Feng, W.; Lin, Y.; Li, X.; Luna, C.; Koch, M.; Bansal, R.; Duffel, M.W.; Puschner, B.; et al. Comparative Analyses of the 12 Most Abundant PCB Congeners Detected in Human Maternal Serum for Activity at the Thyroid Hormone Receptor and Ryanodine Receptor. Environ. Sci. Technol. 2019, 53, 3948–3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.; Chainy, G.B. Thyroid Hormone Influences Antioxidant Defense System in Adult Rat Brain. Neurochem. Res. 2004, 29, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, V.; Tapia, G.; Varela, P.; Romanque, P.; Cartier-Ugarte, D.; Videla, L.A. Thyroid Hormone-Induced Oxidative Stress in Rodents and Humans: A Comparative View and Relation to Redox Regulation of Gene Expression. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 2006, 142, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abe, K.; Milanesi, A.; Liu, Y.-Y.; Brent, G.A. Thyroid Hormone Protects Primary Cortical Neurons Exposed to Hypoxia by Reducing DNA Methylation and Apoptosis. Endocrinology 2019, 160, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular Aspects of Thyroid Hormone Actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [Green Version]
- Davis, P.J.; Lin, H.Y.; Mousa, S.A.; Luidens, M.K.; Hercbergs, A.A.; Wehling, M.; Davis, F.B. Overlapping Nongenomic and Genomic Actions of Thyroid Hormone and Steroids. Steroids 2011, 76, 829–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flamant, F. Futures Challenges in Thyroid Hormone Signaling Research. Front. Endocrinol. 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Thyroid Hormone Action in Mitochondria. J. Mol. Endocrinol. 2001, 26, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Liu, G.; Allen, P.D.; Pessah, I.N. Transmembrane Redox Sensor of Ryanodine Receptor Complex. Int. J. Biol. Chem. 2000, 275, 35902–35907. [Google Scholar] [CrossRef] [Green Version]
- Donoso, P.; Sanchez, G.; Bull, R.; Hidalgo, C. Modulation of Cardiac Ryanodine Receptor Activity by ROS and RNS. Front. Biosci. 2011, 16, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, V.; Csordás, G.; Hajnóczky, G. Interactions between Sarco-Endoplasmic Reticulum and Mitochondria in Cardiac and Skeletal Muscle—Pivotal Roles in Ca2+ and Reactive Oxygen Species Signaling. J. Cell Sci. 2013, 126, 2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Partin, J. Evidence for Mitochondrial Control of Neuronal Polarity. J. Neurosci. Res. 1999, 56, 8–20. [Google Scholar] [CrossRef]
- Wayman, G.A.; Yang, D.; Bose, D.D.; Lesiak, A.; Ledoux, V.; Bruun, D.; Pessah, I.N.; Lein, P.J. PCB-95 Promotes Dendritic Growth via Ryanodine Receptor-Dependent Mechanisms. Environ. Health Perspect. 2012, 120, 997–1002. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Kania-Korwel, I.; Ghogha, A.; Chen, H.; Stamou, M.; Bose, D.D.; Pessah, I.N.; Lehmler, H.J.; Lein, P.J. PCB 136 Atropselectively Alters Morphometric and Functional Parameters of Neuronal Connectivity in Cultured Rat Hippocampal Neurons via Ryanodine Receptor-Dependent Mechanisms. Toxicol. Sci. 2014, 138, 379–392. [Google Scholar] [CrossRef]
- Yui, K.; Sato, A.; Imataka, G. Mitochondrial Dysfunction and Its Relationship with mTOR Signaling and Oxidative Damage in Autism Spectrum Disorders. Mini-Rev. Med. Chem. 2015, 15, 373–389. [Google Scholar] [CrossRef]
- Song, G.; Napoli, E.; Wong, S.; Hagerman, R.; Liu, S.; Tassone, F.; Giulivi, C. Altered Redox Mitochondrial Biology in the Neurodegenerative Disorder Fragile X-Tremor/Ataxia Syndrome: Use of Antioxidants in Precision Medicine. Mol. Med. 2016, 22, 548–559. [Google Scholar] [CrossRef] [Green Version]
- Chevrier, J.; Harley, K.G.; Bradman, A.; Gharbi, M.; Sjodin, A.; Eskenazi, B. Polybrominated Diphenyl Ether (PBDE) Flame Retardants and Thyroid Hormone during Pregnancy. Environ. Health Perspect. 2010, 118, 1444–1449. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Carty, R.K.; Bautista, A.C.; Hayakawa, K.A.; Lein, P.J. Triiodothyronine or Antioxidants Block the Inhibitory Effects of BDE-47 and BDE-49 on Axonal Growth in Rat Hippocampal Neuron-Glia Co-Cultures. Toxics 2022, 10, 92. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10020092
Chen H, Carty RK, Bautista AC, Hayakawa KA, Lein PJ. Triiodothyronine or Antioxidants Block the Inhibitory Effects of BDE-47 and BDE-49 on Axonal Growth in Rat Hippocampal Neuron-Glia Co-Cultures. Toxics. 2022; 10(2):92. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10020092
Chicago/Turabian StyleChen, Hao, Rhianna K. Carty, Adrienne C. Bautista, Keri A. Hayakawa, and Pamela J. Lein. 2022. "Triiodothyronine or Antioxidants Block the Inhibitory Effects of BDE-47 and BDE-49 on Axonal Growth in Rat Hippocampal Neuron-Glia Co-Cultures" Toxics 10, no. 2: 92. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10020092