
Water-Rock Interaction Processes: A Local Scale Study on Arsenic Sources and Release Mechanisms from a Volcanic Rock Matrix


Abstract
:1. Introduction
- Sampling and analysis of water samples from 69 private wells and springs tapping a volcanic aquifer located just north of Rome for the geochemical characterization of groundwater.
- Selective Sequential Extraction operated on selected tuff samples extracted from three quarries within the study area for defining the As distribution among the different solid phases that potentially constitute the aquifer matrix.
- Two batch tests realized on one of the samples analyzed by SSE in order to simulate water-rock interaction processes in different conditions.
2. Materials and Methods
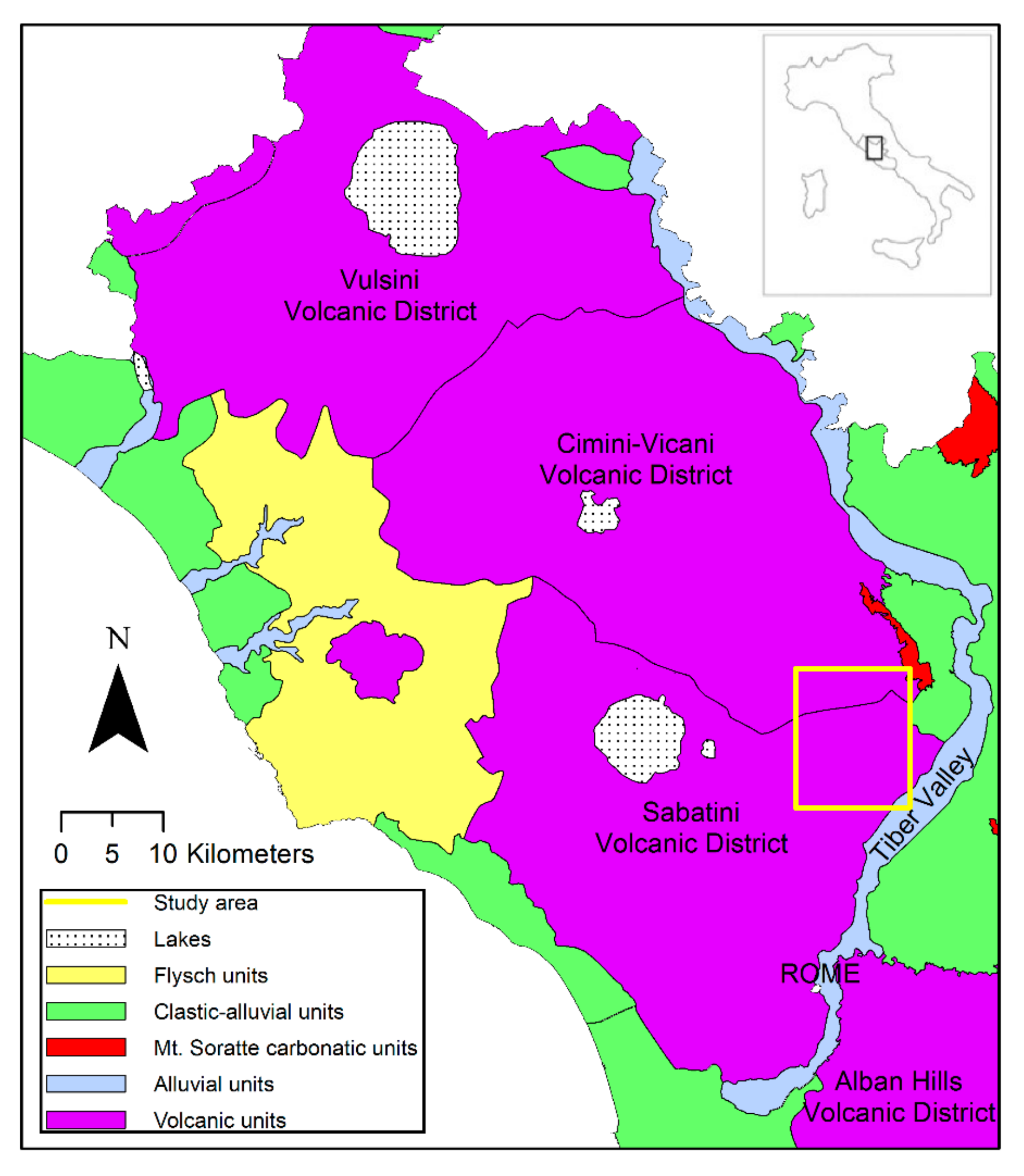
2.1. Study Area
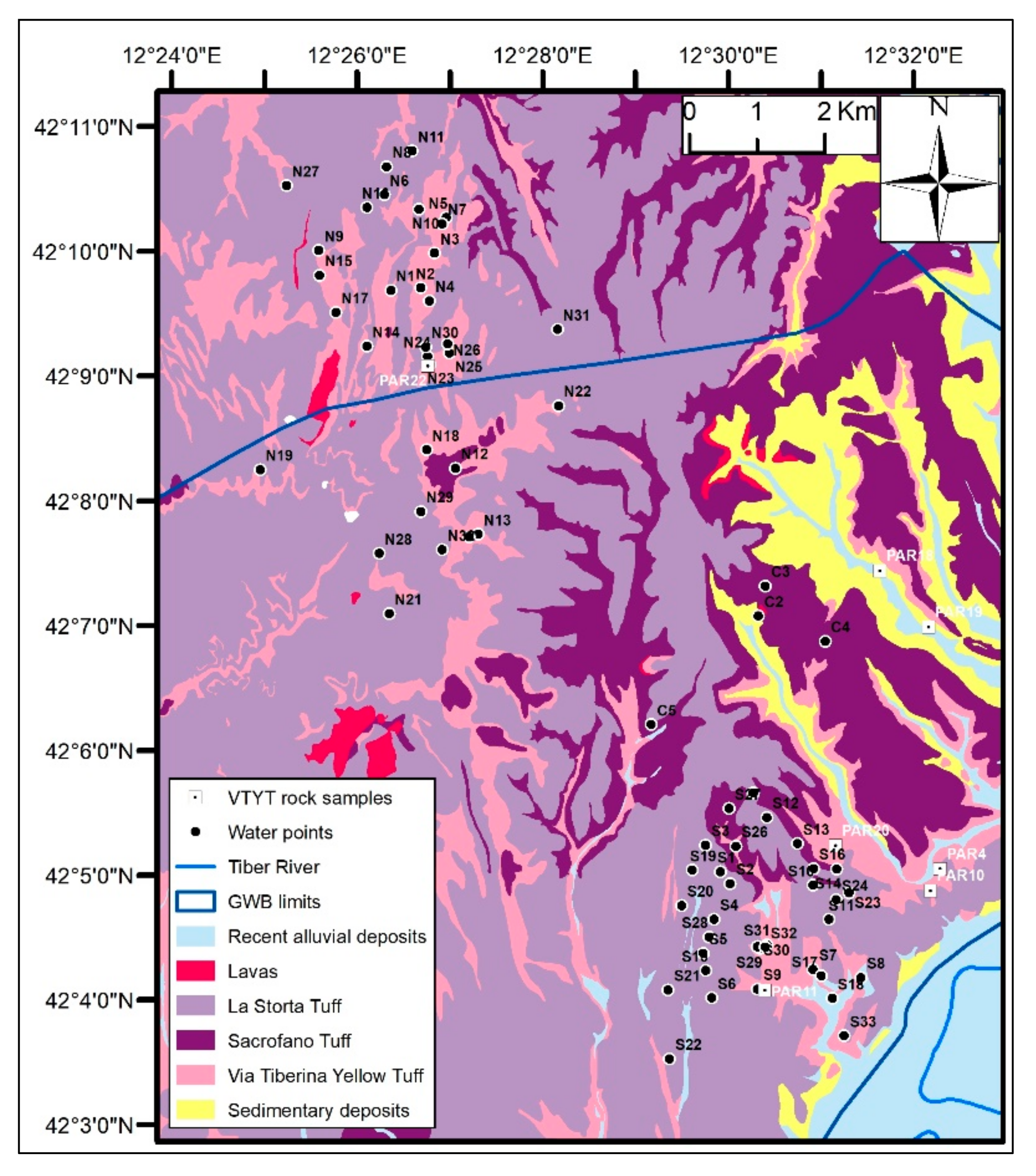
2.2. Groundwater Sampling and Analysis
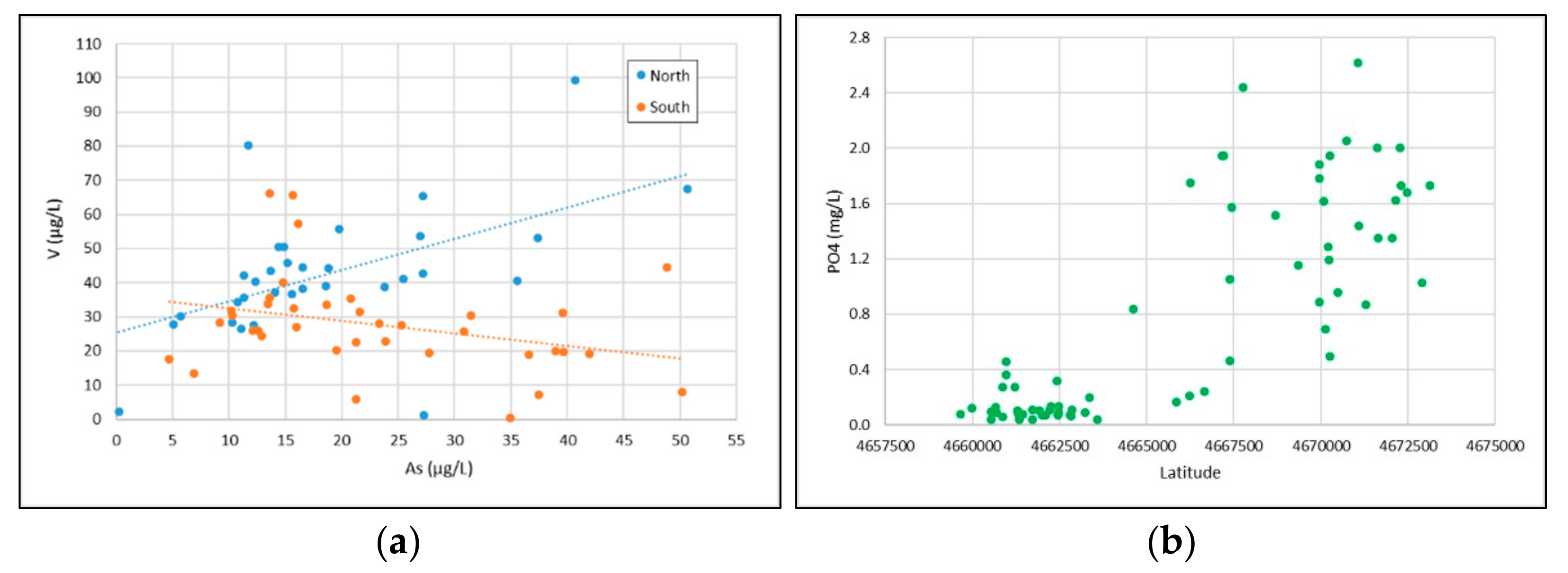
2.3. Statistical Analysis of Groundwater Chemical Data
2.4. Outcropping Rock Sampling and Mineralogical Analysis
2.5. Selective Sequential Extraction
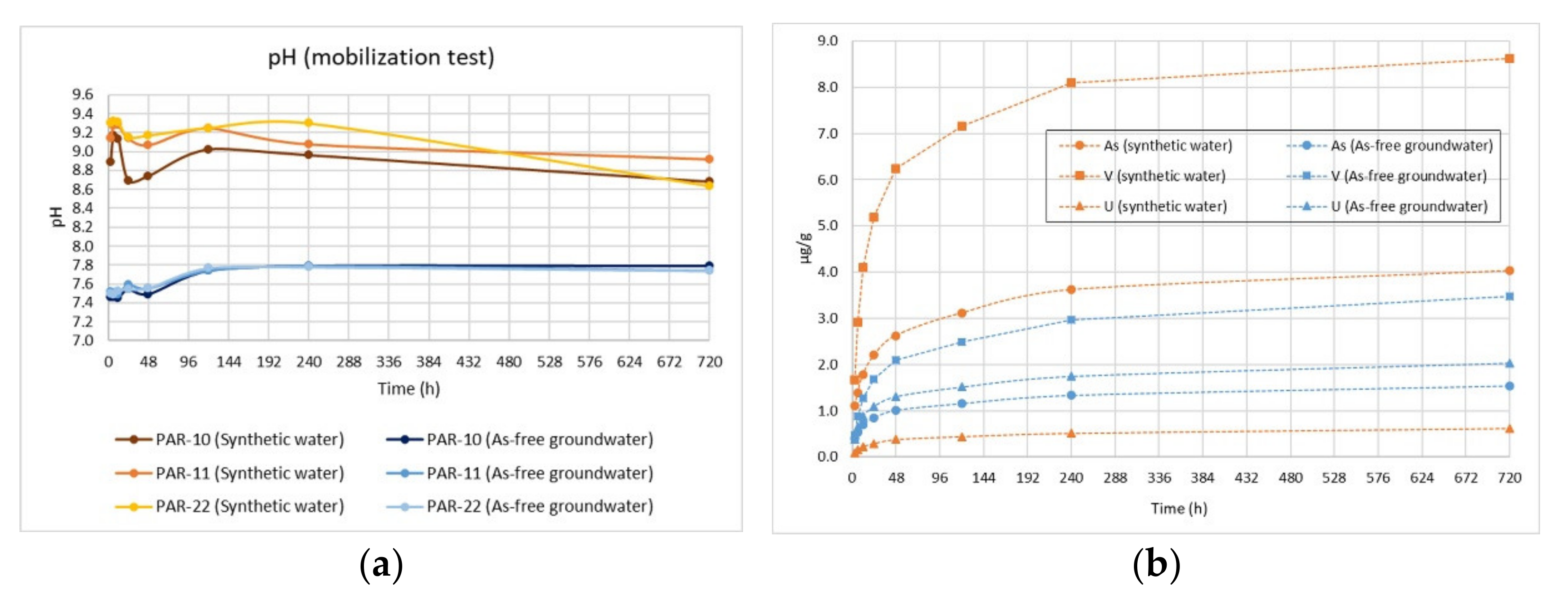
2.6. Batch Tests
- Synthetic water consisting of MilliQ (18.2 MΩ cm at 25 °C) to which KNO3 was added until obtaining a low ionic strength solution (0.5 mM) to simulate contact with rainwater.
- As-free groundwater with ionic strength and physical-chemical characteristics similar to those of groundwater circulating in the study area, simulating the water-rock interaction processes.
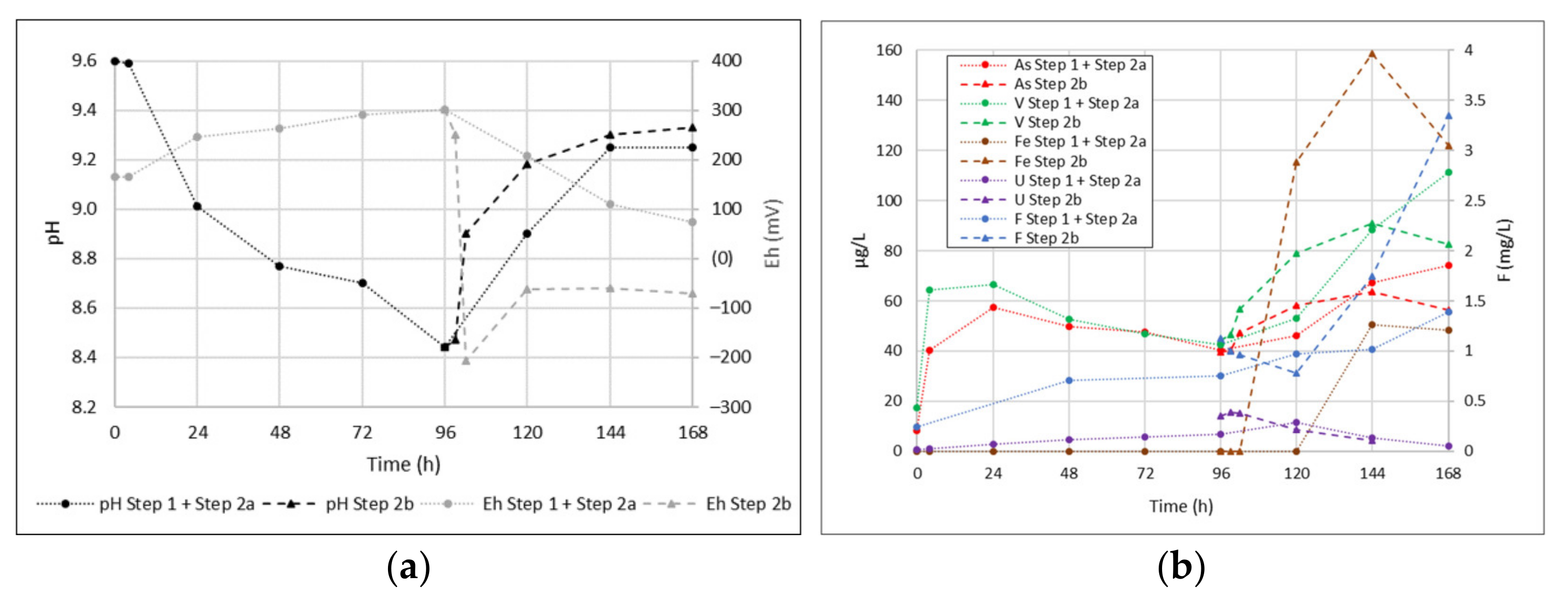
- STEP 1 (dissolution under aerobic conditions): The sample was in solution under oxygenated conditions. We carried out water sampling after 0, 4, 24, 48, 72, and 96 h.
- STEP 2a (induced anaerobic conditions): subsequently, the test continued by blowing N2 within the system in order to eliminate the oxygen present. In this phase, we collected water samples after 24, 48, and 72 h.
- STEP 2b (induced anaerobic condition with sulfide presence): After repeating STEP 1 (4 days in oxygenated conditions with no sampling), the test continued under N2 flux and adding sulfide to reach 0.5 mg/L final concentration. We then collected aliquots at 0, 3, 6, 24, 48, and 72 h.
3. Results and Discussion
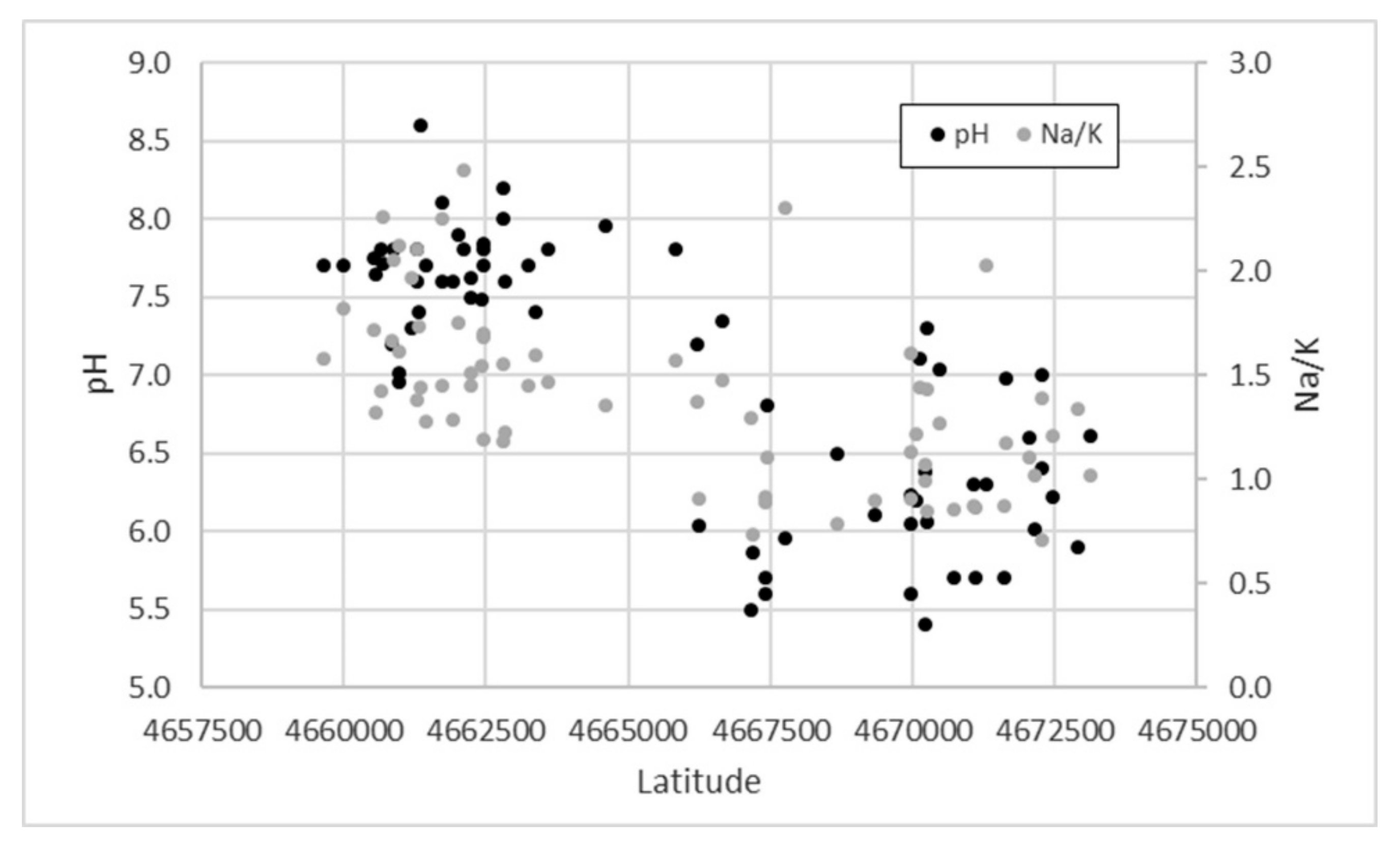
3.1. Groundwater Geochemistry
3.2. Mineralogical Characterization of the Rocks
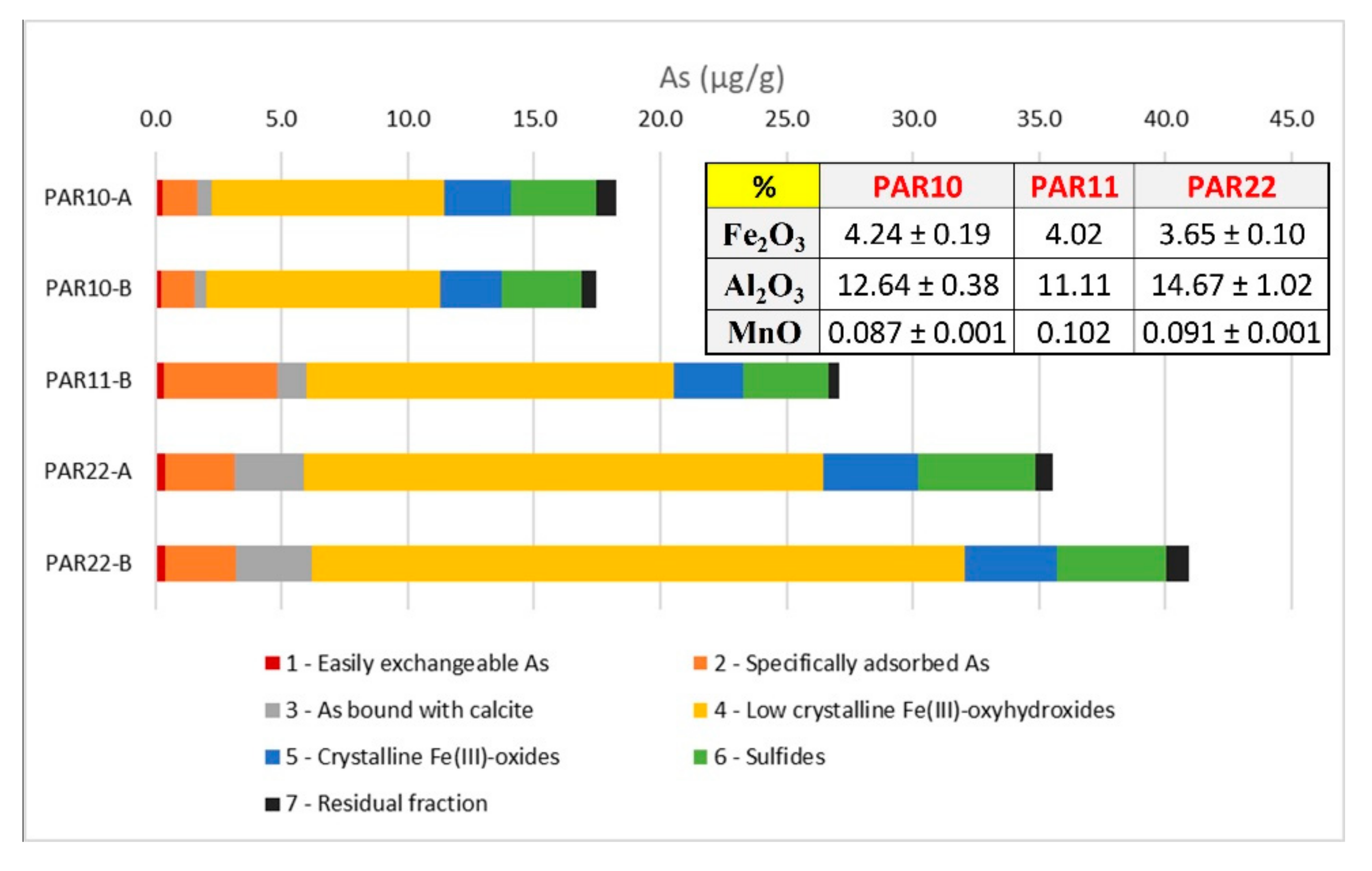
3.3. As Fractioning within the Selected Tuffs
3.4. Batch Experiments
3.5. Arsenic and Other PTEs: Linking Solid Matrix Analysis with Groundwater Geochemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cubadda, F.; D’Amato, M.; Mancini, F.R.; Aureli, F.; Raggi, A.; Busani, L.; Mantovani, A. Assessing Human Exposure to Inorganic Arsenic in High-Arsenic Areas of Latium: A Biomonitoring Study Integrated with Indicators of Dietary Intake. Annu. Ig. Med. Prev. Comun. 2015, 27, 7458. [Google Scholar] [CrossRef]
- Karim, M.M. Arsenic in Groundwater and Health Problems in Bangladesh. Water Res. 2000, 34, 128. [Google Scholar] [CrossRef]
- Rossman, T.G.; Uddin, A.N.; Burns, F.J. Evidence That Arsenite Acts as a Cocarcinogen in Skin Cancer. Toxicol. Appl. Pharmacol. 2004, 198, 457. [Google Scholar] [CrossRef] [PubMed]
- WHO. Guidelines for Drinking-Water Quality, 4th ed.; Incorporating the First Addendum; World Health Organization: Geneva, Switzerland, 2017; p. 4. [Google Scholar]
- European Union. 98/83/EC. In Quality of Water Intented for Human Consumption; European Union: Brussels, Belgium, 1998; p. 3. [Google Scholar]
- Anawar, H.M.; Akai, J.; Komaki, K.; Terao, H.; Yoshioka, T.; Ishizuka, T.; Safiullah, S.; Kato, K. Geochemical Occurrence of Arsenic in Groundwater of Bangladesh: Sources and Mobilization Processes. J. Geochem. Explor. 2003, 77, 273. [Google Scholar] [CrossRef]
- McArthur, J.M.; Banerjee, D.M.; Hudson-Edwards, K.A.; Mishra, R.; Purohit, R.; Ravenscroft, P.; Cronin, A.; Howarth, R.J.; Chatterjee, A.; Talukder, T.; et al. Natural Organic Matter in Sedimentary Basins and Its Relation to Arsenic in Anoxic Ground Water: The Example of West Bengal and Its Worldwide Implications. Appl. Geochem. 2004, 19, 56. [Google Scholar] [CrossRef]
- Nickson, R.T.; Mcarthur, J.M.; Ravenscroft, P.; Burgess, W.G.; Ahmed, K.M. Mechanism of Arsenic Release to Groundwater, Bangladesh and West Bengal. Appl. Geochem. 2000, 15, 86–90. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G. A Review of the Source, Behaviour and Distribution of Arsenic in Natural Waters. Appl. Geochem. 2002, 17, 754. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Verma, S.; Gupta, S.; Henke, K.R.; Bhattacharya, P. Influence of Tectonics, Sedimentation and Aqueous Flow Cycles on the Origin of Global Groundwater Arsenic: Paradigms from Three Continents. J. Hydrol. 2014, 518, 44. [Google Scholar] [CrossRef]
- BGS. Arsenic Contamination of Groundwater. Available online: https://www.bgs.ac.uk/research/groundwater/health/arsenic/home.html (accessed on 12 April 2022).
- Cullen, W.R.; Reimer, K.J. Arsenic Speciation in the Environment. Chem. Rev. 1989, 89, 94. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Bang, S.; Korfiatis, G.P. Effects of Silicate, Sulfate, and Carbonate on Arsenic Removal by Ferric Chloride. Water Res. 2000, 34, 272–283. [Google Scholar] [CrossRef]
- Nicholas, D.R.; Ramamoorthy, S.; Palace, V.; Spring, S.; Moore, J.N.; Rosenzweig, R.F. Biogeochemical Transformations of Arsenic in Circumneutral Freshwater Sediments. Biodegradation 2003, 14, 695. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mulligan, C.N. Effect of Natural Organic Matter on Arsenic Release from Soils and Sediments into Groundwater. Environ. Geochem. Health 2006, 28, 45. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Claesson, M.; Bundschuh, J.; Sracek, O.; Fagerberg, J.; Jacks, G.; Martin, R.A.; Storniolo, A.D.R.; Thir, J.M. Distribution and Mobility of Arsenic in the Río Dulce Alluvial Aquifers in Santiago Del Estero Province, Argentina. Sci. Total Environ. 2006, 358, 48. [Google Scholar] [CrossRef] [PubMed]
- Currell, M.; Cartwright, I.; Raveggi, M.; Han, D. Controls on Elevated Fluoride and Arsenic Concentrations in Groundwater from the Yuncheng Basin, China. Appl. Geochem. 2011, 26, 12. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, K.; Ko, K.S.; Kim, Y.; Lee, K.S. Co-Contamination of Arsenic and Fluoride in the Groundwater of Unconsolidated Aquifers under Reducing Environments. Chemosphere 2012, 87, 25. [Google Scholar] [CrossRef]
- Molinari, A.; Guadagnini, L.; Marcaccio, M.; Straface, S.; Sanchez-Vila, X.; Guadagnini, A. Arsenic Release from Deep Natural Solid Matrices under Experimentally Controlled Redox Conditions. Sci. Total Environ. 2013, 444, 93. [Google Scholar] [CrossRef]
- Nicolli, H.B.; García, J.W.; Falcón, C.M.; Smedley, P.L. Mobilization of Arsenic and Other Trace Elements of Health Concern in Groundwater from the Salí River Basin, Tucumán Province, Argentina. Environ. Geochem. Health 2012, 34, 9429. [Google Scholar] [CrossRef] [Green Version]
- Alarcón-Herrera, M.T.; Bundschuh, J.; Nath, B.; Nicolli, H.B.; Gutierrez, M.; Reyes-Gomez, V.M.; Nuñez, D.; Martín-Dominguez, I.R.; Sracek, O. Co-Occurrence of Arsenic and Fluoride in Groundwater of Semi-Arid Regions in Latin America: Genesis, Mobility and Remediation. J. Hazard. Mater. 2013, 262, 5. [Google Scholar] [CrossRef]
- Francisca, F.M.; Carro Perez, M.E. Assessment of Natural Arsenic in Groundwater in Cordoba Province, Argentina. Environ. Geochem. Health 2009, 31, 9245. [Google Scholar] [CrossRef]
- Morales, I.; Villanueva-Estrada, R.E.; Rodríguez, R.; Armienta, M.A. Geological, Hydrogeological, and Geothermal Factors Associated to the Origin of Arsenic, Fluoride, and Groundwater Temperature in a Volcanic Environment “El Bajío Guanajuatense”, Mexico. Environ. Earth Sci. 2015, 74, 4554–4563. [Google Scholar] [CrossRef]
- Ren, M.; Rodríguez-Pineda, J.A.; Goodell, P. Arsenic Mineral in Volcanic Tuff, a Source of Arsenic Anomaly in Groundwater: City of Chihuahua, Mexico. Geosciences 2022, 12, 69. [Google Scholar] [CrossRef]
- Winkel, L.H.E.; Casentini, B.; Bardelli, F.; Voegelin, A.; Nikolaidis, N.P.; Charlet, L. Speciation of Arsenic in Greek Travertines: Co-Precipitation of Arsenate with Calcite. Geochim. Cosmochim. Acta 2013, 106, 49. [Google Scholar] [CrossRef]
- Di Benedetto, F.; Costagliola, P.; Benvenuti, M.; Lattanzi, P.; Romanelli, M.; Tanelli, G. Arsenic Incorporation in Natural Calcite Lattice: Evidence from Electron Spin Echo Spectroscopy. Earth Planet. Sci. Lett. 2006, 246, 47. [Google Scholar] [CrossRef]
- Baiocchi, A.; Coletta, A.; Espositi, L.; Lotti, F.; Piscopo, V. Sustainable Groundwater Development in a Naturally Arsenic-Contaminated Aquifer: The Case of the Cimino-Vico Volcanic Area (Central Italy). Ital. J. Eng. Geol. Environ. 2013, 13, 5–18. [Google Scholar] [CrossRef]
- Preziosi, E.; Giuliano, G.; Vivona, R. Natural Background Levels and Threshold Values Derivation for Naturally As, V and F Rich Groundwater Bodies: A Methodological Case Study in Central Italy. Environ. Earth Sci. 2010, 61, 404. [Google Scholar] [CrossRef]
- Vivona, R.; Preziosi, E.; Madé, B.; Giuliano, G. Occurrence of Minor Toxic Elements in Volcanic-Sedimentary Aquifers: A Case Study in Central Italy. Hydrogeol. J. 2007, 15, 169. [Google Scholar] [CrossRef]
- Angelone, M.; Cremisini, C.; Piscopo, V.; Proposito, M.; Spaziani, F. Influence of Hydrostratigraphy and Structural Setting on the Arsenic Occurrence in Groundwater of the Cimino-Vico Volcanic Area (Central Italy). Hydrogeol. J. 2009, 17, 403. [Google Scholar] [CrossRef]
- Armiento, G.; Baiocchi, A.; Cremisini, C.; Crovato, C.; Lotti, F.; Lucentini, L.; Mazzuoli, M.; Nardi, E.; Piscopo, V.; Proposito, M.; et al. An Integrated Approach to Identify Water Resources for Human Consumption in an Area Affected by High Natural Arsenic Content. Water 2015, 7, 5091. [Google Scholar] [CrossRef] [Green Version]
- Cinti, D.; Poncia, P.P.; Brusca, L.; Tassi, F.; Quattrocchi, F.; Vaselli, O. Spatial Distribution of Arsenic, Uranium and Vanadium in the Volcanic-Sedimentary Aquifers of the Vicano-Cimino Volcanic District (Central Italy). J. Geochem. Explor. 2015, 152, 8. [Google Scholar] [CrossRef] [Green Version]
- Casentini, B.; Pettine, M.; Millero, F.J. Release of Arsenic from Volcanic Rocks through Interactions with Inorganic Anions and Organic Ligands. Aquat. Geochem. 2010, 16, 9090. [Google Scholar] [CrossRef]
- Lim, M.S.; Yeo, I.W.; Prabhakar Clement, T.; Roh, Y.; Lee, K.K. Mathematical Model for Predicting Microbial Reduction and Transport of Arsenic in Groundwater Systems. Water Res. 2007, 41, 17. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.P.; Itoi, R.; Yamashiro, R. Influence of Redox Potential on Arsenic Release from Soil in the Presence of Iron Oxyhydroxide. Mem. Fac. Eng. Kyushu Univ. 2008, 68, 754. [Google Scholar]
- Razzak, A.; Jinno, K.; Hiroshiro, Y.; Halim, M.A.; Oda, K. Mathematical Modeling of Biologically Mediated Redox Processes of Iron and Arsenic Release in Groundwater. Environ. Geol. 2009, 58, 1517. [Google Scholar] [CrossRef]
- Gao, Y.; Mucci, A. Acid Base Reaction, Phosphate and Arsenate Complexation, and Thier Competitive Adsorption at the Surface of Geothite in 0.7 M Nacl Solution. Geochim. Cosmochim. Acta 2001, 65, 589. [Google Scholar] [CrossRef]
- Frohne, T.; Rinklebe, J.; Diaz-Bone, R.A.; du Laing, G. Controlled Variation of Redox Conditions in a Floodplain Soil: Impact on Metal Mobilization and Biomethylation of Arsenic and Antimony. Geoderma 2011, 160, 12. [Google Scholar] [CrossRef]
- Kumar, M.; Das, N.; Goswami, R.; Sarma, K.P.; Bhattacharya, P.; Ramanathan, A.L. Coupling Fractionation and Batch Desorption to Understand Arsenic and Fluoride Co-Contamination in the Aquifer System. Chemosphere 2016, 164, 75. [Google Scholar] [CrossRef]
- Deuel, L.E.; Swoboda, A.R. Arsenic Solubility in a Reduced Environment. Soil Sci. Soc. Am. J. 1972, 36, 22. [Google Scholar] [CrossRef]
- Hess, R.E.; Blanchar, R.W. Dissolution of Arsenic from Waterlogged and Aerated Soil. Soil Sci. Soc. Am. J. 1977, 41, 9. [Google Scholar] [CrossRef]
- McGeehan, S.L. Arsenic Sorption and Redox Reactions: Relevance to Transport and Remediation. J. Environ. Sci. Health Part A Toxic Hazard. Subst. Environ. Eng. 1996, 31, 6493. [Google Scholar] [CrossRef]
- Tingzong, G.; DeLaune, R.D.; Patrick, W.H. The Influence of Sediment Redox Chemistry on Chemically Active Forms of Arsenic, Cadmium, Chromium, and Zinc in Estuarine Sediment. Environ. Int. 1997, 23, 33. [Google Scholar] [CrossRef]
- Yu, K.; Böhme, F.; Rinklebe, J.; Neue, H.-U.; DeLaune, R.D. Major Biogeochemical Processes in Soils-A Microcosm Incubation from Reducing to Oxidizing Conditions. Soil Sci. Soc. Am. J. 2007, 71, 155. [Google Scholar] [CrossRef] [Green Version]
- Matis, K.A.; Zouboulis, A.I.; Malamas, F.B.; Ramos Afonso, M.D.; Hudson, M.J. Flotation Removal of As (V) onto Goethite. Environ. Pollut. 1997, 97, 91–93. [Google Scholar] [CrossRef]
- Jönsson, J.; Sherman, D.M. Sorption of As (III) and As (V) to Siderite, Green Rust (Fougerite) and Magnetite: Implications for Arsenic Release in Anoxic Groundwaters. Chem. Geol. 2008, 255, 36. [Google Scholar] [CrossRef]
- National Research Council (US) Committee on Medical and Biological Effects of Environmental Pollutants Arsenic. Medical and Biologic Effects of Environmental Pollutants; National Academies Press: Washington, DC, USA, 1977.
- Bombach, G.; Pierra, A.; Klemm, W. Arsenic in Contaminated Soil and River Sediment. Fresenius’ J. Anal. Chem. 1994, 350, 6252. [Google Scholar] [CrossRef]
- Voigt, D.E.; Brantley, S.L.; Hennet, R.J.C. Chemical Fixation of Arsenic in Contaminated Soils. Appl. Geochem. 1996, 11, 9–10. [Google Scholar] [CrossRef]
- Kavanagh, P.J.; Farago, M.E.; Thornton, I.; Braman, R.S. Bioavailability of Arsenic in Soil and Mine Wastes of the Tamar Valley, SW England. Chem. Speciat. Bioavailab. 1997, 9, 3291. [Google Scholar] [CrossRef] [Green Version]
- Hudson-Edwards, K.A.; Houghton, S.L.; Osborn, A. Extraction and Analysis of Arsenic in Soils and Sediments. TrAC Trends Anal. Chem. 2004, 23, 10. [Google Scholar] [CrossRef]
- Bacon, J.R.; Davidson, C.M. Is There a Future for Sequential Chemical Extraction? Analyst 2008, 133, 45. [Google Scholar] [CrossRef]
- Tessier, A.; Campbell, P.G.C.; Bisson, M. Sequential Extraction Procedure for the Speciation of Particulate Trace Metals. Anal. Chem. 1979, 51, 17. [Google Scholar] [CrossRef]
- Shuman, L.M. Fractionation Method for Soil Microelements. Soil Sci. 1985, 140, 3. [Google Scholar] [CrossRef]
- Gleyzes, C.; Tellier, S.; Sabrier, R.; Astruc, M. Arsenic Characterisation in Industrial Soils by Chemical Extractions. Environ. Technol. 2001, 22, 8313. [Google Scholar] [CrossRef] [PubMed]
- Keon, N.E.; Swartz, C.H.; Brabander, D.J.; Harvey, C.; Hemond, H.F. Validation of an Arsenic Sequential Extraction Method for Evaluating Mobility in Sediments. Environ. Sci. Technol. 2001, 35, 1511. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, W.W.; Kirchbaumer, N.; Prohaska, T.; Stingeder, G.; Lombi, E.; Adriano, D.C. Arsenic Fractionation in Soils Using an Improved Sequential Extraction Procedure. Anal. Chim. Acta 2001, 436, 924–926. [Google Scholar] [CrossRef]
- Pagnanelli, F.; Moscardini, E.; Giuliano, V.; Toro, L. Sequential Extraction of Heavy Metals in River Sediments of an Abandoned Pyrite Mining Area: Pollution Detection and Affinity Series. Environ. Pollut. 2004, 132, 2. [Google Scholar] [CrossRef] [PubMed]
- Claff, S.R.; Sullivan, L.A.; Burton, E.D.; Bush, R.T. A Sequential Extraction Procedure for Acid Sulfate Soils: Partitioning of Iron. Geoderma 2010, 155, 2. [Google Scholar] [CrossRef]
- Matera, V.; le Hécho, I.; Laboudigue, A.; Thomas, P.; Tellier, S.; Astruc, M. A Methodological Approach for the Identification of Arsenic Bearing Phases in Polluted Soils. Environ. Pollut. 2003, 126, 146–151. [Google Scholar] [CrossRef]
- Costagliola, P.; Bardelli, F.; Benvenuti, M.; di Benedetto, F.; Lattanzi, P.; Romanelli, M.; Paolieri, M.; Rimondi, V.; Vaggelli, G. Arsenic-Bearing Calcite in Natural Travertines: Evidence from Sequential Extraction, ΜxAS, and ΜxRF. Environ. Sci. Technol. 2013, 47, 4953a. [Google Scholar] [CrossRef]
- Dold, B. Speciation of the Most Soluble Phases in a Sequential Extraction Procedure Adapted for Geochemical Studies of Copper Sulfide Mine Waste. J. Geochem. Explor. 2003, 80, 182–183. [Google Scholar] [CrossRef]
- Ahn, J.S.; Park, Y.S.; Kim, J.Y.; Kim, K.W. Mineralogical and Geochemical Characterization of Arsenic in an Abandoned Mine Tailings of Korea. Environ. Geochem. Health 2005, 27, 121–129. [Google Scholar] [CrossRef]
- Damris, M.; O’Brien, G.A.; Price, W.E.; Chenhall, B.E. Fractionation of Sedimentary Arsenic from Port Kembla Harbour, NSW, Australia. J. Environ. Monit. 2005, 7, 393b. [Google Scholar] [CrossRef] [Green Version]
- Van Elteren, J.T.; Šlejkovec, Z.; Arčon, I.; Glass, H.J. An Interdisciplinary Physical-Chemical Approach for Characterization of Arsenic in a Calciner Residue Dump in Cornwall (UK). Environ. Pollut. 2006, 139, 3. [Google Scholar] [CrossRef] [PubMed]
- Parrone, D.; Ghergo, S.; Preziosi, E. A Multi-Method Approach for the Assessment of Natural Background Levels in Groundwater. Sci. Total Environ. 2019, 659, 350. [Google Scholar] [CrossRef] [PubMed]
- Conticelli, S.; Francalanci, L.; Manetti, P.; Cioni, R.; Sbrana, A. Petrology and Geochemistry of the Ultrapotassic Rocks from the Sabatini Volcanic District, Central Italy: The Role of Evolutionary Processes in the Genesis of Variably Enriched Alkaline Magmas. J. Volcanol. Geotherm. Res. 1997, 75, 107–136. [Google Scholar] [CrossRef]
- Sottili, G.; Palladino, D.M.; Marra, F.; Jicha, B.; Karner, D.B.; Renne, P. Geochronology of the Most Recent Activity in the Sabatini Volcanic District, Roman Province, Central Italy. J. Volcanol. Geotherm. Res. 2010, 196, 20–30. [Google Scholar] [CrossRef]
- Masotta, M.; Gaeta, M.; Gozzi, F.; Marra, F.; Palladino, D.M.; Sottili, G. H2O- and Temperature-Zoning in Magma Chambers: The Example of the Tufo Giallo Della Via Tiberina Eruptions (Sabatini Volcanic District, Central Italy). Lithos 2010, 118, 119–130. [Google Scholar] [CrossRef]
- Capelli, G.; Mazza, R.; Gazzetti, C. Strumenti e Strategie per La Tutela e l’Uso Compatibile della Risorsa Idrica nel Lazio. Gli Acquiferi Vulcanici; Quaderni di Tecniche di Protezione Ambientale; Pitagora Editrice: Bologna, Italy, 2005; Volume 78. [Google Scholar]
- Ventriglia, U. Idrogeologia Della Provincia di Roma, II Volume: Regione Vulcanica Sabatina; Amministrazione Provinciale di Roma—Assessorato LL.PP.: Rome, Italy, 1989.
- Nappi, G.; de Casa, G.; Volponi, E. Geologia e Caratteristiche Del Tufo Giallo Della Via Tiberina. Boll. Soc. Geol. It. 1979, 98, 431–445. [Google Scholar]
- De Rita, D.; Di Filippo, M.; Funiciello, R.; Parotto, M.; Sposato, A. Carta Geologica Del Complesso Vulcanico Sabatino. In Sabatini Volcanic Complex; Di Filippo, M., Ed.; CNR, Quaderni de La Ricerca Scientifica: Rome, Italy, 1993; Volume 114. [Google Scholar]
- Campobasso, C.; Cioni, R.; Salvati, L.; Sbrana, A. Geology and Paleogeographic Evolution of the Peripheral Sector of the Vico and Sabatini Volcanic Complex, between Civita Castellana and Mazzano Romano (Latium Italy). Mem. Descr. Carta Geol. It. 1994, 49, 277–290. [Google Scholar]
- Lombardi, G.; Meucci, C. Il Tufo Giallo Della Via Tiberina (Roma) Utilizzato Nei Monumenti Romani. Rend. Lincei 2006, 17, 263–287. [Google Scholar] [CrossRef]
- Jackson, M.D.; Marra, F.; Hay, R.L.; Cawood, C.; Winkler, E.M. The Judicious Selection and Preservation of Tuff and Travertine Building Stone in Ancient Rome. Archaeometry 2005, 47, 485–510. [Google Scholar] [CrossRef]
- Shapiro, S.S.; Wilk, M.B. An Analysis of Variance Test for Normality (Complete Samples). Biometrika 1965, 52, 591. [Google Scholar] [CrossRef]
- Rosner, B. Percentage Points for a Generalized Esd Many-Outlier Procedure. Technometrics 1983, 25, 7848. [Google Scholar] [CrossRef]
- Huber, P.J.; Ronchetti, E.M. Robust Statistics, 2nd ed.; Wiley: West Maitland, FL, USA, 2009. [Google Scholar]
- USEPA ProUCL. Statistical Software for Environmental Applications for Data Sets with and without Nondetect Observations; Version 5.1.002 Technical Guide, EPA/600/R-07/041; USEPA: Washington, DC, USA, 2015.
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 455. [Google Scholar]
- Coetzee, P.P.; Gouws, K.; Pluddermann, S.; Yacoby, M.; Howell, S.; den Drijver, L. Evaluation of Sequential Extraction Procedures for Metal Speciation in Model Sediments. Water SA 1995, 21, 458. [Google Scholar]
- Kubová, J.; Streško, V.; Bujdoš, M.; Matúš, P.; Medved, J. Fractionation of Various Elements in CRMs and in Polluted Soils. Anal. Bioanal. Chem. 2004, 379, 56. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.; Auleda, M. A Sequential Extraction Procedure for Sediments Affected by Acid Mine Drainage. J. Geochem. Explor. 2013, 128, 12. [Google Scholar] [CrossRef]
- Dinelli, E.; Lima, A.; Albanese, S.; Birke, M.; Cicchella, D.; Giaccio, L.; Valera, P.; de Vivo, B. Major and Trace Elements in Tap Water from Italy. J. Geochem. Explor. 2012, 112, 9. [Google Scholar] [CrossRef]
- Alexandratos, V.G.; Elzinga, E.J.; Reeder, R.J. Arsenate Uptake by Calcite: Macroscopic and Spectroscopic Characterization of Adsorption and Incorporation Mechanisms. Geochim. Cosmochim. Acta 2007, 71, 55. [Google Scholar] [CrossRef]
- Pfeifer, H.R.; Gueye-Girardet, A.; Reymond, D.; Schlegel, C.; Temgoua, E.; Hesterberg, D.L.; Chou, J.W. Dispersion of Natural Arsenic in the Malcantone Watershed, Southern Switzerland: Field Evidence for Repeated Sorption-Desorption and Oxidation-Reduction Processes. Geoderma 2004, 122, 765. [Google Scholar] [CrossRef]
- Bissen, M.; Frimmel, F.H. Arsenic–A Review. Part I: Occurrence, Toxicity, Speciation, Mobility. Acta Hydrochim. Hydrobiol. 2003, 31, 25. [Google Scholar] [CrossRef]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modeling: Hydrous Ferric Oxide; John Wiley & Sons: Toronto, ON, Canada, 1991. [Google Scholar]
- Fuller, C.C.; Dadis, J.A.; Waychunas, G.A. Surface Chemistry of Ferrihydrite: Part 2. Kinetics of Arsenate Adsorption and Coprecipitation. Geochim. Cosmochim. Acta 1993, 57, 568. [Google Scholar] [CrossRef]
- Deutsch, W.J. Groundwater Geochemistry: Fundamentals and Applications to Contamination; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Bowles, J.F.W.; Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties Reactions Occurrence and Uses. Mineral. Mag. 1997, 61, 20. [Google Scholar] [CrossRef]
- Hinkle, S.R.; Polette, D.J. Arsenic in Ground Water of the Willamette Basin, Oregon; Water-Resources Investigations Report 98–4205; USGS: Portland, OR, USA, 1999.
- Molinari, A.; Guadagnini, L.; Marcaccio, M.; Guadagnini, A. Arsenic Fractioning in Natural Solid Matrices Sampled in a Deep Groundwater Body. Geoderma 2015, 247, 11. [Google Scholar] [CrossRef]
- Dodge, C.J.; Francis, A.J.; Gillow, J.B.; Halada, G.P.; Eng, C.; Clayton, C.R. Association of Uranium with Iron Oxides Typically Formed on Corroding Steel Surfaces. Environ. Sci. Technol. 2002, 36, 1450. [Google Scholar] [CrossRef] [PubMed]
- Parrone, D.; Ghergo, S.; Frollini, E.; Rossi, D.; Preziosi, E. Arsenic-Fluoride Co-Contamination in Groundwater: Background and Anomalies in a Volcanic-Sedimentary Aquifer in Central Italy. J. Geochem. Explor. 2020, 217, 6590. [Google Scholar] [CrossRef]
- Zhu, H.; Xiao, X.; Guo, Z.; Han, X.; Liang, Y.; Zhang, Y.; Zhou, C. Adsorption of Vanadium (V) on Natural Kaolinite and Montmorillonite: Characteristics and Mechanism. Appl. Clay Sci. 2018, 161, 35. [Google Scholar] [CrossRef]
- Gäbler, H.E.; Glüh, K.; Bahr, A.; Utermann, J. Quantification of Vanadium Adsorption by German Soils. J. Geochem. Explor. 2009, 103, 2. [Google Scholar] [CrossRef]
- Luo, X.; Yu, L.; Wang, C.; Yin, X.; Mosa, A.; Lv, J.; Sun, H. Sorption of Vanadium (V) onto Natural Soil Colloids under Various Solution PH and Ionic Strength Conditions. Chemosphere 2017, 169, 105. [Google Scholar] [CrossRef]
- Mikkonen, A.; Tummavuori, J. Retention of Vanadium (V) by Three Finnish Mineral Soils. Eur. J. Soil Sci. 1994, 45, 520. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, S.; Fernandez-Marcos, M.L. Sorption and Desorption of Vanadate, Arsenate and Chromate by Two Volcanic Soils of Equatorial Africa. Soil Syst. 2021, 5, 22. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Extractant | Time/Temperature | As Fraction | Reference |
|---|---|---|---|---|
| 1 | (NH4)2SO4 0.05M | 4 h/25 °C | Easily exchangeable As | [57] |
| 2 | (NH4)H2PO4 0.05M | 16 h/20 °C | Specifically adsorbed As | [57] |
| 3 | Acetic buffer 1M (pH 5) | 12 h/25 °C | As bound with calcite | [61] |
| 4 | NH4-oxalate buffer 0.2M (pH 3.2) | 4 h/20 °C + 10 min (wash) | Low crystalline Fe(III)-oxyhydroxides | [57] |
| 5 | NH4-oxalate buffer 0.2M + ascorbic acid 0.1M (pH 3.2) | 30 min/96 °C + 10 min (wash) | Crystalline Fe(III)-oxides | [57] |
| 6 | HNO3 8M | 3 h/80 °C | Sulfides | [84] |
| 7 | Microwave digestion (HNO3 + H2O2) | 30 min/180 °C | Residual fraction | [57] |
| Parameter | North | South | ||
|---|---|---|---|---|
| r | rs | r | rs | |
| Eh | 0.083 | −0.175 | −0.255 | −0.338 |
| T | 0.185 | 0.109 | 0.291 | 0.318 |
| pH | 0.291 | 0.379 | 0.242 | 0.302 |
| DO | −0.066 | −0.040 | −0.618 | −0.619 |
| Cond | −0.066 | 0.183 | 0.156 | 0.121 |
| F | 0.658 | 0.690 | 0.741 | 0.805 |
| Cl | −0.157 | −0.121 | −0.167 | −0.180 |
| PO4 | −0.107 | 0.025 | −0.223 | −0.301 |
| SO4 | −0.327 | −0.264 | 0.288 | 0.449 |
| HCO3 | 0.165 | 0.261 | 0.161 | 0.161 |
| Na | 0.216 | 0.280 | 0.630 | 0.488 |
| K | 0.055 | 0.194 | 0.541 | 0.542 |
| Mg | −0.250 | −0.172 | −0.356 | −0.449 |
| Ca | −0.076 | 0.241 | −0.105 | −0.225 |
| Al | 0.002 | 0.036 | −0.070 | 0.213 |
| Mn | −0.189 | 0.028 | 0.034 | 0.244 |
| Fe | 0.114 | −0.040 | 0.156 | −0.013 |
| U | 0.613 | 0.536 | 0.230 | 0.399 |
| Li | 0.230 | 0.326 | 0.711 | 0.686 |
| B | 0.366 | 0.111 | 0.365 | 0.497 |
| V | 0.533 | 0.574 | −0.326 | −0.309 |
| Ni | −0.217 | −0.041 | −0.116 | 0.011 |
| Cu | −0.192 | −0.324 | 0.083 | 0.029 |
| Zn | 0.397 | 0.060 | −0.219 | −0.195 |
| Ba | 0.045 | 0.003 | −0.161 | −0.177 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrone, D.; Ghergo, S.; Preziosi, E.; Casentini, B. Water-Rock Interaction Processes: A Local Scale Study on Arsenic Sources and Release Mechanisms from a Volcanic Rock Matrix. Toxics 2022, 10, 288. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10060288
Parrone D, Ghergo S, Preziosi E, Casentini B. Water-Rock Interaction Processes: A Local Scale Study on Arsenic Sources and Release Mechanisms from a Volcanic Rock Matrix. Toxics. 2022; 10(6):288. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10060288
Chicago/Turabian StyleParrone, Daniele, Stefano Ghergo, Elisabetta Preziosi, and Barbara Casentini. 2022. "Water-Rock Interaction Processes: A Local Scale Study on Arsenic Sources and Release Mechanisms from a Volcanic Rock Matrix" Toxics 10, no. 6: 288. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10060288