Conserved Expression of Nav1.7 and Nav1.8 Contribute to the Spontaneous and Thermally Evoked Excitability in IL-6 and NGF-Sensitized Adult Dorsal Root Ganglion Neurons In Vitro

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Adult DRG Culture
2.2. Primary Embryonic Cortical Culture
2.3. Extracellular Recordings
2.4. Pharmacology
2.5. Immunostaining and Image Analysis
2.6. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
2.7. Statistical Analysis
3. Results
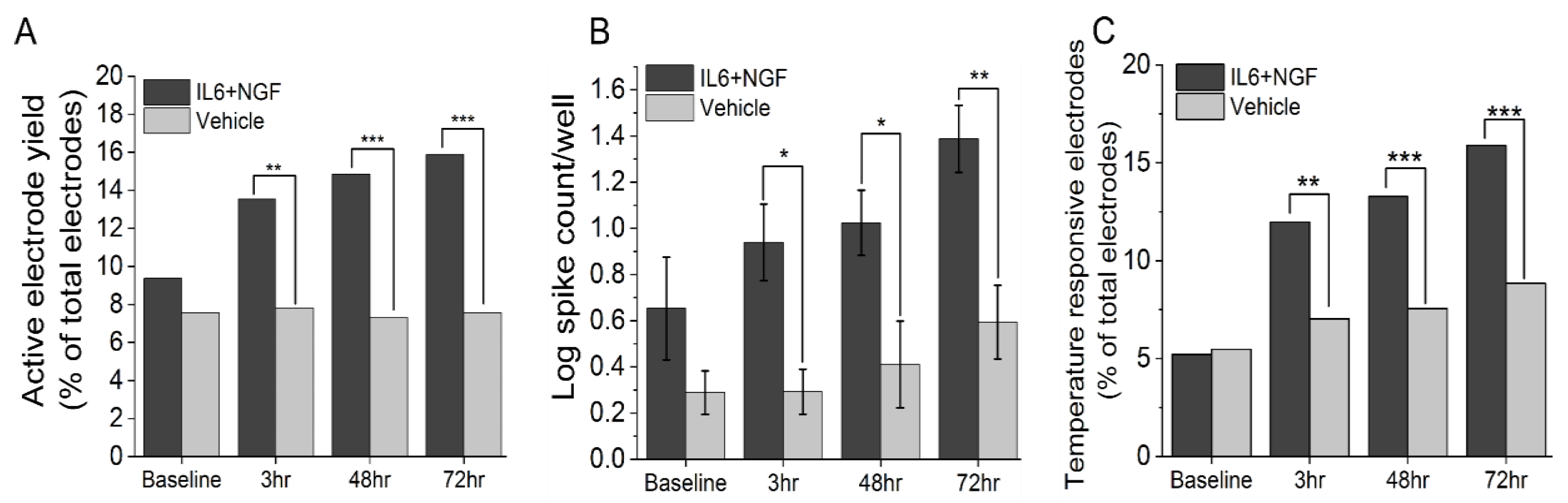
3.1. IL-6 and NGF Increase Spontaneous and Stimulus Evoked Excitability
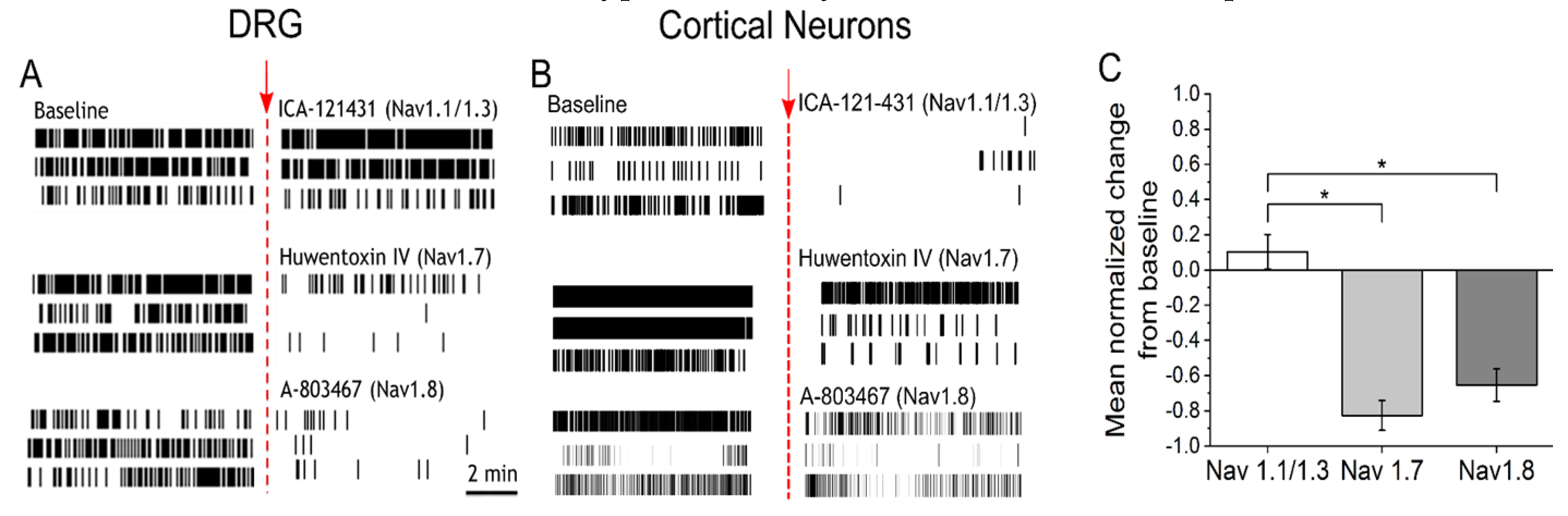
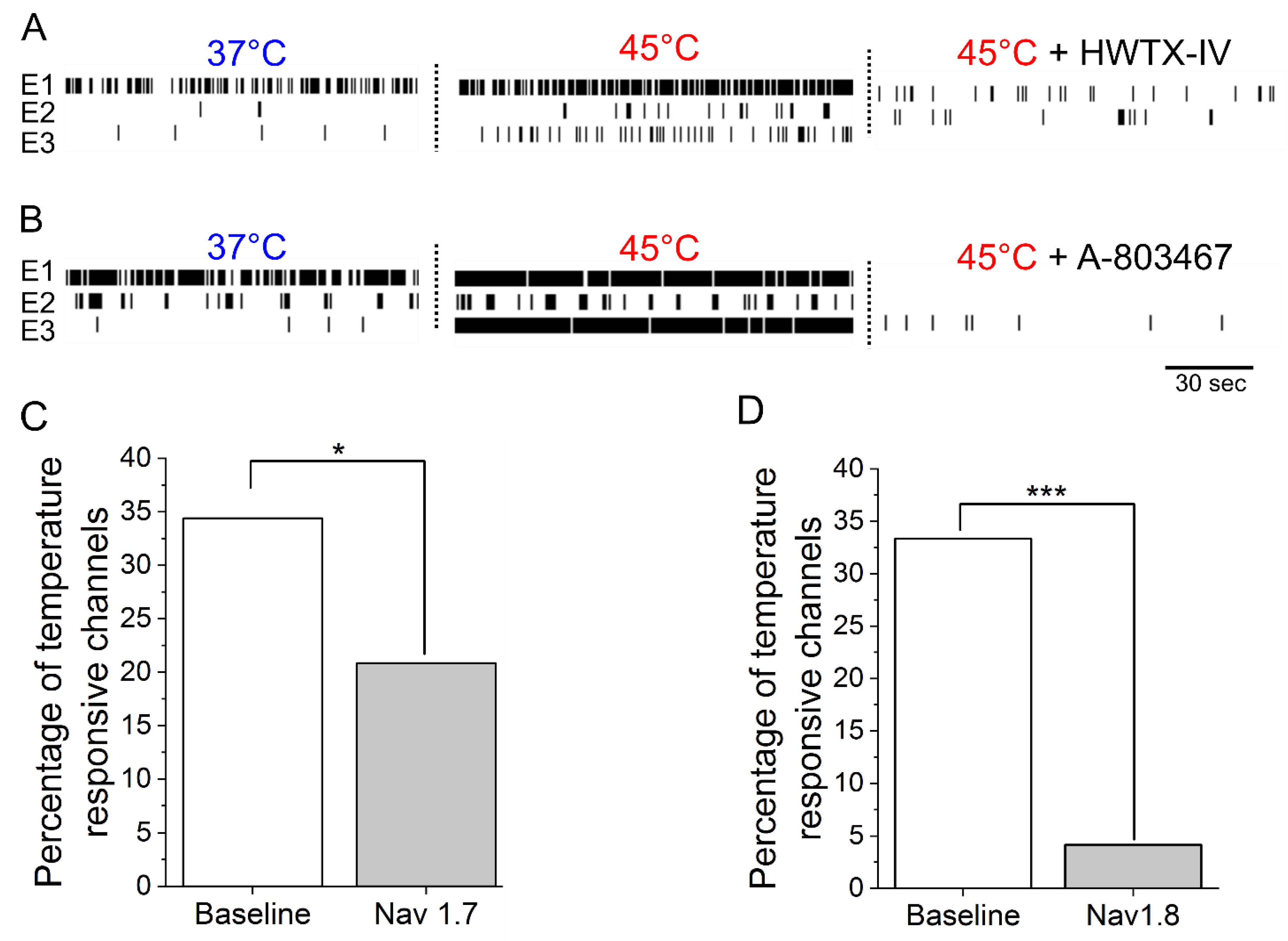
3.2. IL-6 and NGF-Mediated Hyperexcitability Is Attenuated by Nav1.7 and Nav1.8 Antagonists
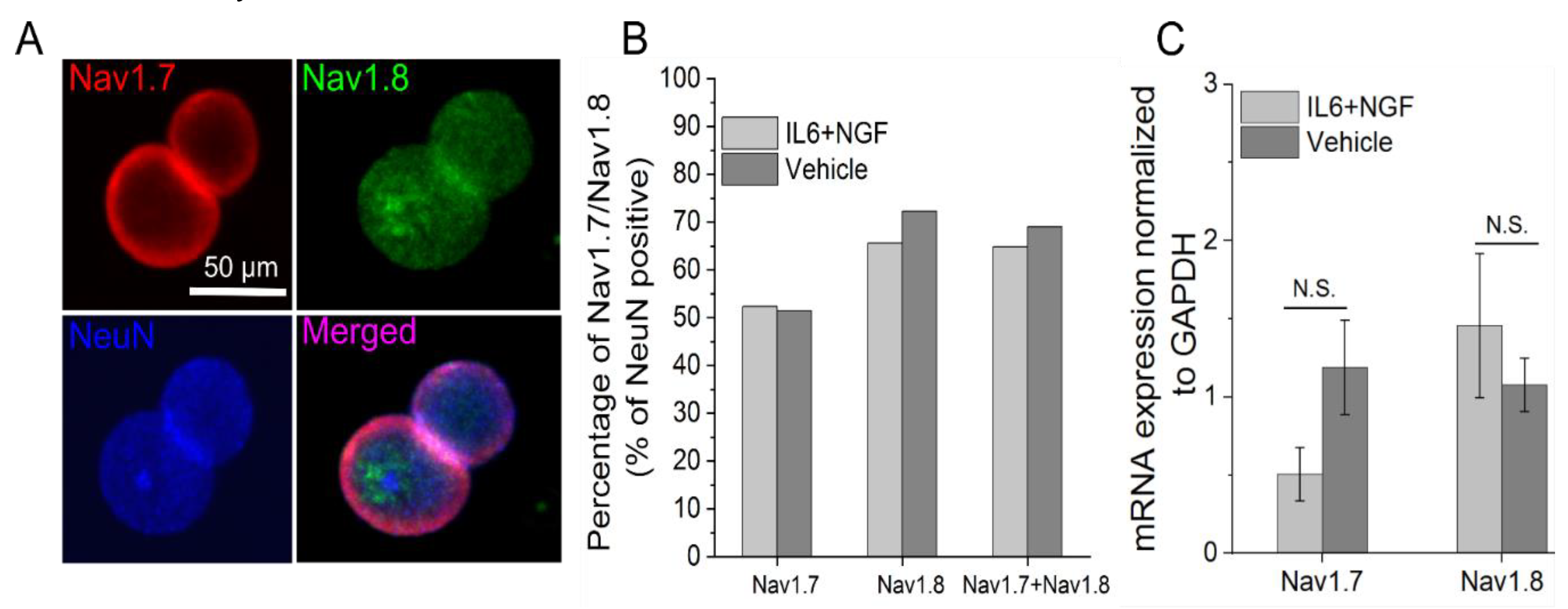
3.3. Nav1.7/1.8 Expression is Unchanged after 72 Hour IL-6 and NGF Treatment
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Okuse, K. Pain signalling pathways: From cytokines to ion channels. Int. J. Biochem. Cell Biol. 2007, 39, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium Channels in Normal and Pathological Pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, M.J.; Hovhannisyan, A.H.; Akopian, A.N. Characteristics of sensory neuronal groups in CGRP-cre-ER reporter mice: Comparison to lines. PLoS ONE 2018, 112747, 1–27. [Google Scholar]
- Levinson, S.R.; Luo, S.; Henry, M.A. The Role of Sodium Channels in Chronic Pain. Muscle Nerve 2012, 2, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Gould, H.J.; Gould, T.N.; Paul, D.; England, J.D.; Liu, Z.P.; Reeb, S.C.; Levinson, S.R. Development of inflammatory hypersensitivity and augmentation of sodium channels in rat dorsal root ganglia. Brain Res. 1999, 824, 296–299. [Google Scholar] [CrossRef]
- Gould, H.J.; England, J.D.; Liu, Z.P.; Levinson, S.R. Rapid sodium channel augmentation in response to inflammation induced by complete Freund’s adjuvant. Brain Res. 1998, 802, 69–74. [Google Scholar] [CrossRef]
- Strickland, I.T.; Martindale, J.C.; Woodhams, P.L.; Reeve, A.J.; Chessell, I.P.; McQueen, D.S. Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain 2008, 12, 564–572. [Google Scholar] [CrossRef]
- Black, J.A.; Liu, S.; Tanaka, M.; Cummins, T.R.; Waxman, S.G. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 2004, 108, 237–247. [Google Scholar] [CrossRef]
- Tamura, R.; Nemoto, T.; Maruta, T.; Onizuka, S.; Yanagita, T.; Wada, A.; Murakami, M.; Tsuneyoshi, I. Up-Regulation of NaV1.7 sodium channels expression by tumor necrosis factor-α in cultured bovine adrenal chromaffin cells and rat dorsal root ganglion neurons. Anesth. Analg. 2014, 118, 318–324. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Fjell, J.; Cummins, T.R.; Zheng, Z.; Fried, K.; Lamotte, R.; Black, J.A.; Waxman, S.G. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 1999, 83, 591–600. [Google Scholar] [CrossRef]
- Stamboulian, S.; Choi, J.-S.; Ahn, H.-S.; Chang, Y.-W.; Tyrrell, L.; Black, J.A.; Waxman, S.G.; Dib-Hajj, S.D. ERK1/2 Mitogen-Activated Protein Kinase Phosphorylates Sodium Channel Nav1.7 and Alters Its Gating Properties. J. Neurosci. 2010, 30, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Hudmon, A.; Choi, J.-S.; Tyrrell, L.; Black, J.A.; Rush, A.M.; Waxman, S.G.; Dib-Hajj, S.D. Phosphorylation of Sodium Channel Nav1.8 by p38 Mitogen-Activated Protein Kinase Increases Current Density in Dorsal Root Ganglion Neurons. J. Neurosci. 2008, 28, 3190–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Gold, M.S.; Kim, C.; Bian, D.; Ossipov, M.H.; Hunter, J.C.; Porreca, F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin- resistant sodium channel, NaV1.8. Pain 2002, 95, 143–152. [Google Scholar] [CrossRef]
- Black, B.J.; Atmaramani, R.; Kumaraju, R.; Plagens, S.; Romero-Ortega, M.; Dussor, G.; Price, T.J.; Campbell, Z.T.; Pancrazio, J.J. Adult Mouse Sensory Neurons on Microelectrode Arrays Exhibit Increased Spontaneous and Stimulus-Evoked Activity in the Presence of Interleukin-6. J. Neurophysiol. 2018, 120, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B. NGF as a mediator of inflammatory pain. Philos. Trans. R. Soc. B Biol. Sci. 1996, 351, 431–440. [Google Scholar]
- Yan, J.; Melemedjian, O.K.; Price, T.J.; Dussor, G. Sensitization of dural afferents underlies migraine-related behavior following meningeal application of interleukin-6 (IL-6). Mol. Pain 2012, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.Q.; Liu, Z.; Liu, Z.H.; Chen, S.P.; Li, M.; Shahveranov, A.; Ye, D.W.; Tian, Y.K. Interleukin-6: An emerging regulator of pathological pain. J. Neuroinflamm. 2016, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Brook, S.; Oncology, M. Identification of PN1, a predominant voltage-dependent sodium. Neurobiology 1997, 94, 1527–1532. [Google Scholar]
- Dib-Hajj, S.D.; Black, J.A.; Cummins, T.R.; Kenney, A.M.; Kocsis, J.D.; Waxman, S.G. Rescue of α-SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo. J. Neurophysiol. 1998, 79, 2668–2676. [Google Scholar] [CrossRef]
- Leffler, A.; Cummins, T.R.; Dib-Hajj, S.D.; Hormuzdiar, W.N.; Black, J.A.; Waxman, S.G. GDNF and NGF reverse changes in repriming of TTX-sensitive Na + currents following axotomy of dorsal root ganglion neurons. J. Neurophysiol. 2002, 88, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Black, J.A.; Dib-Hajj, S.D.; Waxman, S.G. Glial-derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J. Neurosci. 2000, 20, 8754–8761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Vasko, M.R.; Nicol, G.D. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J. Physiol. 2002, 544, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Krafte, D.S.; Bannon, A.W. Sodium channels and nociception: Recent concepts and therapeutic opportunities. Curr. Opin. Pharmacol. 2008, 8, 50–56. [Google Scholar] [CrossRef]
- Linley, J.E.; Rose, K.; Ooi, L.; Gamper, N. Understanding inflammatory pain: Ion channels contributing to acute and chronic nociception. Pflugers Arch. Eur. J. Physiol. 2010, 459, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Liu, B.; Zang, K.; Yang, L.; Xu, H.; Pan, H.; Zhao, Z.; Zhang, Y. Dexmedetomidine inhibits Tetrodotoxin-resistant Nav1.8 sodium channel activity through G i/o-dependent pathway in rat dorsal root ganglion neurons. Mol. Brain 2015, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Li, Y.; Fu, Q.; Chao, Y.F.; Liu, Z.Y. Stromal Cell-Derived Factor 1 Increases Tetrodotoxin-Resistant Sodium Currents Nav1.8 and Nav1.9 in Rat Dorsal Root Ganglion Neurons via Different Mechanisms. Neurochem. Res. 2016, 41, 1587–1603. [Google Scholar] [CrossRef]
- Schink, M.; Leipold, E.; Schirmeyer, J.; Schönherr, R.; Hoshi, T.; Heinemann, S.H. Reactive species modify NaV 1.8 channels and affect action potentials in murine dorsal root ganglion neurons. Eur. J. Physiol. 2016, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Belkouch, M.; Dansereau, M.A.; Réaux-Le Goazigo, A.; Van Steenwinckel, J.; Beaudet, N.; Chraibi, A.; Melik-Parsadaniantz, S.; Sarret, P. The chemokine CCL2 increases Nav 1.8 sodium channel activity in primary sensory neurons through a Gβγ-dependent mechanism. J. Neurosci. 2011, 31, 18381–18390. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Xia, Z.; Wu, Y.; Zhao, B. Sodium channel Nav 1.7 expression is upregulated in the dorsal root ganglia in a rat model of paclitaxel-induced peripheral neuropathy. Springerplus 2016, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; North, R.Y.; Rhines, X.L.D.; Tatsui, X.C.E.; Rao, G.; Edwards, D.D.; Cassidy, X.R.M.; Harrison, X.D.S.; Johansson, X.C.A.; Zhang, X.H.; et al. Neurobiology of Disease DRG Voltage-Gated Sodium Channel 1.7 Is Upregulated in Paclitaxel-Induced Neuropathy in Rats and in Humans with Neuropathic Pain. J. Neurosci. 2018, 38, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Berta, T.; Kim, Y.H.; Lee, S.; Lee, S.Y.; Ji, R.R. Expression and Role of Voltage-Gated Sodium Channels in Human Dorsal Root Ganglion Neurons with Special Focus on Nav1.7, Species Differences, and Regulation by Paclitaxel. Neurosci. Bull. 2018, 34, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Cao, J.; Ren, X.; Qiao, L.; Chen, X.; Li, M.; Zang, W. shRNA mediated knockdown of Nav1.7 in rat dorsal root ganglion attenuates pain following burn injury. BMC Anesthesiol. 2016, 16, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Pérez, J.V.; Adamek, P.; Palecek, J.; Vizcaychipi, M.; Nagy, I.; Varga, A. The NAv1.7 blocker protoxin II reduces burn injury-induced spinal nociceptive processing. J. Mol. Med. 2018, 96, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salas, M.M.; McIntyre, M.K.; Petz, L.N.; Korz, W.; Wong, D.; Clifford, J.L. Tetrodotoxin suppresses thermal hyperalgesia and mechanical allodynia in a rat full thickness thermal injury pain model. Neurosci. Lett. 2015, 607, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, M.F.; Honore, P.; Shieh, C.-C.; Chapman, M.; Joshi, S.; Zhang, X.-F.; Kort, M.; Carroll, W.; Marron, B.; Atkinson, R.; et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 2007, 104, 8520–8525. [Google Scholar] [CrossRef] [Green Version]
- Sidders, B.; Karlsson, A.; Kitching, L.; Torella, R.; Karila, P.; Phelan, A. Network-Based Drug Discovery: Coupling Network Pharmacology with Phenotypic Screening for Neuronal Excitability. J. Mol. Biol. 2018, 430, 3005–3015. [Google Scholar] [CrossRef]
- Vincent, A.M.; Feldman, E.V.A.L. Can Drug Screening Lead to Candidate Therapies for Testing ABSTRACT FOR DISCOVERY. Antioxid. Redox Signal. 2008, 10. [Google Scholar] [CrossRef] [Green Version]
- Rana, P.; Luerman, G.; Hess, D.; Rubitski, E.; Adkins, K.; Somps, C. Toxicology in Vitro Utilization of iPSC-derived human neurons for high-throughput drug- induced peripheral neuropathy screening. Toxicol. In Vitro 2017, 45, 111–118. [Google Scholar] [CrossRef]
- Bourque, K.; Jones-tabah, J.; Mnasri, N.; Martin, R.D.; Terence, E.H. Combining Optical Approaches with Human Inducible Pluripotent Stem Cells in G Protein-Coupled Receptor Drug Screening and Development. Biomolecules 2018, 8, 180. [Google Scholar] [CrossRef] [Green Version]
- Martínez, A.L.; Brea, J.; Monroy, X.; Merlos, M.; Burgueño, J.; Loza, M.I. A New Model of Sensorial Neuron-Like Cells for HTS of Novel Analgesics for Neuropathic Pain. SLAS Discov. 2019, 24, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Melli, G. Dorsal Root Ganglia Sensory Neuronal Cultures: A tool for drug discovery for peripheral neuropathies. Expert Opin. Drug Discov. 2014, 4, 1035–1045. [Google Scholar] [CrossRef]
- Stacey, P.; Wassermann, A.M.; Kammonen, L.; Impey, E.; Wilbrey, A.; Cawkill, D. Plate-Based Phenotypic Screening for Pain Using Human iPSC-Derived Sensory Neurons. SLAS Discov. 2018, 23, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Newberry, K.; Wang, S.; Hoque, N.; Ahlijanian, M.K.; Kiss, L.; Herrington, J.; Graef, J.D. Development of a Spontaneously Active Dorsal Root Ganglia Assay using Multi-well Multielectrode Arrays. J. Neurophysiol. 2016, 115, 3217–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benn, S.C.; Costigan, M.; Tate, S.; Fitzgerald, M.; Woolf, C.J. Developmental Expression of the TTX-Resistant Voltage-Gated Sodium Channels Nav1.8 (SNS) and Nav1.9 (SNS2) in Primary Sensory Neurons. J. Neurosci. 2001, 21, 6077–6085. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R.; Guthrie, R.; Roskoski, L.M. Degradation of Rat Brain Cholinergic Muscarinic Receptors In Vitro: Enhancement by Agonists and Inhibition by Antagonists. J. Neurochem. 1985, 45, 1096–1100. [Google Scholar] [CrossRef]
- Brunner, F.; Kukovetz, W.R. Radioligand binding to muscarinic receptors of bovine aortic endothelial cells. Br. J. Pharmacol. 1991, 102, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moy, J.K.; Khoutorsky, A.; Asiedu, M.N.; Black, B.J.; Kuhn, J.L.; Barragán-Iglesias, P.; Megat, S.; Burton, M.D.; Burgos-Vega, C.C.; Melemedjian, O.K.; et al. The MNK—eIF4E signaling axis contributes to injury-induced nociceptive plasticity and the development of chronic pain. J. Neurosci. 2017, 37, 7481–7499. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Weir, G.A.; Schiavo, G. A simple, step-by-step dissection protocol for the rapid isolation of mouse dorsal root ganglia. BMC Res. Notes 2016, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Charkhkar, H.; Frewin, C.; Nezafati, M.; Knaack, G.L.; Peixoto, N.; Saddow, S.E.; Pancrazio, J.J. Use of cortical neuronal networks for in vitro material biocompatibility testing. Biosens. Bioelectron. 2014, 53, 316–323. [Google Scholar] [CrossRef]
- Laedermann, C.J.; Pertin, M.; Suter, M.R.; Decosterd, I. Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol. Pain 2014, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, D.L.; Clark, X.A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, B.J.; Souslova, V.; Mcmahon, S.B.; Wood, J.N. A role for the TTX-resistant sodium channel Nav1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 2001, 12, 3077–3080. [Google Scholar] [CrossRef]
- Ma, Z.; Li, Y.; Zhang, Y.P.; Shields, L.B.E.; Xie, Q.; Yan, G.; Liu, W.; Chen, G.; Zhang, Y.; Brommer, B.; et al. Thermal nociception using a modified Hargreaves method in primates and humans. Funct. Neurol. 2015, 30, 229–236. [Google Scholar] [CrossRef]
- Barragán-Iglesias, P.; Lou, T.F.; Bhat, V.D.; Megat, S.; Burton, M.D.; Price, T.J.; Campbell, Z.T. Inhibition of Poly(A)-binding protein with a synthetic RNA mimic reduces pain sensitization in mice. Nat. Commun. 2018, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Peña, J.B.I.; Song, J.J.; Campbell, Z.T. RNA control in pain: Blame it on the messenger. Wiley Interdiscip. Rev. RNA 2019, 0, 1546. [Google Scholar] [CrossRef]
- Melemedjian, O.K.; Asiedu, M.N.; Tillu, D.V.; Peebles, K.A.; Yan, J.; Ertz, N.; Dussor, G.O.; Price, T.J. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J. Neurosci. 2010, 30, 15113–15123. [Google Scholar] [CrossRef] [Green Version]
- Gould, H.J.; England, J.D.; Soignier, R.D.; Nolan, P.; Minor, L.D.; Liu, Z.P.; Levinson, S.R.; Paul, D. Ibuprofen Blocks Changes in Na. J. Pain 2004, 5, 270–280. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Tyrrell, L.; Cummins, T.R.; Black, J.A.; Wood, P.M.; Waxman, S.G. Two tetrodotoxin-resistant sodium channels in human dorsal root ganglion neurons. FEBS Lett. 1999, 462, 117–120. [Google Scholar] [CrossRef] [Green Version]
- Tate, S.; Benn, S.; Hick, C.; Trezise, D.; John, V.; Mannion, R.J.; Costigan, M.; Plumpton, C.; Grose, D.; Gladwell, Z.; et al. Two sodium channels contribute to the TTX-R sodium current in primary sensory neurons. Nat. Neurosci. 1998, 1, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Iii, H.J.G.; Levinson, S.R. A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Res. 2000, 854, 19–29. [Google Scholar]
- Shu, X.; Mendell, L.M. Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci. Lett. 1999, 274, 159–162. [Google Scholar] [CrossRef]
- Gumy, L.F.; Yeo, G.S.H.; Tung, Y.-C.L.; Zivraj, K.H.; Willis, D.; Coppola, G.; Lam, B.Y.H.; Twiss, J.L.; Holt, C.E.; Fawcett, J.W. Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 2011, 17, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Friedel, R.H.; Schnürch, H.; Stubbusch, J.; Barde, Y.-A. Identification of genes differentially expressed by nerve growth factor- and neurotrophin-3-dependent sensory neurons. Proc. Natl. Acad. Sci. USA 1997, 94, 12670–12675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuoka, T.; Noguchi, K. Comparative study of voltage-gated sodium channel α-subunits in non-overlapping four neuronal populations in the rat dorsal root ganglion. Neurosci. Res. 2011, 70, 164–171. [Google Scholar] [CrossRef]
- Bennett, G.J. What is spontaneous pain and who has it? J. Pain 2012, 13, 921–929. [Google Scholar] [CrossRef]
- Djouhri, L. Spontaneous Pain, Both Neuropathic and Inflammatory, Is Related to Frequency of Spontaneous Firing in Intact C-Fiber Nociceptors. J. Neurosci. 2006, 26, 1281–1292. [Google Scholar] [CrossRef] [Green Version]
- Ørstavik, K.; Weidner, C.; Schmidt, R.; Schmelz, M.; Hilliges, M.; Jørum, E.; Handwerker, H.; Torebjörk, E. Pathological C-fibres in patients with a chronic painful condition. Brain 2003, 126, 567–578. [Google Scholar] [CrossRef]
- O’Donnell, C.; van Rossum, M.C.W. Systematic analysis of the contributions of stochastic voltage gated channels to neuronal noise. Front. Comput. Neurosci. 2014, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Atmaramani, R.; Pancrazio, J.J.; Black, B.J. Adaptation of robust Z’ factor for assay quality assessment in microelectrode array based screening using adult dorsal root ganglion neurons. J. Neurosci. Methods 2020, 339, 108699. [Google Scholar] [CrossRef] [PubMed]
- Mogil, J.S. Animal models of pain: Progress and challenges. Nat. Rev. Neurosci. 2009, 10, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Rostock, C.; Schrenk-Siemens, K.; Pohle, J.; Siemens, J. Human vs. Mouse Nociceptors—Similarities and Differences. Neuroscience 2018, 387, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Otto, W.R.; Casula, M.A.; Day, N.C.; Davis, J.B.; Bountra, C.; Birch, R.; Anand, P. The effect of neurotrophic factors on morphology, TRPV1 expression and capsaicin responses of cultured human DRG sensory neurons. Neurosci. Lett. 2006, 399, 51–56. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atmaramani, R.R.; Black, B.J.; de la Peña, J.B.; Campbell, Z.T.; Pancrazio, J.J. Conserved Expression of Nav1.7 and Nav1.8 Contribute to the Spontaneous and Thermally Evoked Excitability in IL-6 and NGF-Sensitized Adult Dorsal Root Ganglion Neurons In Vitro. Bioengineering 2020, 7, 44. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020044
Atmaramani RR, Black BJ, de la Peña JB, Campbell ZT, Pancrazio JJ. Conserved Expression of Nav1.7 and Nav1.8 Contribute to the Spontaneous and Thermally Evoked Excitability in IL-6 and NGF-Sensitized Adult Dorsal Root Ganglion Neurons In Vitro. Bioengineering. 2020; 7(2):44. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020044
Chicago/Turabian StyleAtmaramani, Rahul R., Bryan J. Black, June Bryan de la Peña, Zachary T. Campbell, and Joseph J. Pancrazio. 2020. "Conserved Expression of Nav1.7 and Nav1.8 Contribute to the Spontaneous and Thermally Evoked Excitability in IL-6 and NGF-Sensitized Adult Dorsal Root Ganglion Neurons In Vitro" Bioengineering 7, no. 2: 44. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020044