Human Mesenchymal Stem Cells Overexpressing Interleukin 2 Can Suppress Proliferation of Neuroblastoma Cells in Co-Culture and Activate Mononuclear Cells In Vitro

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Differentiation of Mesenchymal Stem Cells (MSCs) into Adipocytes, Chondrocytes and Osteoblasts
2.3. Lentivirus Production
2.4. Genetic Modification and Selection
2.5. Quantitative Polymerase Chain Reaction (qPCR)
2.6. Immunofluorescence
2.7. Western Blot Analysis
2.8. Immunophenotyping
2.9. Conditioned Medium Collection
2.10. Cell Proliferation Assays
2.11. Transmission Electron Microscopy
2.12. Cytokine Multiplex Analysis
2.13. Peripheral Blood Mononuclear Cell (PBMC) Activation
2.14. Co-Culture of Cancer Cells and Human Adipose Tissue-Derived Mesenchymal Stem Cells (hADSCs) on Plastic
2.15. Co-Culture of Cancer Cells and hADSCs on Matrigel
2.16. Statistical Analysis
3. Results
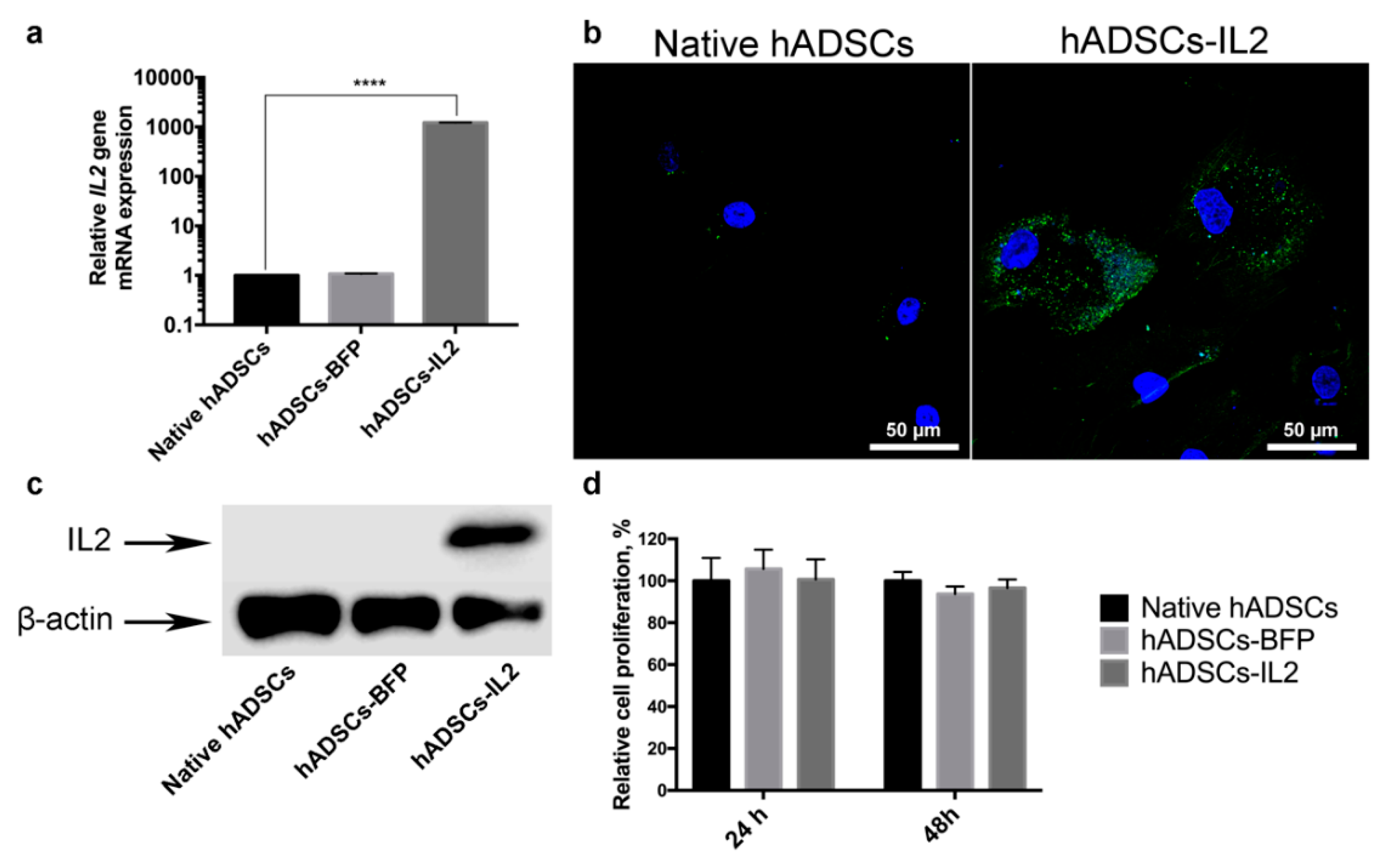
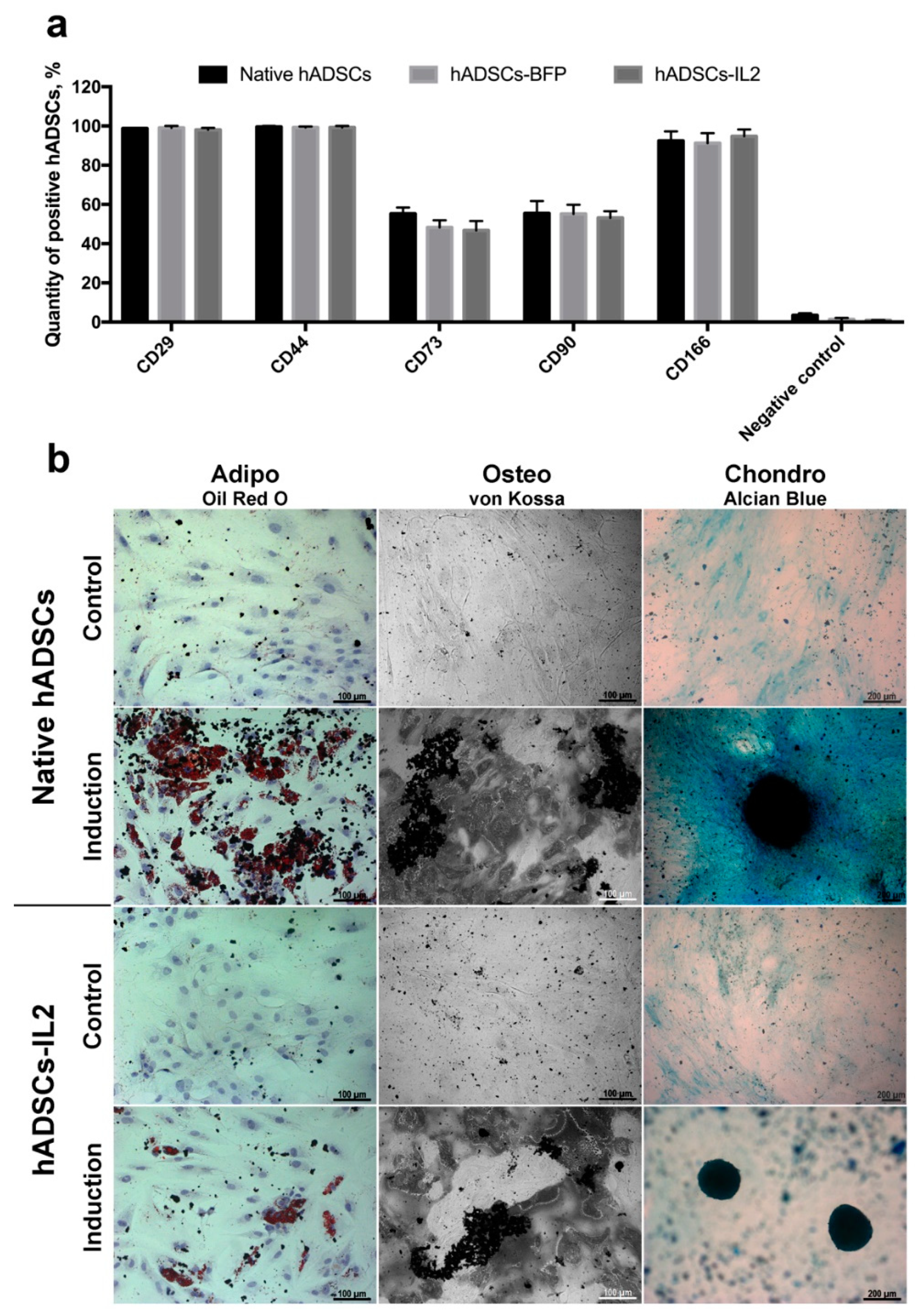
3.1. hADSCs-IL2 Overexpress Interleukin 2 (IL2) mRNA and Protein and Retain a Mesenchymal Stem Cell Phenotype
3.2. IL2 Expression Affects the hADSC Ultrastructure
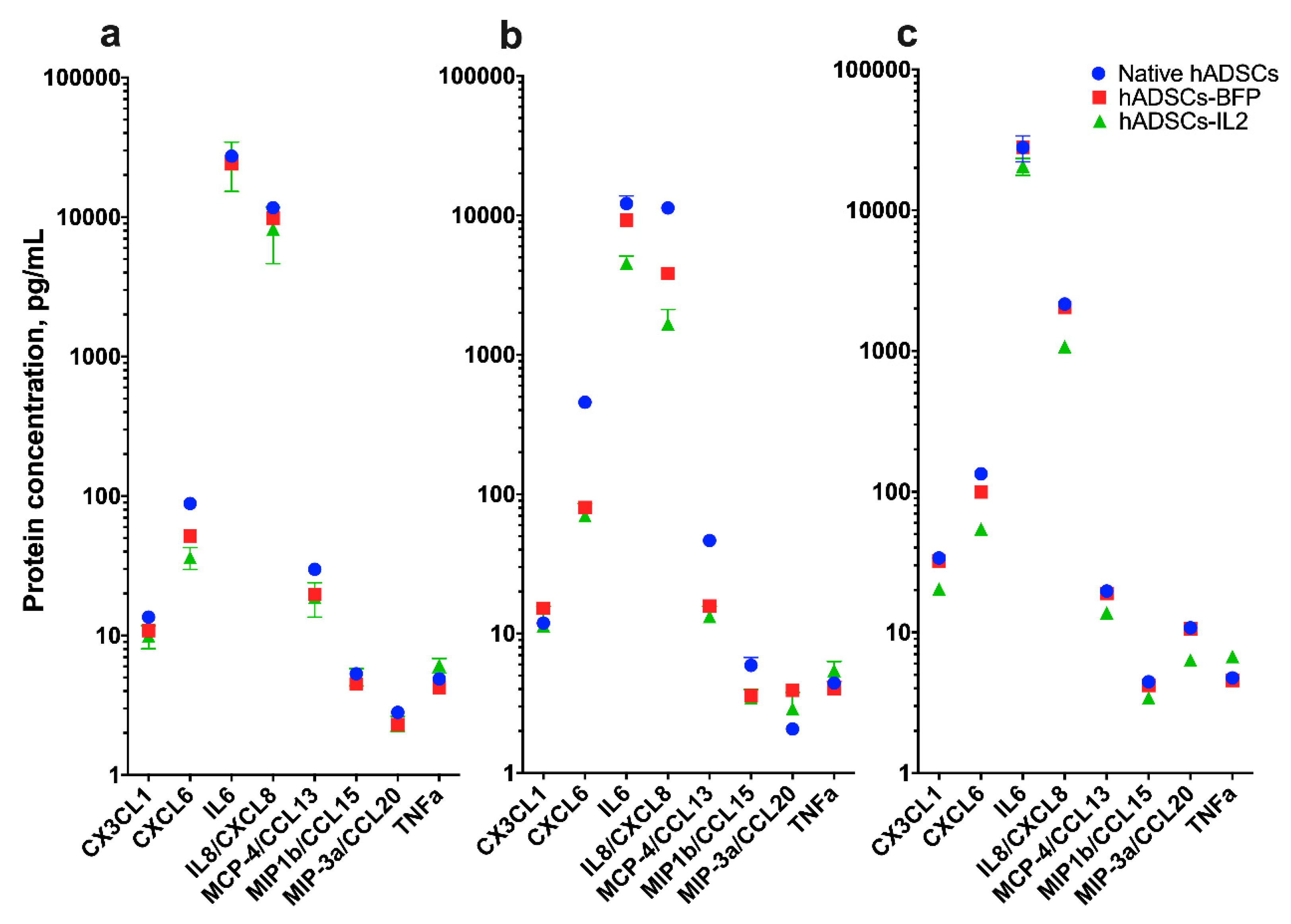
3.3. IL2 Overexpression Leads to a Modest Change in Cytokine Production
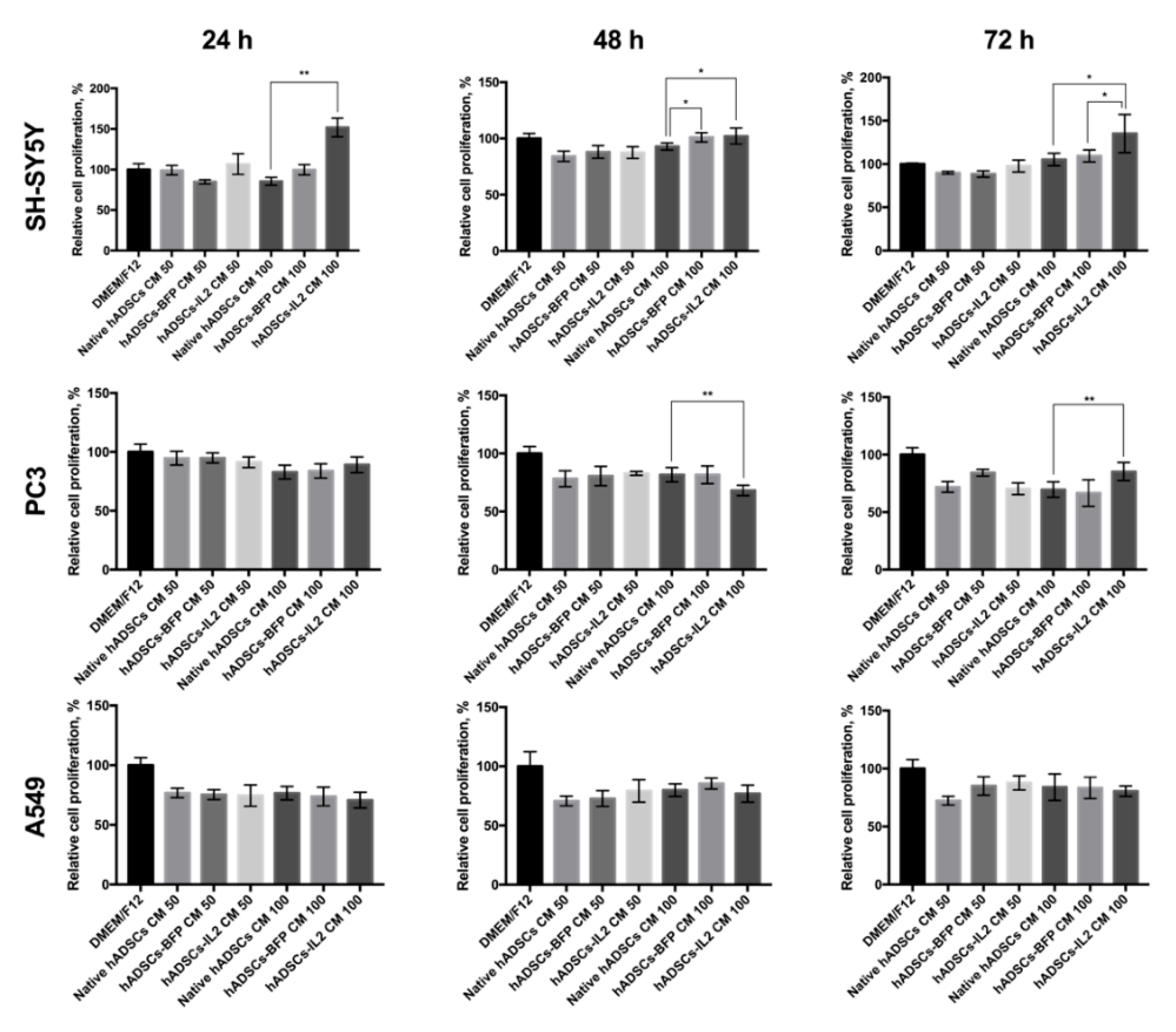
3.4. hADSC-IL2 Conditioned Medium (CM) Can Stimulate the Proliferation of Tumor Cells
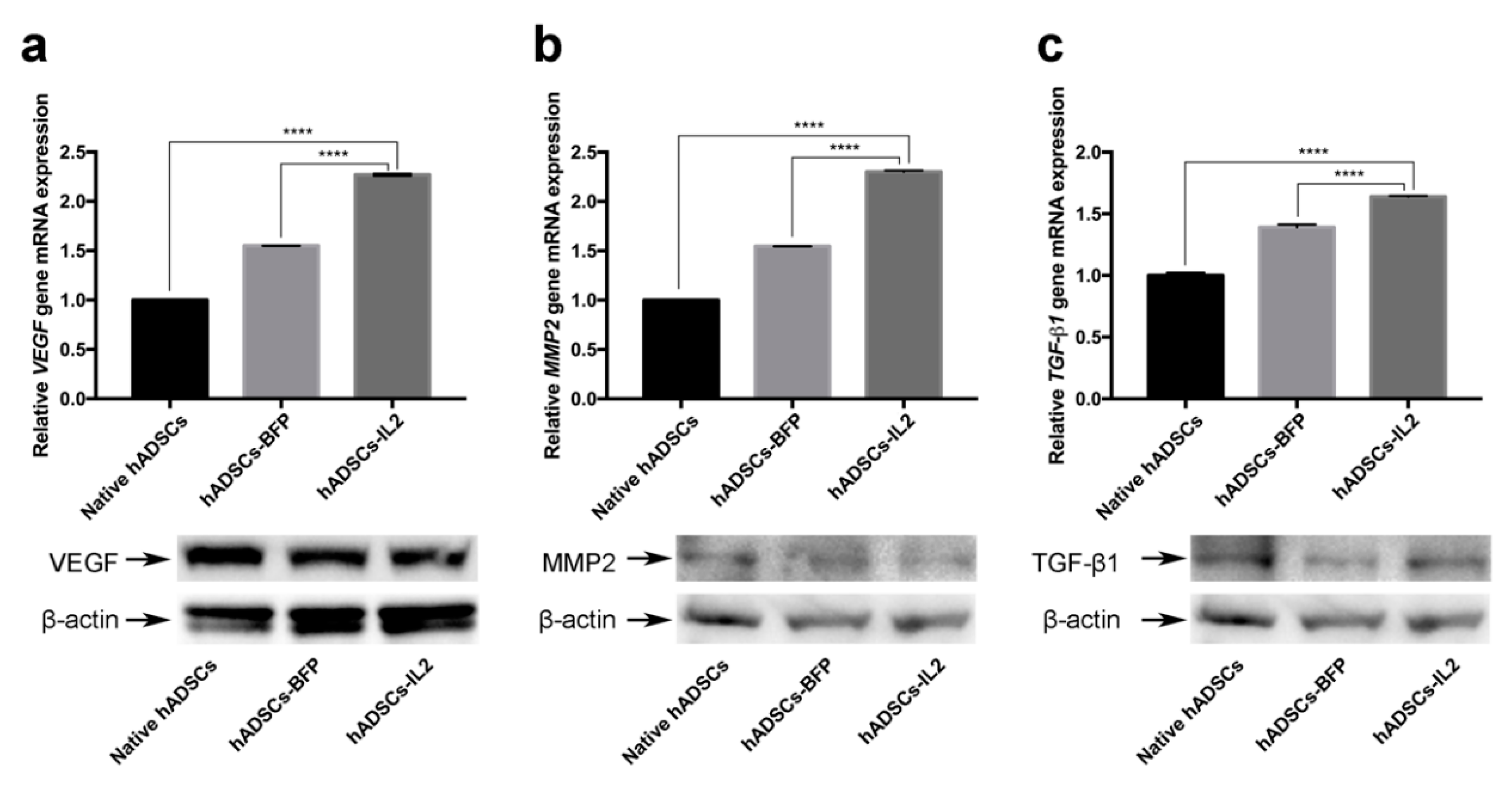
3.5. hADSCs-IL2 Can Promote Angiogenesis and Increase Tumor Cell Invasion
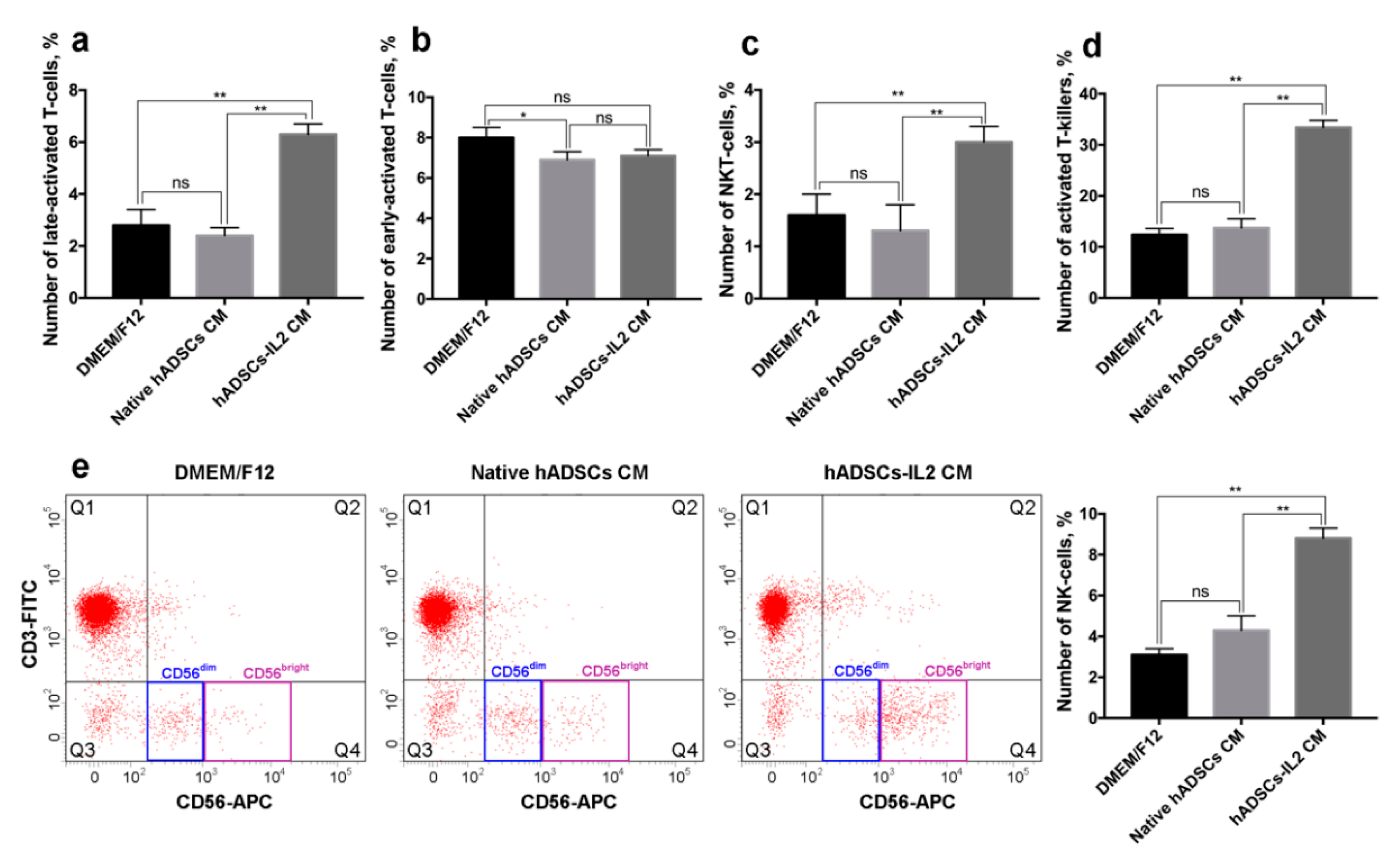
3.6. hADSCs-IL2 Activates Mononuclear Blood Cells In Vitro
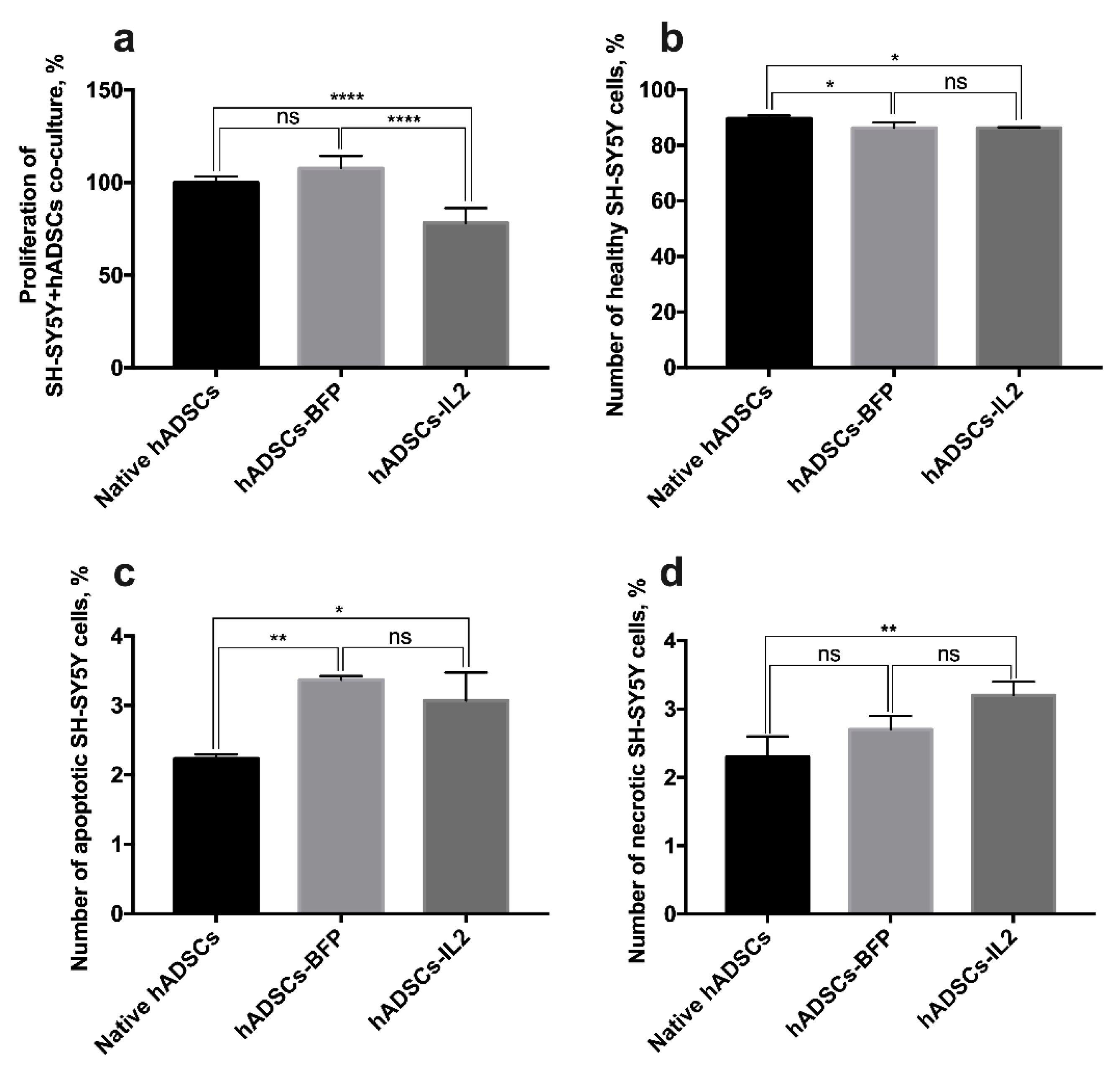
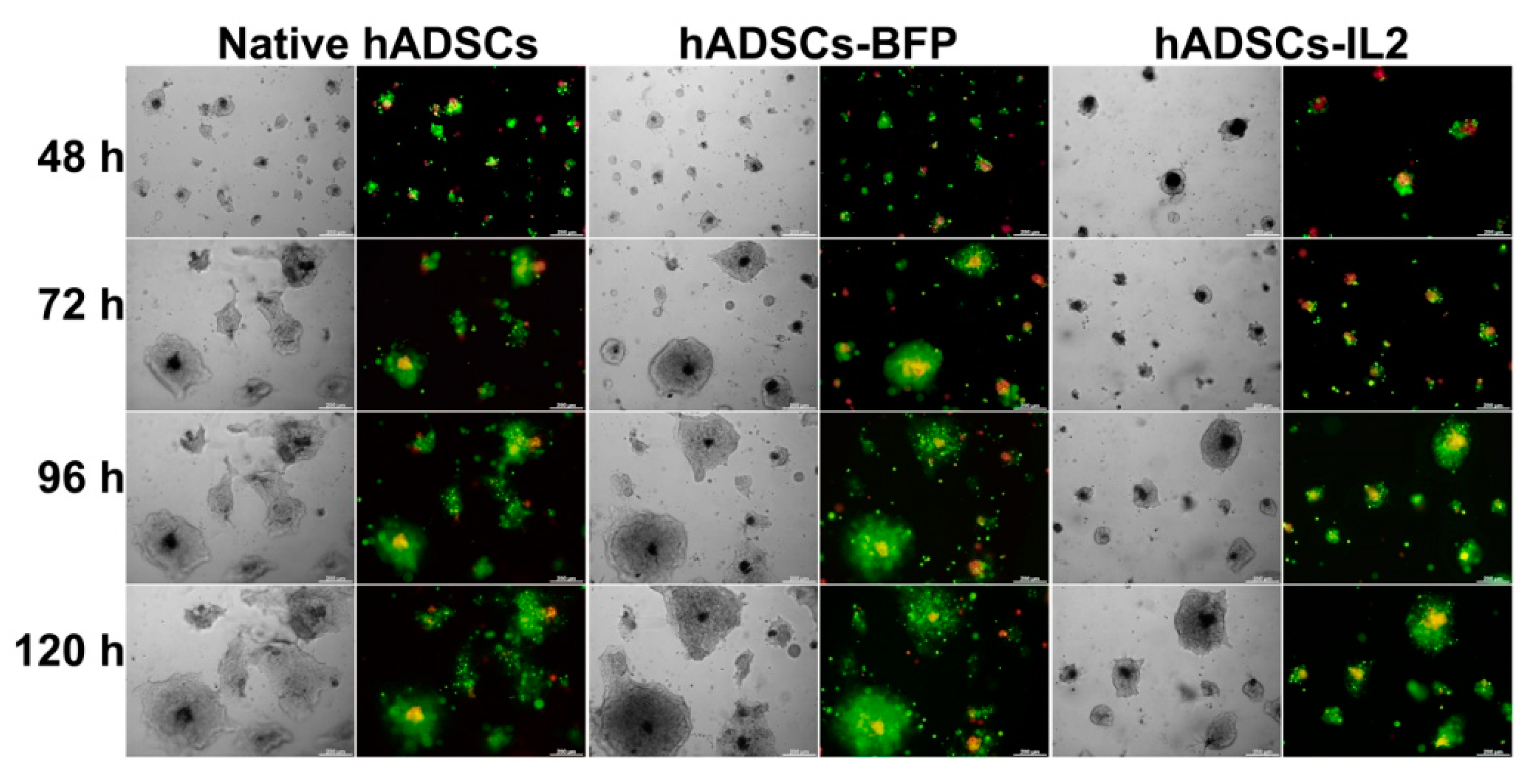
3.7. Co-Culture of hADSCs-IL2 Reduces Proliferation and Mediates Apoptosis of SH-SY5Y
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Statement
References
- Choudhry, H.; Helmi, N.; Abdulaal, W.H.; Zeyadi, M.; Zamzami, M.A.; Wu, W.; Mahmoud, M.M.; Warsi, M.K.; Rasool, M.; Jamal, M.S. Prospects of il-2 in cancer immunotherapy. BioMed Res. Int. 2018, 2018, 9056173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacalano, N.A.; Johnston, J.A. Interleukin-2 signaling and inherited immunodeficiency. Am. J. Hum. Genet. 1999, 65, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; White, D.E.; Steinberg, S.M. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: Identification of the antigens mediating response. Ann. Surg. 1998, 228, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. Jama 1994, 271, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of t cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef] [Green Version]
- Chavez, A.R.; Buchser, W.; Basse, P.H.; Liang, X.; Appleman, L.J.; Maranchie, J.K.; Zeh, H.; de Vera, M.E.; Lotze, M.T. Pharmacologic administration of interleukin-2. Ann. N. Y. Acad Sci. 2009, 1182, 14–27. [Google Scholar] [CrossRef]
- McDermott, D.F.; Atkins, M.B. Application of il-2 and other cytokines in renal cancer. Expert Opin. Biol. Ther. 2004, 4, 455–468. [Google Scholar] [CrossRef]
- Lissoni, P. Therapy implications of the role of interleukin-2 in cancer. Expert Rev. Clin. Immunol. 2017, 13, 491–498. [Google Scholar] [CrossRef]
- Donohue, J.H.; Rosenberg, S.A. The fate of interleukin-2 after in vivo administration. J. Immunol. 1983, 130, 2203–2208. [Google Scholar]
- Yang, J.C.; Sherry, R.M.; Steinberg, S.M.; Topalian, S.L.; Schwartzentruber, D.J.; Hwu, P.; Seipp, C.A.; Rogers-Freezer, L.; Morton, K.E.; White, D.E.; et al. Randomized study of high-dose and low-dose interleukin-2 in patients with metastatic renal cancer. J. Clin. Oncol. 2003, 21, 3127–3132. [Google Scholar] [CrossRef] [Green Version]
- Den Otter, W.; Jacobs, J.J.; Battermann, J.J.; Hordijk, G.J.; Krastev, Z.; Moiseeva, E.V.; Stewart, R.J.; Ziekman, P.G.; Koten, J.W. Local therapy of cancer with free il-2. Cancer Immunol. Immunother. 2008, 57, 931–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.J.; Sparendam, D.; Den Otter, W. Local interleukin 2 therapy is most effective against cancer when injected intratumourally. Cancer Immunol. Immunother. 2005, 54, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of msc with tumor cells. Cell Commun. Signal. CCS 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamizo, A.; Marini, F.; Amano, T.; Khan, A.; Studeny, M.; Gumin, J.; Chen, J.; Hentschel, S.; Vecil, G.; Dembinski, J.; et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005, 65, 3307–3318. [Google Scholar] [CrossRef] [Green Version]
- Kalimuthu, S.; Zhu, L.; Oh, J.M.; Gangadaran, P.; Lee, H.W.; Baek, S.H.; Rajendran, R.L.; Gopal, A.; Jeong, S.Y.; Lee, S.W.; et al. Migration of mesenchymal stem cells to tumor xenograft models and in vitro drug delivery by doxorubicin. Int. J. Med. Sci. 2018, 15, 1051–1061. [Google Scholar] [CrossRef] [Green Version]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Therapeutic prospects of extracellular vesicles in cancer treatment. Front. Immunol. 2018, 9, 1534. [Google Scholar] [CrossRef] [Green Version]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of mesenchymal stem cells for therapeutic agent delivery in anti-tumor treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Solovyeva, V.V.; Kitaeva, K.V.; Dunham, S.P.; Khaiboullina, S.F.; Rizvanov, A.A. Recombinant viruses for cancer therapy. Biomedicines 2018, 6, 94. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Kumar, S.; Chanda, D.; Chen, J.; Mountz, J.D.; Ponnazhagan, S. Therapeutic potential of mesenchymal stem cells producing interferon-alpha in a mouse melanoma lung metastasis model. Stem. Cells 2008, 26, 2332–2338. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.X.; Zhan, Y.A.; Hu, H.L.; Wang, Y.; Fu, B. Mesenchymal stem cells modified to express interferon-beta inhibit the growth of prostate cancer in a mouse model. J. Int. Med. Res. 2012, 40, 317–327. [Google Scholar] [CrossRef]
- Han, J.; Zhao, J.; Xu, J.; Wen, Y. Mesenchymal stem cells genetically modified by lentivirus-mediated interleukin-12 inhibit malignant ascites in mice. Exp. Ther. Med. 2014, 8, 1330–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Ito, Y.; Kawano, Y.; Kurozumi, K.; Kobune, M.; Tsuda, H.; Bizen, A.; Honmou, O.; Niitsu, Y.; Hamada, H. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004, 11, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Marini, I.; Siegemund, M.; Hutt, M.; Kontermann, R.E.; Pfizenmaier, K. Antitumor activity of a mesenchymal stem cell line stably secreting a tumor-targeted tnf-related apoptosis-inducing ligand fusion protein. Front. Immunol. 2017, 8, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cafforio, P.; Viggiano, L.; Mannavola, F.; Pelle, E.; Caporusso, C.; Maiorano, E.; Felici, C.; Silvestris, F. Pil6-trail-engineered umbilical cord mesenchymal/stromal stem cells are highly cytotoxic for myeloma cells both in vitro and in vivo. Stem Cell Res. Ther. 2017, 8, 206. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Miller, C.; Savant-Bhonsale, S.; Kalkanis, S.N. Antitumor treatment using interleukin- 12-secreting marrow stromal cells in an invasive glioma model. Neurosurgery 2009, 64, 1139–1146. [Google Scholar] [CrossRef]
- Zhao, W.H.; Cheng, J.X.; Shi, P.F.; Huang, J.Y. [human umbilical cord mesenchymal stem cells with adenovirus-mediated interleukin 12 gene transduction inhibits the growth of ovarian carcinoma cells both in vitro and in vivo]. Nan Fang Yi Ke Da Xue Xue Bao J. South. Med. Univ. 2011, 31, 903–907. [Google Scholar]
- You, Q.; Yao, Y.; Zhang, Y.; Fu, S.; Du, M.; Zhang, G. Effect of targeted ovarian cancer therapy using amniotic fluid mesenchymal stem cells transfected with enhanced green fluorescent protein-human interleukin-2 in vivo. Mol. Med. Rep. 2015, 12, 4859–4866. [Google Scholar] [CrossRef]
- Kansy, B.A.; Dissmann, P.A.; Hemeda, H.; Bruderek, K.; Westerkamp, A.M.; Jagalski, V.; Schuler, P.; Kansy, K.; Lang, S.; Dumitru, C.A.; et al. The bidirectional tumor--mesenchymal stromal cell interaction promotes the progression of head and neck cancer. Stem Cell Res. Ther. 2014, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.G.; Joyce, C.; et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the il-6/gp130/stat3 pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [Green Version]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Nwabo Kamdje, A.H.; Kamga, P.T.; Simo, R.T.; Vecchio, L.; Seke Etet, P.F.; Muller, J.M.; Bassi, G.; Lukong, E.; Goel, R.K.; Amvene, J.M.; et al. Mesenchymal stromal cells’ role in tumor microenvironment: Involvement of signaling pathways. Cancer Biol. Med. 2017, 14, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Roh, K.H.; Park, J.R.; Lee, S.R.; Park, S.B.; Jung, J.W.; Kang, S.K.; Lee, Y.S.; Kang, K.S. Therapeutic potential of mesenchymal stromal cells in a mouse breast cancer metastasis model. Cytotherapy 2009, 11, 289. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, A.Y.; Pati, S.; Anderson, S.A.; Reid, W.; Elshal, M.F.; Rovira, I.I.; Nguyen, A.T.; Malide, D.; Combs, C.A.; Hall, G.; et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of kaposi’s sarcoma. J. Exp. Med. 2006, 203, 1235–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, C.; Maurya, D.K.; Pyle, M.M.; Troyer, D.; Tamura, M. Cytotherapy with naive rat umbilical cord matrix stem cells significantly attenuates growth of murine pancreatic cancer cells and increases survival in syngeneic mice. Cytotherapy 2010, 12, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Ramdasi, S.; Sarang, S.; Viswanathan, C. Potential of mesenchymal stem cell based application in cancer. Int. J. Hematol. Oncol. Stem Cell Res. 2015, 9, 95–103. [Google Scholar]
- Solovyeva, V.V.; Salafutdinov, I.I.; Tazetdinova, L.G.; Masgutov, R.F.; Khaiboullina, S.F.; Rizvanov, A.A. Genetic modification of adipose derived stem cells with recombinant plasmid DNA pbud-vegf-fgf2 results in increased of il-8 and mcp-1 secretion. J. Pure Appl. Microbiol. 2014, 8, 523–528. [Google Scholar]
- Solovyeva, V.V.; Chulpanova, D.S.; Tazetdinova, L.G.; Salafutdinov, I.I.; Bozo, I.Y.; Isaev, A.A.; Deev, R.V.; Rizvanov, A.A. In vitro angiogenic properties of plasmid DNA encoding sdf-1alpha and vegf165 genes. Appl. Biochem. Biotechnol. 2020, 190, 773–788. [Google Scholar] [CrossRef]
- Islamov, R.R.; Rizvanov, A.A.; Mukhamedyarov, M.A.; Salafutdinov, I.I.; Garanina, E.E.; Fedotova, V.Y.; Solovyeva, V.V.; Mukhamedshina, Y.O.; Safiullov, Z.Z.; Izmailov, A.A.; et al. Symptomatic improvement, increased life-span and sustained cell homing in amyotrophic lateral sclerosis after transplantation of human umbilical cord blood cells genetically modified with adeno-viral vectors expressing a neuro-protective factor and a neural cell adhesion molecule. Curr. Gene Ther. 2015, 15, 266–276. [Google Scholar]
- Kolobynina, K.; Solovyeva, V.; Gomzikova, M.; Tazetdinova, L.; Rizvanov, A. Generation of human adipose-derived stem cell lines with expression of tesc gene. BioNanoSci 2017, 7, 92–96. [Google Scholar] [CrossRef]
- Kitaeva, K.V.; Prudnikov, T.S.; Gomzikova, M.O.; Kletukhina, S.K.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Analysis of the interaction and proliferative activity of adenocarcinoma, peripheral blood mononuclear and mesenchymal stromal cells after co-cultivation in vitro. BioNanoSci 2019, 9, 502–509. [Google Scholar] [CrossRef]
- Gilazieva, Z.E.; Tazetdinova, L.G.; Arkhipova, S.S.; Solovyeva, V.V.; Rizvanov, A.A. Effect of cisplatin on ultrastructure and viability of adipose-derived mesenchymal stem cells. BioNanoSci 2016, 6, 534–539. [Google Scholar] [CrossRef]
- Chekhonin, V.P.; Shein, S.A.; Korchagina, A.A.; Gurina, O.I. Vegf in tumor progression and targeted therapy. Curr. Cancer Drug Targets 2013, 13, 423–443. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Chen, Z.; Sternlicht, M.D.; Hidalgo, M.; Steffensen, B. Matrix metalloproteinase-2 contributes to cancer cell migration on collagen. Cancer Res. 2005, 65, 130–136. [Google Scholar] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Vicious cycle of tgf-beta signaling in tumor progression and metastasis. Am. J. Clin. Exp. Urol. 2014, 2, 149–155. [Google Scholar] [PubMed]
- Amlot, P.L.; Tahami, F.; Chinn, D.; Rawlings, E. Activation antigen expression on human t cells. I. Analysis by two-colour flow cytometry of umbilical cord blood, adult blood and lymphoid tissue. Clin. Exp. Immunol. 1996, 105, 176–182. [Google Scholar] [CrossRef]
- Salgado, F.J.; Lojo, J.; Fernandez-Alonso, C.M.; Vinuela, J.; Cordero, O.J.; Nogueira, M. Interleukin-dependent modulation of hla-dr expression on cd4and cd8 activated t cells. Immunol. Cell Biol. 2002, 80, 138–147. [Google Scholar] [CrossRef]
- Fei, F.; Lim, M.; George, A.A.; Kirzner, J.; Lee, D.; Seeger, R.; Groffen, J.; Abdel-Azim, H.; Heisterkamp, N. Cytotoxicity of cd56-positive lymphocytes against autologous b-cell precursor acute lymphoblastic leukemia cells. Leukemia 2015, 29, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Sandoval-Montes, C.; Santos-Argumedo, L. Cd38 is expressed selectively during the activation of a subset of mature t cells with reduced proliferation but improved potential to produce cytokines. J. Leukoc Biol. 2005, 77, 513–521. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Capsomidis, A.; Smits, E.L.; Van Tendeloo, V.F. Cd56 in the immune system: More than a marker for cytotoxicity? Front. Immunol. 2017, 8, 892. [Google Scholar] [CrossRef]
- Hussain, H.; Maldonado-Agurto, R.; Dickson, A.J. The endoplasmic reticulum and unfolded protein response in the control of mammalian recombinant protein production. Biotechnol. Lett. 2014, 36, 1581–1593. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of bip and er stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Kikuchi, T.; Andarini, S.; Ohkouchi, S.; Suzuki, T.; Nukiwa, T.; Huqun; Hagiwara, K.; Honjo, T.; Saijo, Y. Antitumor immune response by cx3cl1 fractalkine gene transfer depends on both nk and t cells. Eur. J. Immunol. 2005, 35, 1371–1380. [Google Scholar] [CrossRef]
- Mendez-Enriquez, E.; Garcia-Zepeda, E.A. The multiple faces of ccl13 in immunity and inflammation. Inflammopharmacology 2013, 21, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Van Coillie, E.; Van Aelst, I.; Wuyts, A.; Vercauteren, R.; Devos, R.; De Wolf-Peeters, C.; Van Damme, J.; Opdenakker, G. Tumor angiogenesis induced by granulocyte chemotactic protein-2 as a countercurrent principle. Am. J. Pathol. 2001, 159, 1405–1414. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Y.; Wang, S.; Cai, J.; Shi, J.; Sui, X.; Cao, Y.; Huang, W.; Chen, X.; Cai, Z.; et al. Bone marrow-derived mesenchymal stem cell-secreted il-8 promotes the angiogenesis and growth of colorectal cancer. Oncotarget 2015, 6, 42825–42837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Shi, L.; Sun, X.; Wang, L.; Wang, X.; Chen, C. Production of ccl20 from lung cancer cells induces the cell migration and proliferation through pi3k pathway. J. Cell Mol. Med. 2016, 20, 920–929. [Google Scholar] [CrossRef]
- Li, Y.; Yu, H.P.; Zhang, P. Ccl15 overexpression predicts poor prognosis for hepatocellular carcinoma. Hepatol. Int. 2016, 10, 488–492. [Google Scholar] [CrossRef]
- Samaniego, R.; Gutierrez-Gonzalez, A.; Gutierrez-Seijo, A.; Sanchez-Gregorio, S.; Garcia-Gimenez, J.; Mercader, E.; Marquez-Rodas, I.; Aviles, J.A.; Relloso, M.; Sanchez-Mateos, P. Ccl20 expression by tumor-associated macrophages predicts progression of human primary cutaneous melanoma. Cancer Immunol. Res. 2018, 6, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Romieu-Mourez, R.; Francois, M.; Boivin, M.N.; Bouchentouf, M.; Spaner, D.E.; Galipeau, J. Cytokine modulation of tlr expression and activation in mesenchymal stromal cells leads to a proinflammatory phenotype. J. Immunol. 2009, 182, 7963–7973. [Google Scholar] [CrossRef] [PubMed]
- Mi, F.; Gong, L. Secretion of interleukin-6 by bone marrow mesenchymal stem cells promotes metastasis in hepatocellular carcinoma. Biosci. Rep. 2017, 37, BSR20170181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhou, Y.; Li, W.; Zhang, B.; Zhang, H.; Zhao, S.; Zheng, P.; Wu, H.; Yang, J. Tumor-derived mesenchymal-stem-cell-secreted il-6 enhances resistance to cisplatin via the stat3 pathway in breast cancer. Oncol. Lett. 2018, 15, 9142–9150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabellini, C.; Trisciuoglio, D.; Desideri, M.; Candiloro, A.; Ragazzoni, Y.; Orlandi, A.; Zupi, G.; Del Bufalo, D. Functional activity of cxcl8 receptors, cxcr1 and cxcr2, on human malignant melanoma progression. Eur. J. Cancer 2009, 45, 2618–2627. [Google Scholar] [CrossRef] [PubMed]
- Palena, C.; Hamilton, D.H.; Fernando, R.I. Influence of il-8 on the epithelial-mesenchymal transition and the tumor microenvironment. Future Oncol. 2012, 8, 713–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. Il-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Kerner, Z.J.; Hong, H.; Sun, J. Targeted cancer therapy with tumor necrosis factor-alpha. Biochem. Insights 2008, 2008, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Michalaki, V.; Syrigos, K.; Charles, P.; Waxman, J. Serum levels of il-6 and tnf-alpha correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 2004, 90, 2312–2316. [Google Scholar] [CrossRef]
- Ferrajoli, A.; Keating, M.J.; Manshouri, T.; Giles, F.J.; Dey, A.; Estrov, Z.; Koller, C.A.; Kurzrock, R.; Thomas, D.A.; Faderl, S.; et al. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood 2002, 100, 1215–1219. [Google Scholar] [CrossRef]
- Leibovich, S.J.; Polverini, P.J.; Shepard, H.M.; Wiseman, D.M.; Shively, V.; Nuseir, N. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-alpha. Nature 1987, 329, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, N.; Ong, L.L.; Nesselmann, C.; Klopsch, C.; Ladilov, Y.; Furlani, D.; Piechaczek, C.; Moebius, J.M.; Lutzow, K.; et al. Bcl-2 engineered mscs inhibited apoptosis and improved heart function. Stem Cells 2007, 25, 2118–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Han, Y.; Yan, C.; Tian, X.; Tao, J.; Kang, J.; Li, S. Overexpressing cellular repressor of e1a-stimulated genes protects mesenchymal stem cells against hypoxia- and serum deprivation-induced apoptosis by activation of pi3k/akt. Apoptosis Int. J. Program. Cell Death 2010, 15, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Beckermann, B.M.; Kallifatidis, G.; Groth, A.; Frommhold, D.; Apel, A.; Mattern, J.; Salnikov, A.V.; Moldenhauer, G.; Wagner, W.; Diehlmann, A.; et al. Vegf expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br. J. Cancer 2008, 99, 622–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.C.; Zhang, H.W.; Zhao, Q.C.; Sun, L.I.; Yang, J.J.; Hong, L.; Feng, F.; Cai, L. Mesenchymal stem cells promote tumor angiogenesis via the action of transforming growth factor beta1. Oncol. Lett. 2016, 11, 1089–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Wang, S.; Lin, Y.; Miao, Y.; Zeng, Y.; Nie, Y.; Guo, P.; Jiang, G.; Wu, J. Epithelial-mesenchymal transition in cancer: Role of the il-8/il-8r axis. Oncol. Lett. 2017, 13, 4577–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almalki, S.G.; Agrawal, D.K. Effects of matrix metalloproteinases on the fate of mesenchymal stem cells. Stem Cell Res. Ther. 2016, 7, 129. [Google Scholar] [CrossRef] [Green Version]
- Bhoopathi, P.; Chetty, C.; Gogineni, V.R.; Gujrati, M.; Dinh, D.H.; Rao, J.S.; Lakka, S.S. Mmp-2 mediates mesenchymal stem cell tropism towards medulloblastoma tumors. Gene Ther. 2011, 18, 692–701. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Curtis, L.A.; Qiu, Y.; Xu, A.; Janowska-Wieczorek, A. Migration, proliferation, and differentiation of cord blood mesenchymal stromal cells treated with histone deacetylase inhibitor valproic acid. Stem Cells Int. 2014, 2014, 610495. [Google Scholar] [CrossRef]
- Nam, H.S.; Kwon, I.; Lee, B.H.; Kim, H.; Kim, J.; An, S.; Lee, O.H.; Lee, P.H.; Kim, H.O.; Namgoong, H.; et al. Effects of mesenchymal stem cell treatment on the expression of matrix metalloproteinases and angiogenesis during ischemic stroke recovery. PLoS ONE 2015, 10, e0144218. [Google Scholar] [CrossRef] [Green Version]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine tgf-beta/zeb/mir-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef]
- Li, J.; Li, H.; Zhang, Z.; Wang, N.; Zhang, Y. The anti-cancerous activity of recombinant trichosanthin on prostate cancer cell pc3. Biol. Res. 2016, 49, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Zhao, L.; Zhong, J.; Gu, H.; Feng, D.; Johnstone, B.H.; March, K.L.; Farlow, M.R.; Du, Y. Adipose stromal cells-secreted neuroprotective media against neuronal apoptosis. Neurosci. Lett. 2009, 462, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Pires, A.O.; Neves-Carvalho, A.; Sousa, N.; Salgado, A.J. The secretome of bone marrow and wharton jelly derived mesenchymal stem cells induces differentiation and neurite outgrowth in sh-sy5y cells. Stem Cells Int. 2014, 2014, 438352. [Google Scholar] [CrossRef]
- Anguiano, M.; Castilla, C.; Maska, M.; Ederra, C.; Pelaez, R.; Morales, X.; Munoz-Arrieta, G.; Mujika, M.; Kozubek, M.; Munoz-Barrutia, A.; et al. Characterization of three-dimensional cancer cell migration in mixed collagen-matrigel scaffolds using microfluidics and image analysis. PLoS ONE 2017, 12, e0171417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, G.; Kleinman, H.K.; George, J.; Arnaoutova, I. Multiple uses of basement membrane-like matrix (bme/matrigel) in vitro and in vivo with cancer cells. Int. J. Cancer 2011, 128, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Price, K.J.; Tsykin, A.; Giles, K.M.; Sladic, R.T.; Epis, M.R.; Ganss, R.; Goodall, G.J.; Leedman, P.J. Matrigel basement membrane matrix influences expression of micrornas in cancer cell lines. Biochem. Biophys. Res. Commun. 2012, 427, 343–348. [Google Scholar] [CrossRef]
- Janeczek Portalska, K.; Leferink, A.; Groen, N.; Fernandes, H.; Moroni, L.; van Blitterswijk, C.; de Boer, J. Endothelial differentiation of mesenchymal stromal cells. PLoS ONE 2012, 7, e46842. [Google Scholar] [CrossRef] [Green Version]
- Pulliero, A.; Marengo, B.; Domenicotti, C.; Longobardi, M.G.; Fazzi, E.; Orcesi, S.; Bianchi, M.; Balottin, U.; Izzotti, A. Inhibition of neuroblastoma cell growth by trex1-mutated human lymphocytes. Oncol. Rep. 2012, 27, 1689–1694. [Google Scholar]
- Rizvanov, A.A.; Yalvac, M.E.; Shafigullina, A.K.; Salafutdinov, I.I.; Blatt, N.L.; Sahin, F.; Kiyasov, A.P.; Palotas, A. Interaction and self-organization of human mesenchymal stem cells and neuro-blastoma sh-sy5y cells under co-culture conditions: A novel system for modeling cancer cell micro-environment. Eur. J. Pharm. Biopharm. 2010, 76, 253–259. [Google Scholar] [CrossRef]
- Jen, E.Y.; Poindexter, N.J.; Farnsworth, E.S.; Grimm, E.A. Il-2 regulates the expression of the tumor suppressor il-24 in melanoma cells. Melanoma Res. 2012, 22, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchi, M.; Vitolo, D.; Sedlmayr, P.; Rabinowich, H.; Johnson, J.T.; Herberman, R.B.; Whiteside, T.L. Induction of tumor regression in experimental model of human head and neck cancer by human a-lak cells and il-2. Int. J. Cancer 1991, 47, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.E.; Grosse, S.M.; Siapati, E.K.; Kritz, A.; Kinnon, C.; Thrasher, A.J.; Hart, S.L. Immunotherapy for neuroblastoma using syngeneic fibroblasts transfected with il-2 and il-12. Br. J. Cancer 2007, 97, 210–217. [Google Scholar] [CrossRef]
- Ladenstein, R.; Potschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with anti-gd2 antibody ch14.18/cho (dinutuximab beta) in patients with high-risk neuroblastoma (hr-nbl1/siopen): A multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1617–1629. [Google Scholar] [CrossRef]
- Ozkaynak, M.F.; Gilman, A.L.; London, W.B.; Naranjo, A.; Diccianni, M.B.; Tenney, S.C.; Smith, M.; Messer, K.S.; Seeger, R.; Reynolds, C.P.; et al. A comprehensive safety trial of chimeric antibody 14.18 with gm-csf, il-2, and isotretinoin in high-risk neuroblastoma patients following myeloablative therapy: Children’s oncology group study anbl0931. Front. Immunol. 2018, 9, 1355. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of il-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, D.P.; Jacinto, A.; Borralho, P.; Braga, S.; Cabral, M.G. Hla-dr in cytotoxic t lymphocytes predicts breast cancer patients’ response to neoadjuvant chemotherapy. Front. Immunol. 2018, 9, 2605. [Google Scholar] [CrossRef] [Green Version]
- Neubert, N.J.; Soneson, C.; Barras, D.; Baumgaertner, P.; Rimoldi, D.; Delorenzi, M.; Fuertes Marraco, S.A.; Speiser, D.E. A well-controlled experimental system to study interactions of cytotoxic t lymphocytes with tumor cells. Front. Immunol. 2016, 7, 326. [Google Scholar] [CrossRef] [Green Version]
- Sereti, I.; Gea-Banacloche, J.; Kan, M.Y.; Hallahan, C.W.; Lane, H.C. Interleukin 2 leads to dose-dependent expression of the alpha chain of the il-2 receptor on cd25-negative t lymphocytes in the absence of exogenous antigenic stimulation. Clin. Immunol. 2000, 97, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. Nk cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef]
- Basse, P.H.; Whiteside, T.L.; Herberman, R.B. Cancer immunotherapy with interleukin-2-activated natural killer cells. Mol. Biotechnol. 2002, 21, 161–170. [Google Scholar] [CrossRef]
- Dubois, S.; Conlon, K.C.; Muller, J.R.; Hsu-Albert, J.; Beltran, N.; Bryant, B.R.; Waldmann, T.A. Il15 infusion of cancer patients expands the subpopulation of cytotoxic cd56(bright) nk cells and increases nk-cell cytokine release capabilities. Cancer Immunol. Res. 2017, 5, 929–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundgren, S.; Elebro, J.; Heby, M.; Nodin, B.; Eberhard, J.; Jirstrom, K. Prognostic significance of tumor infiltrating natural killer cells in periampullary adenocarcinoma. J. Clin. Oncol. 2016, 34, 267. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Takeuchi, H.; Maehara, Y.; Tokunaga, E.; Koga, T.; Kakeji, Y.; Sugimachi, K. Prognostic significance of natural killer cell activity in patients with gastric carcinoma: A multivariate analysis. Am. J. Gastroenterol. 2001, 96, 574–578. [Google Scholar] [CrossRef]
- Levi, I.; Amsalem, H.; Nissan, A.; Darash-Yahana, M.; Peretz, T.; Mandelboim, O.; Rachmilewitz, J. Characterization of tumor infiltrating natural killer cell subset. Oncotarget 2015, 6, 13835–13843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 2013, 15, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Euhus, D.M.; Kimura, L.; Arnold, B. Expansion of cd3+cd56+ lymphocytes correlates with induction of cytotoxicity by interleukin-2 gene transfer in human breast tumor cultures. Ann. Surg. Oncol. 1997, 4, 432–439. [Google Scholar] [CrossRef]
- Engel, B.C.; Laws, H.J.; Buttlies, B.; Kahn, T.; Gobel, U.; Burdach, S.E. Induction of a cd3+/cd56+ lymphocyte population following gene therapy with transgenic il-2 secreting fibroblasts in a child with peripheral neuroectodermal malignancy. Med. Pediatr. Oncol. 1998, 31, 56–60. [Google Scholar] [CrossRef]
- Pan, K.; Wang, Q.J.; Liu, Q.; Zheng, H.X.; Li, Y.Q.; Weng, D.S.; Li, J.J.; Huang, L.X.; He, J.; Chen, S.P.; et al. The phenotype of ex vivo generated cytokine-induced killer cells is associated with overall survival in patients with cancer. Tumour Biol. 2014, 35, 701–707. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer (5′−3′) | Reverse Primer (5′−3′) | TaqMan Probe (5′−3′) |
|---|---|---|---|
| 18S rRNA | GCCGCTAGAGGTGAAATTCTTG | CATTCTTGGCAAATGCTTTCG | [HEX] ACCGGCGCAAGACGGACCAG [BH2] |
| IL2 | CACCAGGATGCTCACATTTAAG | GTCCCTGGGTCTTAAGTGAAAG | [FAM] CCCAAGAAGGCCACAGAACTGAAACA [BH1] |
| VEGF | ATCACCATGCAGATTATGGC | TGCATTCACATTTGTTGTGC | [FAM] TCAAACCTCACCAAGGCCAGCA [BH1] |
| MMP2 | ACCCATTTACACCTACACCAAG | TGTTTGCAGATCTCAGGGTC | [FAM] TCAATGTCAGGAGGCCCCATAGA [BH1] |
| TGF-β1 | GCCTTTCCTGCTTCTCATGG | TCCTTGCGGAAGTCAATGTAC | [FAM] CCGACCCTGGACACCAACTAT [BH1] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chulpanova, D.S.; Solovyeva, V.V.; James, V.; Arkhipova, S.S.; Gomzikova, M.O.; Garanina, E.E.; Akhmetzyanova, E.R.; Tazetdinova, L.G.; Khaiboullina, S.F.; Rizvanov, A.A. Human Mesenchymal Stem Cells Overexpressing Interleukin 2 Can Suppress Proliferation of Neuroblastoma Cells in Co-Culture and Activate Mononuclear Cells In Vitro. Bioengineering 2020, 7, 59. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020059
Chulpanova DS, Solovyeva VV, James V, Arkhipova SS, Gomzikova MO, Garanina EE, Akhmetzyanova ER, Tazetdinova LG, Khaiboullina SF, Rizvanov AA. Human Mesenchymal Stem Cells Overexpressing Interleukin 2 Can Suppress Proliferation of Neuroblastoma Cells in Co-Culture and Activate Mononuclear Cells In Vitro. Bioengineering. 2020; 7(2):59. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020059
Chicago/Turabian StyleChulpanova, Daria S., Valeriya V. Solovyeva, Victoria James, Svetlana S. Arkhipova, Marina O. Gomzikova, Ekaterina E. Garanina, Elvira R. Akhmetzyanova, Leysan G. Tazetdinova, Svetlana F. Khaiboullina, and Albert A. Rizvanov. 2020. "Human Mesenchymal Stem Cells Overexpressing Interleukin 2 Can Suppress Proliferation of Neuroblastoma Cells in Co-Culture and Activate Mononuclear Cells In Vitro" Bioengineering 7, no. 2: 59. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020059