
Integrated Process Model Applications Linking Bioprocess Development to Quality by Design Milestones

 ,
, 
Abstract
:1. Introduction
- Risk assessment, often in the form of a failure mode and effects analysis (FMEA), which are conducted by process experts in order to assess relationships between potential critical process parameters (pCPPs) and critical quality attributes (CQAs), which then are iteratively reinforced by subject matter expertise and experimental results;
- Preliminary control strategy establishment for the CPPs by determining the CPPs’ proven acceptable range (PAR) for all CQAs.
1.1. Risk Assessment Severity
1.2. Preliminary Control Strategy
- For each CPP, there exists a worst-case condition for each response. The worst case for CQA 1 might not be the worst case for CQA 2.
- The worst case is not the most likely condition. Processes are usually performed at set point conditions and the normal operating ranges represent the uncontrollable variation, meaning that the most probable condition is the set point.
- For the uncertainty of the model predictions, the lower or upper 95% confidence interval is used as a worst case. This does not take into account that the most likely prediction is the model mean at set point.
- The models are only based on small-scale data and manufacturing data is not considered.
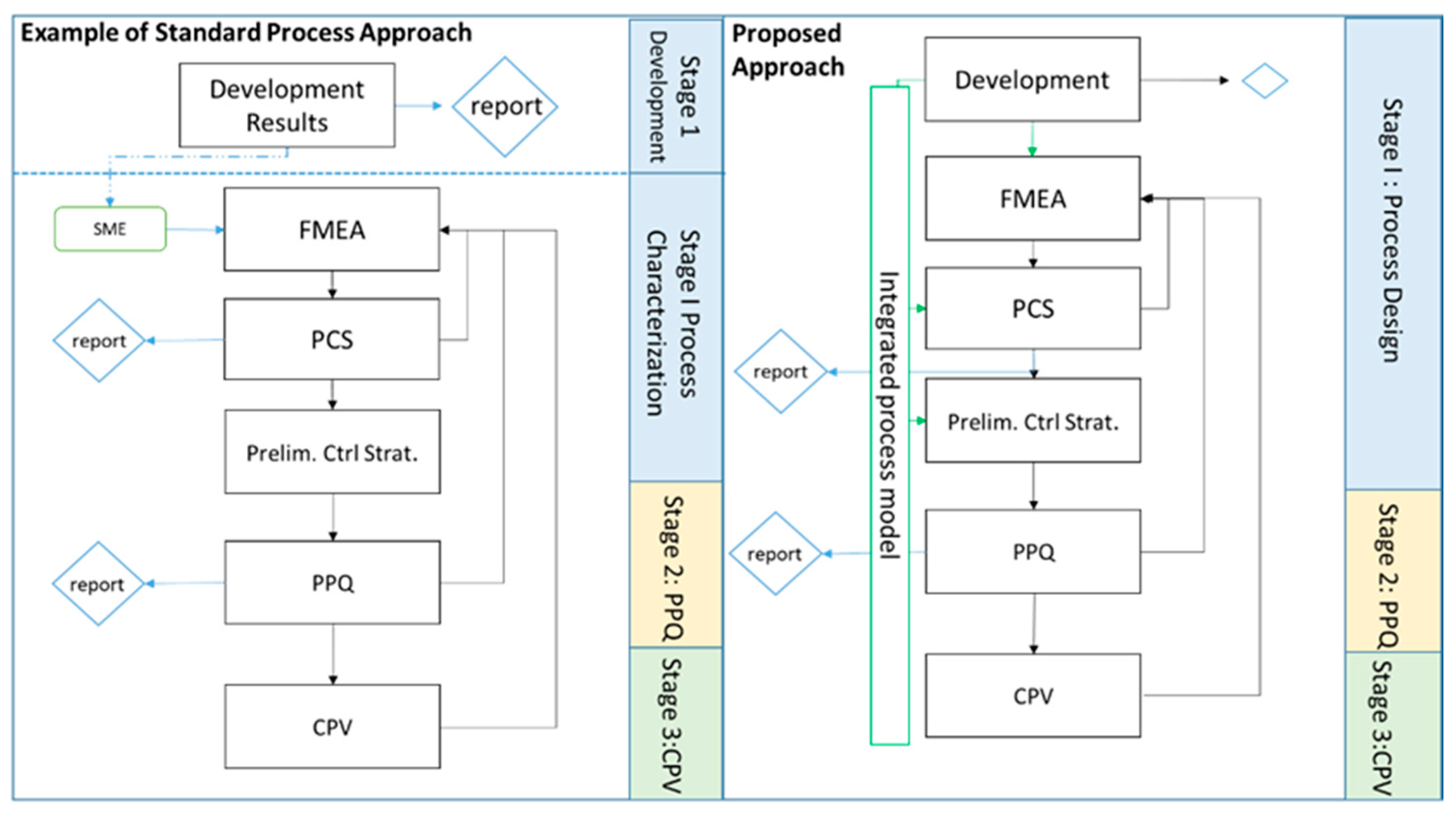
- Create an IPM by concatenating development generated statistical models, thereby establishing an in silico version of the process [21].
- Assess risk assessment severity rankings by application of an IPM parameter sensitivity analysis and rank linearization algorithm that quantifies the behavior of each parameter with regard to its OOA probability, and compares this against a predefined critical OOA rate, assigning an FMEA severity ranking based on the impact at drug substance.
- Propose PAR limits per CPP and CQA by detecting increases in simulated drug substance OOA results across the CPP screening range and assigning a cut-off representative of a predefined acceptable OOA probability.
2. Materials and Methods
2.1. Candidate Process for Case Study
- Eight downstream unit operations, consisting of a capture step (CAP), an acid treatment step (AT), an anion exchange chromatography step (AEX), and a cation exchange chromatography step (CEX), for which process development activities were carried-out. Additional data exists at set point for the following unit operations: depth filtration (DF), ultra-diafiltration (UFDF), viral filtration (VF), and the resulting drug substance (DS).
- DoE-based ordinary least square models, which were fitted, discussed, and selected with subject matter experts before inclusion in the IPM. The experiments were carried out in small-scale systems representative of the manufacturing scale. Subject matter experts evaluated the suitability of these systems prior to experimentation. Model variable selection was based on a standard procedure of selecting the model with lowest Akaike information criterion, and the following diagnostics were then assessed for model significance: R2adj, Q2, RMSE, and partial p-values, as well as the model residuals. All selected models were then discussed with the process experts for process plausibility before acceptance. Acceptable regression models were found for the unit operations CAP, AT, AEX, and CEX.
- Manufacturing data for specific clearance models and yield/clearance calculations as required for the unit operation linkage described elsewhere for all unit operations from two industrial scales [21]:
- ○
- Two manufacturing-scale runs;
- ○
- Three pilot-scale runs.
- Four CQAs typical of monoclonal antibody products, depicting three impurities (CQA1imp, CQA2imp, CQA3imp) and one desired product attribute (CQA1prod):
- ○
- Each of the above CQAs has an acceptance limit at drug substance in place.
2.2. Data for the Integrated Process Model
2.3. Parameter Sensitivity Analysis
3. Results
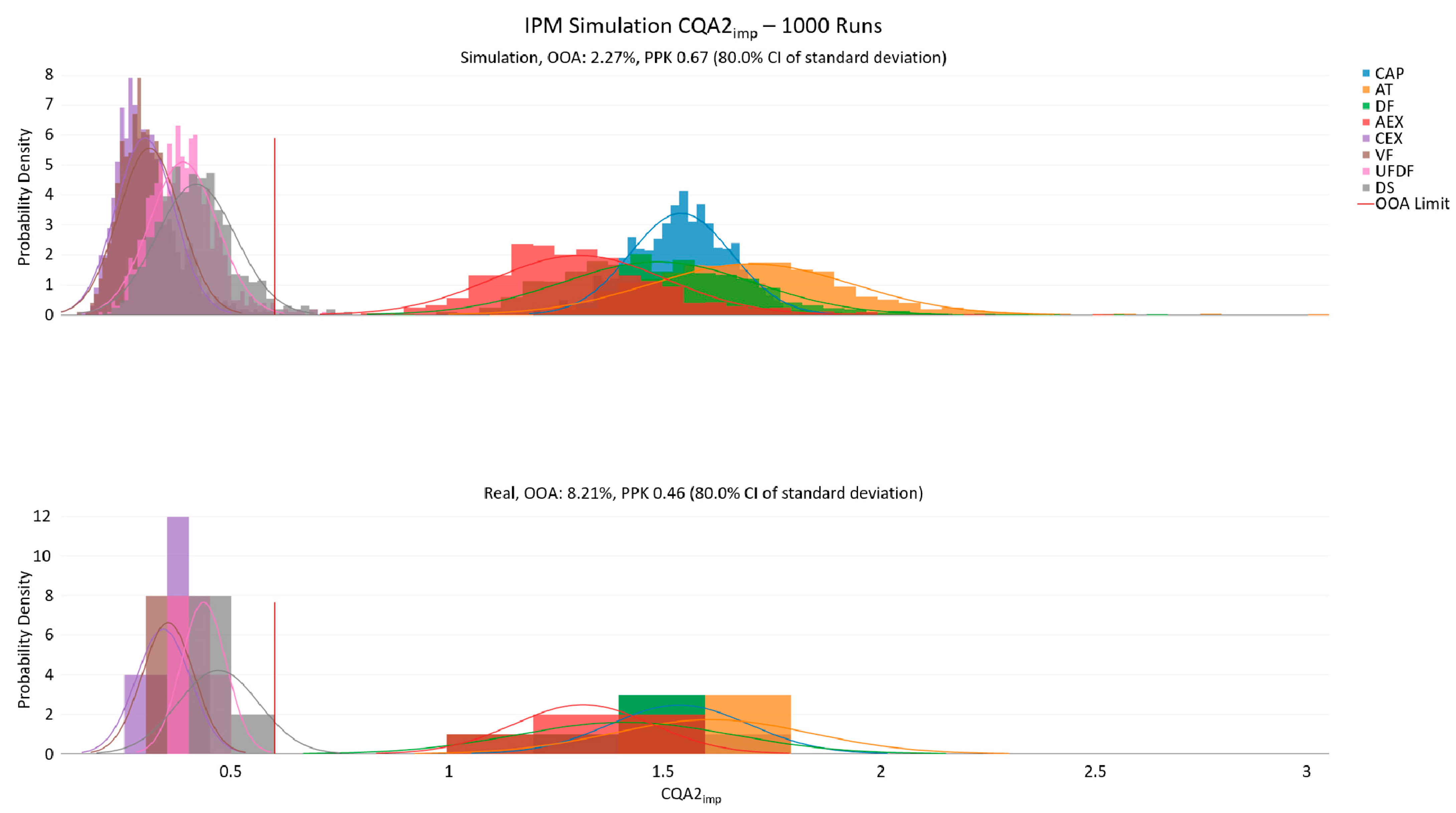
3.1. IPM Plausibility Check
3.2. Automated Generation of FMEA Severity Rankings
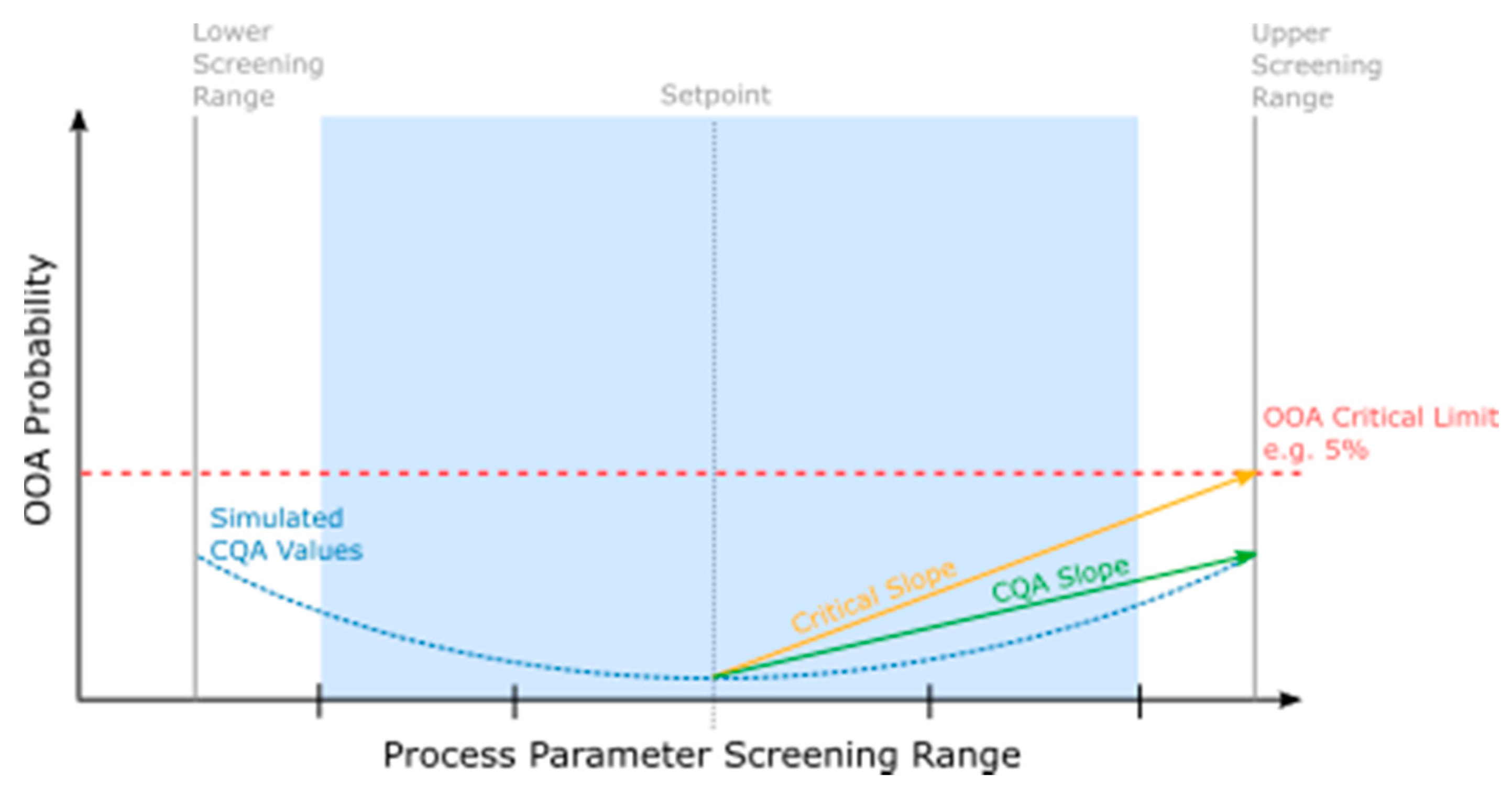
3.2.1. Critical Effect Slope Determination
3.2.2. CQA Slope Determination
3.2.3. FMEA Ranking Algorithm
3.2.4. Preliminary Control Strategy Setting Procedure
4. Discussion
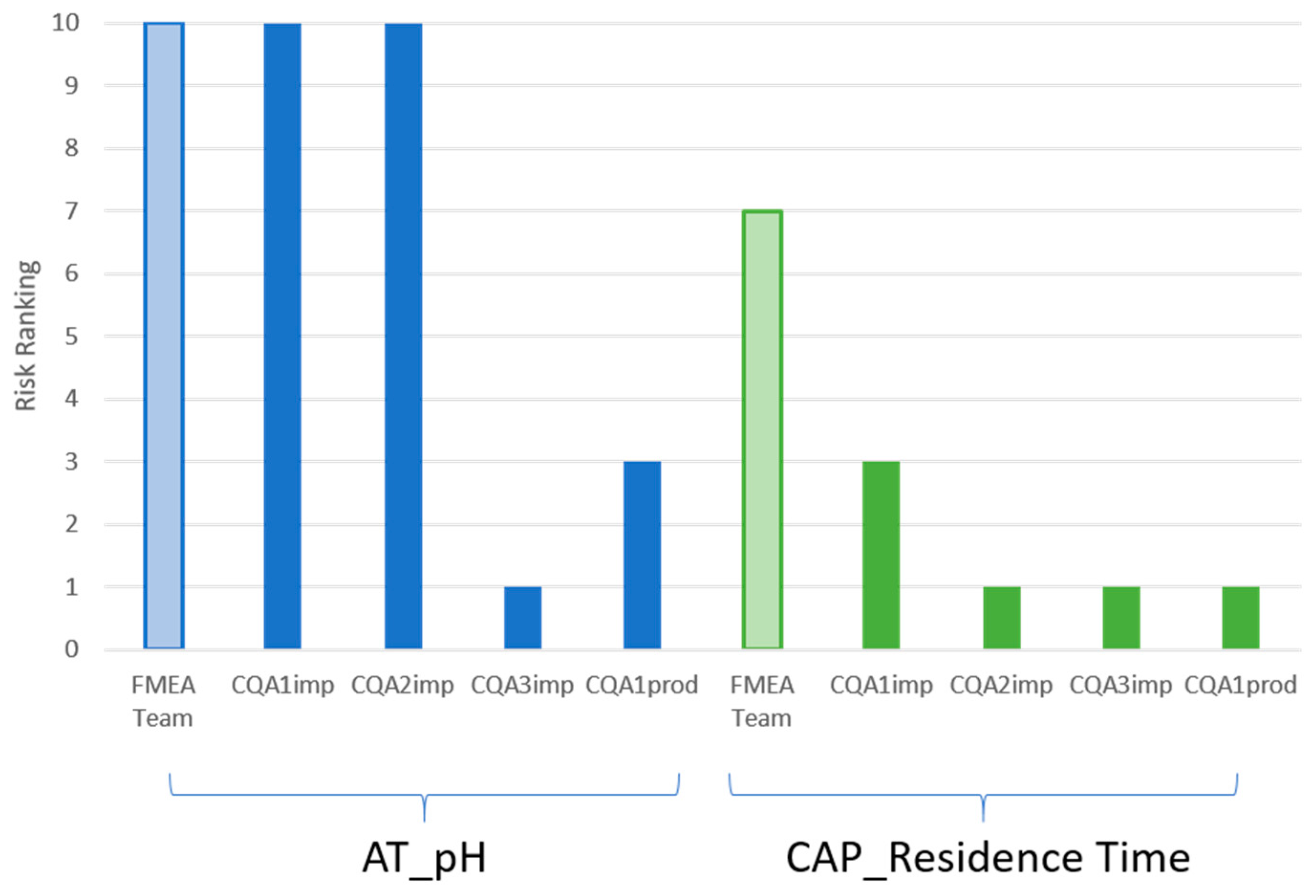
4.1. Automated FMEA Severity Ranking Linearization
- −
- The overall FMEA severity ranking;
- −
- The CQA(s) most likely to be impacted by the pCPP.
4.2. PSA Established Control Strategy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FDA Process Validation: General Principles and Practices Guidance for Industry; FDA, January 2011. Available online: https://www.fda.gov/media/71021/download (accessed on 10 August 2021).
- Lim, L.P.L.; Garnsey, E.; Gregory, M. Product and process innovation in biopharmaceuticals: A new perspective on development. R&D Manag. 2006, 36, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, H.; Luna, M.F.; Von Stosch, M.; Bournazou, M.N.C.; Polotti, G.; Morbidelli, M.; Butté, A.; Sokolov, M. Bioprocessing in the Digital Age: The Role of Process Models. Biotechnol. J. 2019, 15, e1900172. [Google Scholar] [CrossRef] [PubMed]
- Bruynseels, K.; De Sio, F.S.; Hoven, J.V.D. Digital Twins in Health Care: Ethical Implications of an Emerging Engineering Paradigm. Front. Genet. 2018, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Borchert, D.; Zahel, T.; Thomassen, Y.E.; Herwig, C.; Suarez-Zuluaga, D.A. Quantitative CPP Evaluation from Risk Assessment Using Integrated Process Modeling. Bioengineering 2019, 6, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-C.; Liu, L.; Liu, N. Risk evaluation approaches in failure mode and effects analysis: A literature review. Expert Syst. Appl. 2013, 40, 828–838. [Google Scholar] [CrossRef]
- Sharma, K.D.; Srivastava, S. Failure Mode and Effect Analysis (FMEA) Implementation: A Literature Review. J. Adv. Res. Aero Space Sci. 2018, 5, 1–17. [Google Scholar]
- Franceschini, F.; Galetto, M. A new approach for evaluation of risk priorities of failure modes in FMEA. Int. J. Prod. Res. 2001, 39, 2991–3002. [Google Scholar] [CrossRef] [Green Version]
- Shahin, A. Integration of FMEA and the Kano modell: An Exploratory Examination. Int. J. Qual. Reliab. Manag. 2004, 21, 731–746. [Google Scholar] [CrossRef]
- Liu, H.-C.; Wang, L.-E.; Li, Z.; Hu, Y.-P. Improving Risk Evaluation in FMEA With Cloud Model and Hierarchical TOPSIS Method. IEEE Trans. Fuzzy Syst. 2018, 27, 84–95. [Google Scholar] [CrossRef]
- ICH. Q8 (R2) Pharmaceutical Development Q8 (R2)—Step 4; The International Conference on Harmonisation: Geneva, Switzerland, 2009. [Google Scholar]
- EMA/213746/2017 EMA-FDA Questions and Answers: Improving the Understanding of NORs, PARs, DSp and Normal Variability of Process Parameters. Questions and Answers 4. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-improving-understanding-normal-operating-range-nor-proven-acceptable-range-par_en.pdf (accessed on 10 August 2021).
- Djuris, J.; Djuric, Z. Modeling in the quality by design environment: Regulatory requirements and recommendations for design space and control strategy appointment. Int. J. Pharm. 2017, 533, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ide, N.D.; Dirat, O.; Subashi, A.K.; Thomson, N.; Vukovinsky, K.; Watson, T.J. Statistical Tools to Aid in the Assessment of Critical Process Parameters. Pharm. Technol. 2016, 40, 36–44. [Google Scholar]
- Glodek, M.; Liebowitz, S.; Squibb, B.-M.; McCarthy, R.; Sundararajan, M.; Vorkapich, R.; Healthcare, B.; Watts, C.; Millili, G. Process Robustness—A PQRI White Paper. Pharm. Eng. 2006, 26, 11. [Google Scholar]
- Abu-Absi, S.F.; Yang, L.; Thompson, P.; Jiang, C.; Kandula, S.; Schilling, B.; Shukla, A.A. Defining process design space for monoclonal antibody cell culture. Biotechnol. Bioeng. 2010, 106, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Vukovinksy, K.E.; Li, F.; Hertz, D. Estimating Process Capability in Development & Low-Volume Manufacturing. Available online: https://ispe.org/pharmaceutical-engineering/january-february-2017/estimating-process-capability-development-low (accessed on 16 November 2020).
- Eon-Duval, A.; Valax, P.; Solacroup, T.; Broly, H.; Gleixner, R.; Le Strat, C.; Sutter, J. Application of the quality by design approach to the drug substance manufacturing process of an Fc fusion protein: Towards a global multi-step design space. J. Pharm. Sci. 2012, 101, 3604–3618. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.J.; Lief, K. The ICH Q8 Definition of Design Space: A Comparison of the Overlapping Means and the Bayesian Predictive Approaches. Stat. Biopharm. Res. 2010, 2, 249–259. [Google Scholar] [CrossRef]
- Burdick, R.; Coffey, T.; Gutka, H.; Gratzl, G.; Conlon, H.D.; Huang, C.-T.; Boyne, M.; Kuehne, H. Statistical Approaches to Assess Biosimilarity from Analytical Data. AAPS J. 2016, 19, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Zahel, T.; Hauer, S.; Mueller, E.M.; Murphy, P.; Abad, S.; Vasilieva, E.; Maurer, D.; Brocard, C.; Reinisch, D.; Sagmeister, P.; et al. Integrated Process Modeling—A Process Validation Life Cycle Companion. Bioengineering 2017, 4, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit Operation | CQA1imp | CQA2imp | CQA3imp | CQA1product |
|---|---|---|---|---|
| CAP | DoE | DoE | DoE | |
| AT | Both | DoE | DoE | DoE |
| DF | SC Model | |||
| AEX | DoE | Both | Both | DoE |
| CEX | Both | DoE | DoE | DoE |
| VF | ||||
| UFDF | SC Model | SC Model |
| % Reference | FMEA Ranking |
|---|---|
| ≥0.8 | 10 |
| 0.5–0.8 | 7 |
| 0.3–0.5 | 3 |
| <0.3 | 1 |
| Unit Operation | Process Parameter | Proposed Control Range (If Any) | Justification of PAR |
|---|---|---|---|
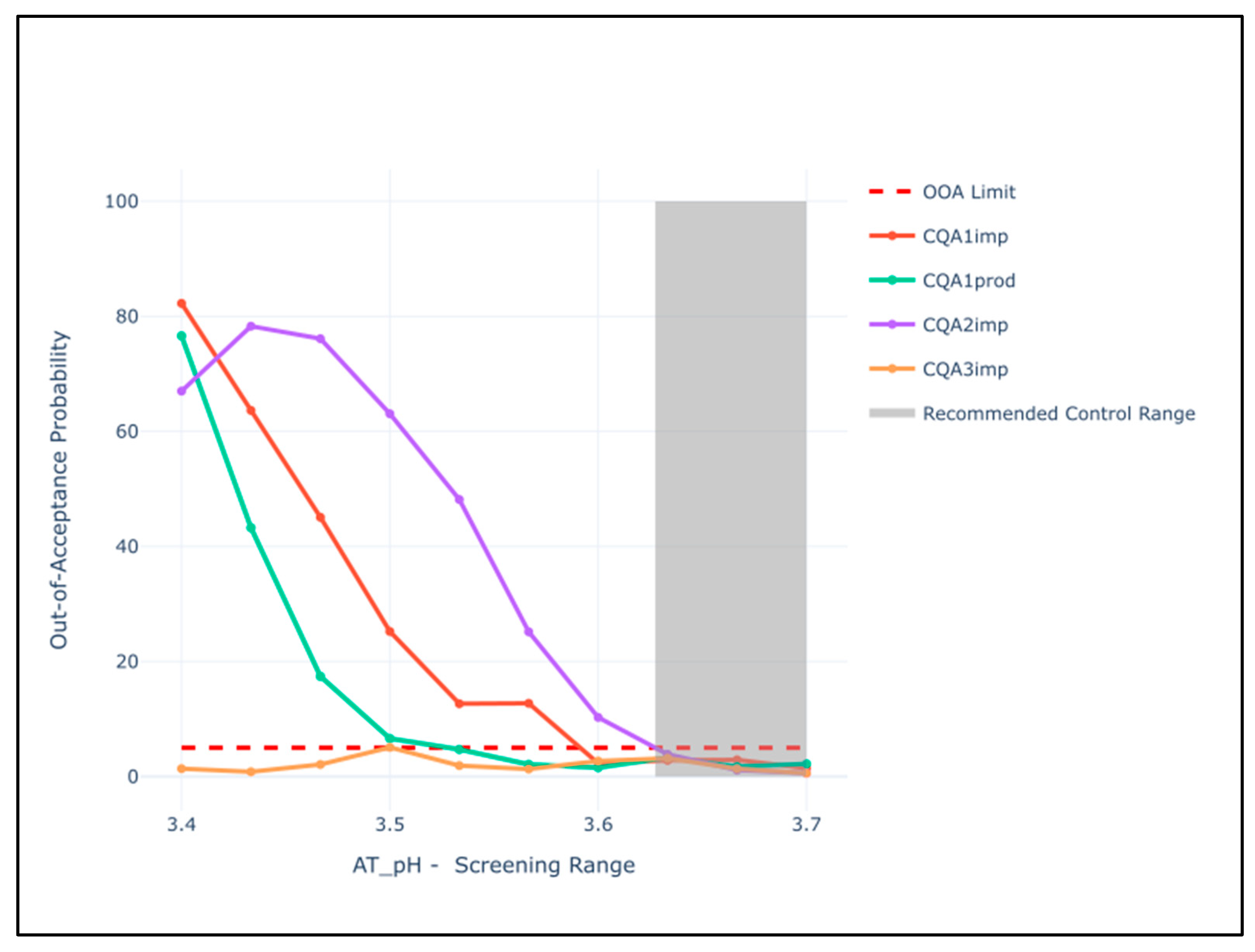
| AT | pH AT | Restriction Low | CQA4prod, CQA1imp, CQA3imp |
| CEX | Conductivity | Restriction High | CQA1imp, CQA2imp, CQA3imp |
| Load Density | Restriction High | CQA1imp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, C.; Marschall, L.; Kunzelmann, M.; Richter, M.; Rudolph, F.; Vajda, J.; Presser, B.; Zahel, T.; Studts, J.; Herwig, C. Integrated Process Model Applications Linking Bioprocess Development to Quality by Design Milestones. Bioengineering 2021, 8, 156. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8110156
Taylor C, Marschall L, Kunzelmann M, Richter M, Rudolph F, Vajda J, Presser B, Zahel T, Studts J, Herwig C. Integrated Process Model Applications Linking Bioprocess Development to Quality by Design Milestones. Bioengineering. 2021; 8(11):156. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8110156
Chicago/Turabian StyleTaylor, Christopher, Lukas Marschall, Marco Kunzelmann, Michael Richter, Frederik Rudolph, Judith Vajda, Beate Presser, Thomas Zahel, Joey Studts, and Christoph Herwig. 2021. "Integrated Process Model Applications Linking Bioprocess Development to Quality by Design Milestones" Bioengineering 8, no. 11: 156. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8110156